INTRODUCTION
Pancreatic cancer is the fourth leading cause of adult cancer related death in USA with five-year survival rates at < 6%. In 2015, an estimated 40,560 Americans (20,710 men and 19,850 women) will die of the disease coupled with an additional 48,960 new cases (24,840 men and 24,120 women) [1]. By 2030, the disease is predicted to be the second leading cause of cancer related deaths [2]. Despite advances in molecular pathogenesis, pancreatic cancer remains a major unsolved health problem [3]. It is a rapidly invasive, metastatic tumor which is resistant to standard therapies [4]. At present, single agent based chemotherapy (e.g. Gemcitabine) is the mainstay treatment for metastatic adenocarcinoma of pancreas, but the tumor response rate is below 10%. Similarly none of the other current chemotherapeutic agents have an objective response rate of over 10% [3, 4]. The magnitude of this problem mandates the need for novel therapeutic agents.
Quinomycin (also called Echinomycin, Figure 1) is a quinoxaline antibiotic that was originally isolated from Streptomyces echinatus [5]. Several studies have shown that it has antitumor activity with the ability to bifunctionally intercalate with double stranded DNA [5]. Quinomycin-induced apoptosis in HT-29 cells occurs via NF-κB activation by modulating IL-8 chemokine expression [6, 7]. In a mouse model of relapsed AML, low dose Quinomycin selectively targets leukemia-initiating cells and spares normal hematopoiesis [8]. Likewise, Quinomycin can be used to treat relapsed AML without affecting host normal hematopoietic stem cells. Moreover, National Cancer Institute sponsored phase II clinical trials has demonstrated anti-tumor efficacy of Quinomycin using various treatment schedules for various cancer types [9–19]. In addition, Quinomycin was shown to suppress leukemia cell growth in association with reduced Notch1 expression [20]. However, none of these studies were performed in pancreatic cancer patients.
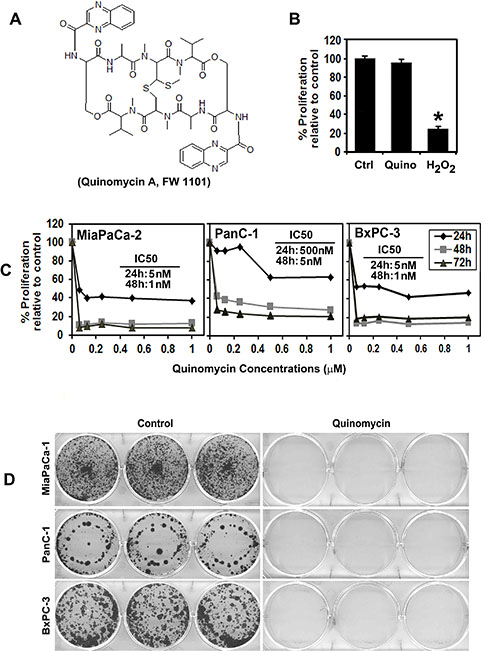
Figure 1: Quinomycin inhibits pancreatic cancer cell proliferation. (A) Chemical structure of Quinomycin. (B) Proliferation of pancreatic ductal epithelial cells is not affected by 50 nM Quinomycin treatment for 48 h. (C) Quinomycin inhibits proliferation of pancreatic cancer cells. Cells were incubated with increasing doses of Quinomycin (0–1 μM) for up to 72 h and analyzed for cell proliferation. Quinomycin treatment resulted in a significant dose and time-dependent decrease in cell proliferation in all three cell lines when compared with untreated controls. (D) Quinomycin inhibits colony formation. Pancreatic cancer cells were incubated with 5 nM Quinomycin for 48 h and allowed to grow into colonies for 10 d. Incubation with Quinomycin inhibits colony formation. Results are representative of three independent experiments.
Notch signaling plays a fundamental role in the differentiation and maintenance of stem cells. Aberrant activation of the Notch signaling has been associated with the development of many cancers, including pancreatic cancers [21, 22]. In fact, Notch signaling has been shown to play a contributing role in the development of pancreatic cancer [23, 24]. Furthermore, the pathway is deemed to be important in maintaining the cancer stem cell population in pancreatic cancer [25]. Interaction of Jagged-1 or Jagged-2 with the Notch-1 receptor promotes a γ-secretase-dependent cleavage of the receptor and release of the Notch intracellular domain (NICD), which translocates to the nucleus and activates transcription of Notch target genes such as Hes-1 and Hey1 [24]. Increased expression of Notch genes and their ligands has been detected in human pancreatic cancer tissues [24]. Overexpression of NICD accelerates the formation of oncogenic K-Ras-induced PanIN lesions [26]. Oral administration of γ-secretase inhibitor in mice blocks the progression of PanIN to ductal adenocarcinoma [27]. γ-secretase is a multiprotein intramembrane-cleaving protease with a growing list of protein substrates, including the Notch receptors. The four components of γ-secretase complex, Presenilin, Nicastrin, Pen2, and Aph1 are all thought to be essential for activity [24]. The catalytic domain resides within presenilin; nicastrin has been suggested to be critical for substrate recognition.
CSCs are the cells within a tumor that exclusively have self-renewal capacities, can give rise to all cancer cell lineages within a tumor, and are exclusively tumorigenic in vivo. They are able of undergoing asymmetric/symmetric cell division, can maintain and expand themselves and also have a distinct profile of surface marker expression that has been linked to poor prognosis [28]. Intriguingly, it has been shown that CSCs are highly resistant to standard therapy [29, 30]. CD44+CD24+EpCAM+ [31], CD133+ [32], ALDH+ are markers for prospectively identifying pancreatic cancer stem cells [33]. We have demonstrated that doublecortin and CaM kinase-like-1 (DCLK1) is an intestinal stem cell marker that is expressed in colon adenocarcinoma [34] and also in pancreatic adenocarcinoma [35]. Recent studies also demonstrated that DCLK1 distinguishes between tumor and normal stem cells in the intestine and could be a therapeutic target for colon cancer [36, 37]. Most recently, DCLK1 has been shown to mark a morphologically distinct subpopulation of cells with stem cell properties in pre-invasive pancreatic cancer [38]. In addition, DCLK1 expression was observed to be occur in early stage pancreatic cancer and in both early and late pancreatic intraepithelial neoplasia (PanIN) and that it increases as disease progresses in genetically engineered mouse models and also in human pancreatic cancer [26]. In this article, we have determined the effect of Quinomycin on pancreatic cancer stem cells and the Notch signaling pathway.
RESULTS
Quinomycin inhibits pancreatic cancer cell proliferation
Previous studies have shown that Quinomycin has antitumor activity with an ability to bifunctionally intercalate with double stranded DNA [5]. We first determined the effect of Quinomycin on proliferation of three pancreatic cancer cell lines, MiaPaCa-2, BxPC-3 and PanC-1 (Figure 1C). Quinomycin significantly suppressed the proliferation of these pancreatic cancer cells in a dose and time dependent manner. This anti-proliferation effect on tumor cells was seen within 24 h at a dose of 5 nM, which continued to significantly increase over the next 72 h (Figure 1C). In contrast, Quinomycin did not affect the proliferation of normal human pancreatic ductal epithelial cells (HPNE) even when treated at 50 nM (Figure 1B). As a positive control for cell death, we used hydrogen peroxide. These data suggest that Quinomycin is not toxic to normal cells. To determine the long-term effect of Quinomycin treatment, cells were treated with 5 nM Quinomycin for 48 h, following which the cells were allowed to grow in normal medium. Quinomycin treatment suppressed colony formation in all pancreatic cancer cell lines (Figure 1D), suggesting that Quinomycin-mediated effects on the tumor cells were irreversible.
Quinomycin treatment induces PreG0/G1 arrest and apoptosis
Given that Quinomycin inhibits proliferation and colony formation, we next determined whether Quinomycin affects cell cycle progression. Treatment with Quinomycin significantly induced Pre-G0/G1 arrest in both MiaPaCa-2 and PanC-1 cells (Figure 2A). Caspase-3 is key effector proteins in the apoptosis pathway [40]. Western blot analyses of MiaPaCa-2 and PanC-1 cell lysates showed a significant increase in activated caspase-3 in cells treated with 5 nM Quinomycin (Figure 2C). In addition, 5 nM Quinomycin inhibited the expression of cyclin D1 and c-Myc (Figure 2C). These data suggest that even at a dose of 5 nM, Quinomycin is a potent inducer of apoptosis of pancreatic cancer cells.
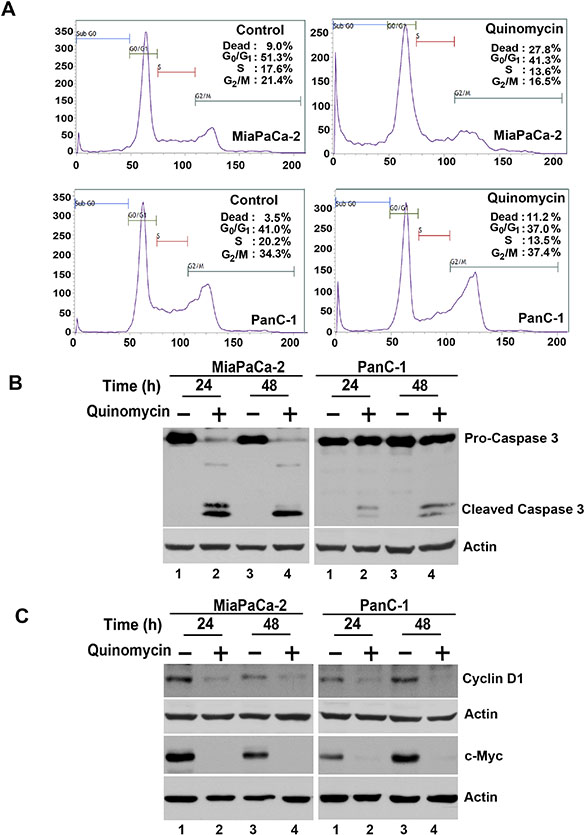
Figure 2: Quinomycin induces cancer cell apoptosis. (A) Cell cycle analysis of Quinomycin treated cells. MiaPaCa-2 or PanC-1 cells were treated with up to 5 nM Quinomycin for 24 h and examined by flow cytometry following propidium iodide staining for DNA content. Quinomycin treatment leads to increased number of cells in the PreG0/G1 arrest. Graphs are representative of data collected from three experiments. (B) Quinomycin induces caspase 3, an apoptosis mediator. Lysates from MiaPaCa-2 or PanC-1 cells incubated with 5 nM Quinomycin were analyzed by western blotting for caspase 3 protein levels using rabbit anti-caspase 3 antibody. Quinomycin treated cells shows cleaved (activated) caspase 3 while untreated cells have no cleaved caspase-3. (C) Lysates from MiaPaCa-2 or PanC-1 cells incubated with 5 nM Quinomycin were analyzed by western blotting for cyclin D1 and c-Myc proteins. Both cyclin D1 and c-myc were reduced following Quinomycin treatment.
Quinomycin inhibits pancosphere formation and expression of cancer stem cell marker proteins
Defining the mechanisms that regulate stem cell fate is critical to increasing our understanding of the neoplastic process. Cancer stem cells are capable of self-renewal and generating tumors resembling the primary tumor [41]. Accordingly, we next determined the effect of Quinomycin on this population of cells. First, we determined the effect of Quinomycin on the ability to form pancreatosphere, a multicellular spheroid that is dependent on presence of stem cells to develop. Quinomycin treatment significantly inhibited PanC-1 pancreatosphere formation (Figure 3A and 3B). In addition, Quinomycin treatment further reduced secondary pancreatosphere (Figure 3B right panel). Flow cytometric analyses showed a significant decrease in DCLK1+ in both MiaPaCa-2 and PanC-1 cells following Quinomycin treatment (Figure 3C). Furthermore, Quinomycin treatment significantly inhibited expression of CSC markers DCLK1, CD44, CD24 and EPCAM in both MiaPaCa-2 and PanC-1 cells (Figure 3D). These data suggest that Quinomycin treatment affects pancreatosphere formation and CSC marker expression and especially DCLK1+ cells.
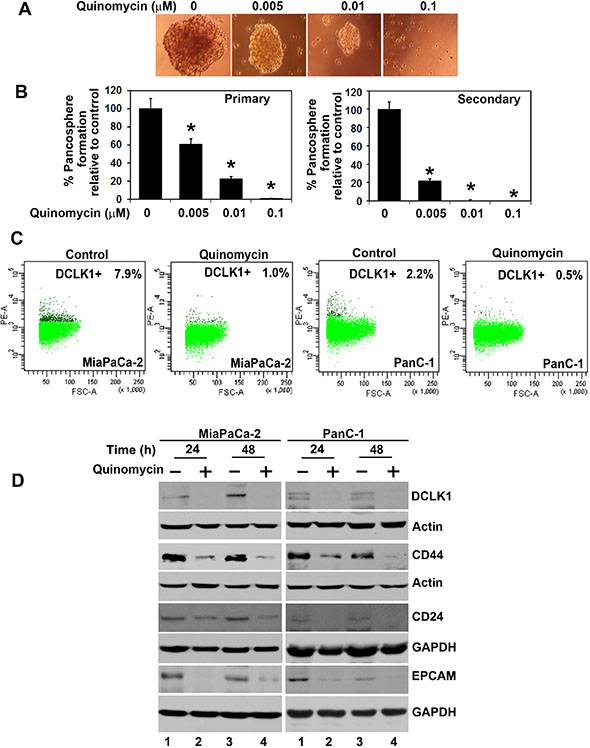
Figure 3: Quinomycin affects cancer stem cell marker expression. (A and B) PanC-1 cells were grown in specific spheroid media in low adherent plates and treated with increasing concentrations of Quinomycin. After 5 days, the pancreatosphere were photographed and counted. The primary spheroids were collected and separated into single cells and replated. The Quinomycin treatment significantly was inhibited in both primary and secondary pancreatospheres formation (right and left panel)(*p < 0.05). (C) Sorting of anti-DCLK1 antibody -tagged phycoerythrin untreated MiaPaCa-2 and PanC-1 cells by flow cytometry. After 24 h, Quinomycin treatment caused significant reduction in the number of DCLK1 expressing cells. (D) Western blot analyses of lysates from Quinomycin treatment showed significant reduction in cancer stem cell marker DCLK1, CD44, CD24 and EPCAM protein levels in both MiaPaCa-2 and PanC-1 cells.
Quinomycin inhibits Notch signaling by downregulating the γ-secretase complex
We next determined the effect of Quinomycin on Notch signaling-related proteins in the pancreatic cancer cell lines. All four Notch receptors (Notch-1 to -4 were downregulated following Quinomycin treatment (Figure 4A). In addition, Notch ligands Jagged-1, 2 and Delta like ligand 1, 3 and 4 were downregulated following Quinomycin treatment (Figure 4B). Further confirmation was obtained when reduced expression of Hes-1 expression was observed (Figure 4C). We next determined whether the γ-secretase complex comprising of Presenilin, Nicastrin, APH1 and PEN2 was affected. Treatment with Quinomycin resulted in downregulation in the expression of all four proteins (Figure 4D). In addition, the co- treatment of Quinomycin in combination with γ-secretase complex inhibitor DAPT further reduced Hes- 1 expression (Figure 5A), and proliferation (left panel) while inducing apoptosis (right panel) (Figure 5B). These data suggest that Quinomycin-mediated downregulation of the Notch signaling pathway occurs at least in part through the inhibition of the γ-secretase complex.
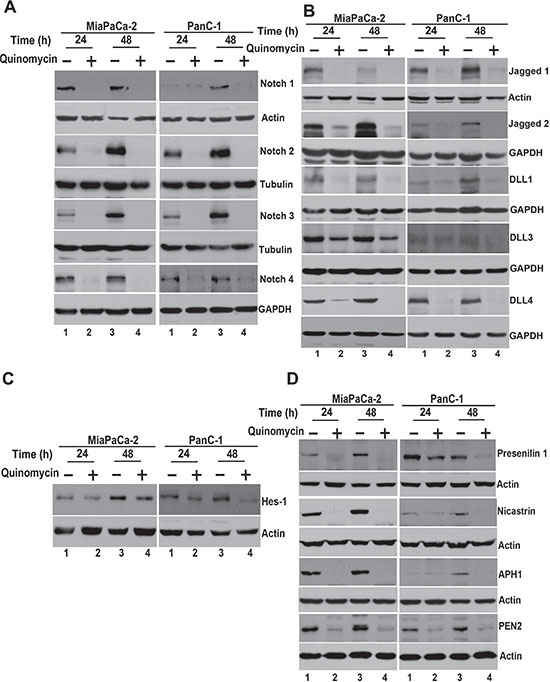
Figure 4: Quinomycin affects Notch signaling. (A) Lysates from cells treated with Quinomycin caused significant reduction in the expression of Notch receptors Notch-1, 2, 3 and 4 in both MiaPaCa-2 and PanC-1 cells. (B) Lysates from cells treated with Quinomycin caused significant reduction in the expression of Notch ligands Jagged-1, 2 and Delta like ligand 1, 3 and 4 in both MiaPaCa-2 and PanC-1 cells. (C) Lysates from cells treated with Quinomycin caused significant reduction in the expression of Notch downstream target gene Hes-1 in both MiaPaCa-2 and PanC-1 cells. (D) Quinomycin also significantly reduced expression of γ-secretase complex proteins Presenilin-1, Nicastrin, APH1 and PEN2 in both MiaPaCa-2 and PanC-1 cells.
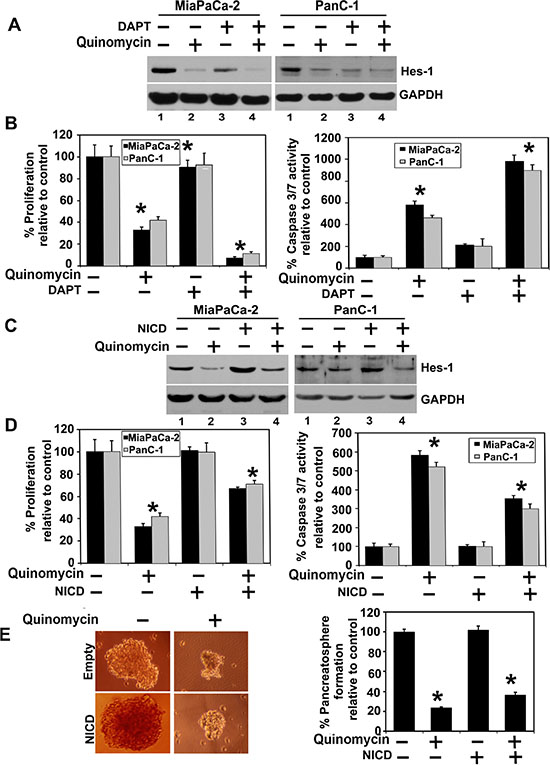
Figure 5: Quinomycin inhibits cell growth through inactivation of the γ-secretase complex. (A) Cells were cotreated with Quinomycin in combination with γ- secretase complex inhibitor DAPT for 24 h. Lysates were analyzed by western blotting. The cotreatment of the Quinomycin in combination with γ- secretase complex inhibitor DAPT further reduced Hes-1 expression, (B) Cells were cotreated with Quinomycin in combination with γ-secretase complex inhibitor DAPT and subsequently measured for proliferation (left panel) and apoptosis (right panel). The cotreatment of the Quinomycin in combination with γ- secretase complex inhibitor DAPT further reduced proliferation (left panel) and further increased apoptosis (right panel). (C) Ectopic expression of NICD overcomes Quinomycin-mediated suppression of Hes-1 expression. Cells transiently expressing NICD was treated with Quinomycin for 24 h. Lysates were analyzed by western blotting. Hes-1 was increased in the NICD expressing cells when compared to vector transfected controls. (D) Cells expressing NICD were treated with Quinomycin and subsequently measured for proliferation (left panel) and apoptosis (right panel). Ectopic expression of NICD rescued Quinomycin mediated inhibition of cell proliferation and apoptosis (*P < 0.05). (E) Cells expressing NICD were treated with Quinomycin and subsequently performed for pancreatosphere formation (left and right panel). Ectopic expression of NICD rescued Quinomycin mediated inhibition of pancreatosphere formation (*P < 0.05).
Cyclin D1 overexpression has been linked to the development and progression of cancer [42]. In addition, a recent study demonstrated that cyclin D1 is a direct downstream target of the Notch signaling pathway [43]. Furthermore, c-Myc is upregulated in cancers and also a direct downstream target of the Notch signaling pathway [44, 45]. In both MiaPaCa-2 and PanC-1 cells, Quinomycin treatment resulted in reduced cyclin D1 and c-Myc expression (Figure 2C), suggesting that Quinomycin mediated downregulation of cyclin D1 and c-Myc occurs in part through the inhibition of the Notch signaling pathway.
Quinomycin inhibits cell growth through inactivation of the γ-secretase complex
We next determined whether lack of Notch-1 activation is the reason for reduced growth of pancreatic cancer cells. For this, we expressed the intracellular domain NICD in MiaPaCa-2 and PanC-1 cells. Western blot analyses demonstrated increased expression of Hes-1 following ectopic expression of NICD in both cell lines (Figure 5C). More importantly, even in the presence of Quinomycin Hes-1 expression remained high in response to ectopic NICD overexpression. In addition, ectopic NICD overexpression reversed Quinomycin mediated inhibition of cell proliferation (Figure 5D). Futhermore, ectopic NICD overexpression resulted in significantly increased PanC-1 pancreatosphere formation, even in the presence of Quinomycin (Figure 5E left and right panel). Together, these data suggest that Quinomycin inhibits the γ-secretase complex thereby affecting Notch signaling.
Quinomycin inhibits pancreatic tumor xenograft growth
To evaluate the role of Quinomycin on tumor growth in vivo, we next examined its effects on growth of pancreatic cancer cell xenografts. MiaPaCa-2 pancreatic cancer xenograft tumors were allowed to develop and grow for one week following which Quinomycin (20 μg/kg bw) mixed with 5% sodium bicarbonate was administered intraperitoneally daily for three weeks. Quinomycin inhibited the growth of the tumor xenografts (Figure 6A and 6B). The excised tumors from control animals weighed ~2000 mg, while those treated with Quinomycin weighed ~300 mg (Figure 6C). In addition, tumor volume was significantly decreased (Figure 6C). There was no apparent change in liver, spleen, or body weight in the animals (data not shown). These data imply that Quinomycin is a potential therapeutic agent for treating pancreatic cancers but is relatively non-toxic to the animals.
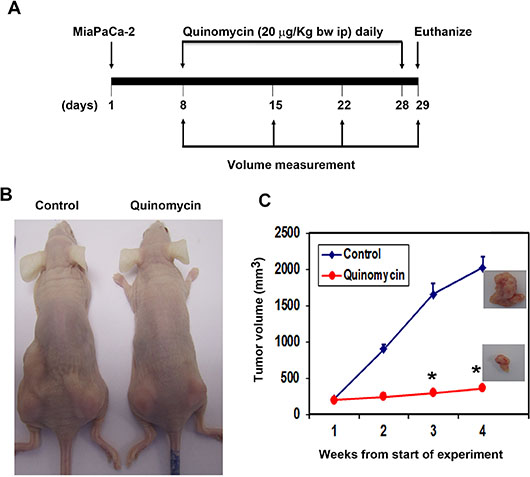
Figure 6: Quinomycin inhibits pancreatic cancer xenografts. (A) MiaPaCa-2 cells were injected in to the flanks of nude mice and palpable tumors were allowed to develop for 7 days. Subsequently, Quinomycin (20 μg/kg bw) was injected daily intraperitoneally every day for 21 days. On day 22, tumors were excised and subject to further analyses. (B) Quinomycin treatment resulted in significantly lower tumor weight when compared to control. Tumor size was measured every week. There was a significant reduction in tumor size from Quinomycin treated animals when compared control (*P < 0.05). (C) Tumor volumes in Quinomycin treated mice were smaller when compared to control (*P < 0.05).
Quinomycin inhibits the expression of cancer stem cell markers and notch signaling proteins in tumor xenograft tissues
To further investigate whether the Quinomycin affects CSCs, we determined specific marker expression in the tumor tissues. Western blot analyses demonstrated that Quinomycin treatment significantly reduced the expression of CSC proteins DCLK1, CD44, CD24 and EPCAM when compared to controls (Figure 7A). This was confirmed by immunohistochemistry (Figure 7B). These data suggest that Quinomycin targets pancreatic CSCs with high potency. Furthermore, we also examined the effects on Notch signaling in the tumor tissues obtained from control and Quinomycin treated mice. Quinomycin treatment resulted in significantly lower levels of activated Notch-1, its ligand Jagged 1 and the downstream target gene Hes-1 (Figure 7C). In addition, Quinomycin treatment resulted in significantly lower levels of Notch-2, 3, and 4 (Figure 8A) and its ligand Jagged 2, DLL-1, 3 and DLL4 (Figure 8C). This was confirmed by immunohistochemistry (Figure 8B and 8D). There was also a significant reduction in the expression of γ-secretase complex proteins, Presenilin 1 and Nicastrin (Figure 7C). Again, further confirmation of the downregulation was obtained by immunohistochemistry for the proteins in the xenograft tissue (Figure 7D). These data suggest that Quinomycin significantly affects the expression of Notch signaling-related proteins, which might contribute to the inhibitory effects of this treatment.
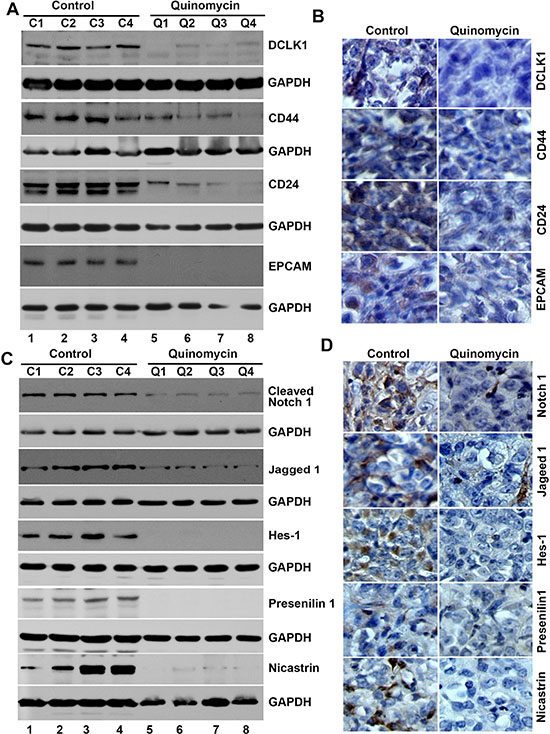
Figure 7: Quinomycin inhibit cancer stem cell marker proteins and Notch signaling in tumor xenografts. (A) Western blot analysis showed that tissue lysates from Quinomycin treated animals have significantly lower levels of cancer stem cell markers. (B) Immunohistochemistry shows that treatment with Quinomycin significantly reduced the expression of cancer stem cell markers. (C)Western blot analysis showed that tissue lysates from Quinomycin treated animals have significantly lower levels of Notch-1, Jagged-1, Hes-1, and γ-secretase complex proteins. (D) Immunohistochemistry shows that Quinomycin treated animals have significantly lower levels of Notch-1, Jagged-1, Hes-1, and γ-secretase complex proteins Presenilin 1 and Nicastrin in the tumor xenograft tissues. (C1-C4: Controls, Q1-Q4: Quinomycin treated).
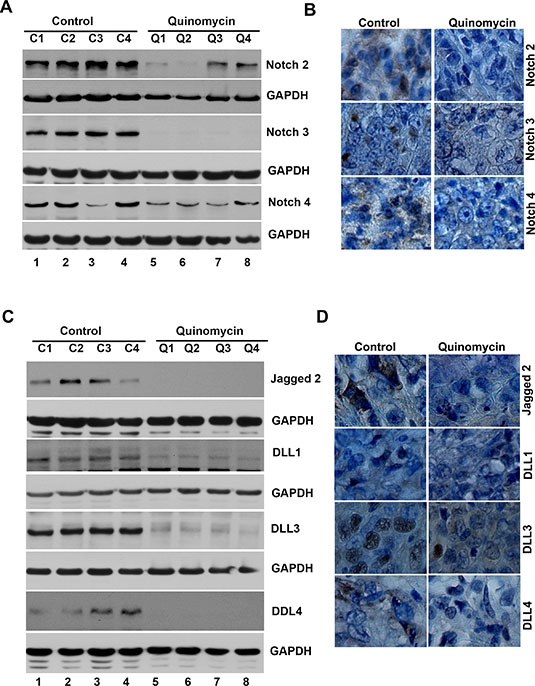
Figure 8: Quinomycin inhibit Notch receptor-2, 3 and 4 and its ligands Jagged 2, DLL-1, 3 and 4 in tumor xenografts. (A) Western blot analysis showed that tissue lysates from Quinomycin treated animals have significantly lower levels of Notch-2, 3 and 4 receptor expression. (B) Immunohistochemistry shows that treatment with Quinomycin significantly reduced the expression of Notch-2, 3 and 4 receptors. (C) Western blot analyses showed that tissue lysates from Quinomycin treated animals have significantly lower levels of Notch ligand Jagged-2, DLL-1, 3 and 4 expressions. (D) Immunohistochemistry shows that treatment with Quinomycin significantly reduced the Notch ligand Jagged-2, DLL-1, 3 and 4 expressions. (C1–C4: Controls, Q1–Q4: Quinomycin treated).
DISCUSSION
We are the first to study and demonstrate that Quinomycin inhibits pancreatic cancer stem cells and the mechanism involves Notch signaling. Our results indicate that Quinomycin possesses great potential as a promising anti-pancreatic cancer therapeutic agent. Pancreatic cancer is one of the most lethal cancers and has emerged as a leading cause of cancer-related death in the western world, with most patients dying within one year of diagnosis. The significant morbidity, toxicity and poor response rates of current chemotherapy regimens have led to searches for less toxic alternative therapies. The data presented in the article show that Quinomycin inhibits the proliferation of pancreatic cancer cells, induces cell cycle arrest and apoptosis, resulting in reduced colony formation. These results were also replicated in vivo, where Quinomycin decreased tumor growth.
Recent studies have suggested that CSCs have the capacity to drive tumor recurrence and resistance to chemotherapeutic agents and radiation [46]. Natural compounds such as curcumin, sulforaphane and honokiol have been suggested to target CSCs [43, 47]. Our results suggest that the Quinomycin is a potent inhibitor CSCs. Markers for prospectively identifying pancreatic cancer stem cells are CD44+CD24+EpCAM+ [31], CD133+ [32], and ALDH+ [33]. We have demonstrated that DCLK1 is stem cell marker whose expression is upregulated in both colon and pancreatic adenocarcinomas [34, 35]. Recent studies demonstrated that DCLK1 distinguishes between tumor and normal stem cells in the intestine and could be a therapeutic target for colon cancer [36, 37]. Similarly, DCLK1 was shown to mark a morphologically distinct subpopulation of cells with stem cell properties in pre-invasive pancreatic cancer [38]. Here, we have demonstrated that Quinomycin inhibits DCLK1 expression. Moreover, we have demonstrated that Quinomycin also inhibits the expression of other stem cell marker proteins such as CD44, CD24 and EPCAM. This was also confirmed in vivo where the Quinomycin significantly reduced the expression of these markers and also the growth of the xenografts. Another method that is commonly used to demonstrate stemness is the growth of spheroids or pancreatospheres. Quinomycin inhibited pancreatosphere formation further suggesting that they target the CSCs.
The Notch pathway plays a critical role in pancreatic cancer. Notch also has been shown to be important in stem cell renewal and vascular development [48]. Notch 2-positive progenitors have the intrinsic ability to give rise to pancreatic ductal cells [49]. Notch3 and HEY-1 have been shown to be prognostic biomarkers in pancreatic adenocarcinoma [50]. Finally, Notch 4 is elevated in pancreatic adenocarcinoma [50]. A recent study demonstrated that this pathway is important in maintaining CSC population in pancreatic cancer [25]. In our studies, we have determined that Quinomycin resulted in downregulation of the Notch ligand Jagged1, 2 and DLL1, 3 and 4 as well as all four essential members of the γ-secretase complex, the critical enzyme that cleaves and releases the NICD from the membrane. Therefore, Quinomycin mediated inhibition of pancreatic cancer growth is partly mediated by inactivating Notch-1. This was further confirmed by the combination of a γ-secretase inhibitor with Quinomycin, which further inhibited proliferation and induced apoptosis. However, ectopic expression of NICD reversed the effects of Quinomycin and partially restored cell growth. It would also be interesting to determine whether there are other clients for the γ-secretase complex and the role of these client proteins in CSC biogenesis.
We believe there is a lot of promise for use of Quinomycin as a therapeutic and preventive agent for pancreatic cancer. It should be noted that the dose of Quinomycin demonstrating efficacy was quite low, because a dose of 20 μg/kg bw corresponds to a calculated human equivalent dose of 57 μg/m2. More importantly, at this dose, while there was significant efficacy against the xenograft tissue, there was no effect on normal tissues in the animals. This is really important an observation because previous human clinical trials have suggested that Quinomycin is toxic. However, the studies showing toxicity utilized doses from 1200 to 2128 μg/m2 administered IV [48]. At lower doses (60 and 120 μg/m2), there was no observed toxicity, with only grade I toxicity encountered at 180 μg/m2 [48]. Our IP dose equivalent of 57 μg/m2 is approximately 1/30th the lowest IV dose studies in solid tumor patients. Tumor growth inhibition at this dose level is also quite consistent with recent reports describing that IP doses of Quinomycin at a Human Equivalent IP Dose of 28.5 μg/m2 eliminated AML leukemia-initiating cells without a significant effect on the hematopoietic stem cells [8]. These studies provide a foundation for further clinical development of the drug for treatment of pancreatic cancers.
In conclusion, our current study provides evidence that treatment with Quinomycin results in growth inhibition in vitro and in vivo. We did not observe any significant toxicity in mice xenografts treated with Quinomycin. Furthermore, Quinomycin treatment was potent against pancreatic CSCs especially DCLK1+ cells. In addition, Quinomycin significantly suppressed Notch-receptor 1–4 activation through γ-secretase complex proteins. Taken together, these data suggest that Quinomycin targets pancreatic CSCs and is an attractive and potentially novel agent for the treatment and prevention of pancreatic cancer. However, it should be noted that while the compound is effective in inhibiting pancreatic cancers, this might not be universal. Hence, additional studies are essential to determine efficacy for different cancers.
MATERIALS AND METHODS
Cells and reagents
Human pancreatic cancer cells PanC-1, MiaPaCa-2, and BxPC-3 (all cell lines obtained from American Type Culture Collection, at passage 4) were grown in RPMI 1640 containing 10% heat inactivated fetal bovine serum (Sigma-Alrich, St. Louis, MO) and 1% antibiotic-antimycotic solution (Corning, Tewksbury, MA) at 37oC in a humidified atmosphere of 5% CO2. HPNE cells were kindly provided by Dr. Anirban Maitra, Johns Hopkins University School of Medicine and grown in DMEM with 4.5 g/L glucose, L-glutamine and Sodium Pyruvate (Corning, Tewksbury, MA) with 5% FBS, 1X N2, 10 ng/ml bFGF and 50 μg/ml Gentamycin. All the cell lines used in this study were within 20 passages after receipt or resuscitation (~3 months of non-continuous culturing). The cell lines were not authenticated as they came from national repositories. Quinomycin was purchased from (AG Scientific, San Diego, CA) and our collaborator. N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) was purchased from (Sigma-Alrich, St. Louis, MO).
Proliferation and apoptosis assays
To assess proliferation, cells were seeded on to 96 well plates and grown overnight before treatment with increasing doses of Quinomycin. Cell proliferation was determined by enzymatic hexoseaminidase assay as described previously [39]. For apoptosis, caspase 3/7 activity was measured using the Apo-one Homogeneous Caspase-3/7 Assay kit (Promega, Madison, WI).
Clonogenicity assay
Briefly, 6 well dishes were seeded with 500 viable cells per well, treated with Quinomycin in 10% FBS containing RPMI1640 for 48 h, then medium containing compound was removed, and the cells were incubated for an additional 10 d in complete medium to allow colonies to form. The colonies were fixed in formalin, followed by staining with crystal violet. Experiments were done in triplicate.
Cell cycle analyses
Cells were treated with Quinomycin for 24 h, and subsequently trypsinized and suspended in phosphate buffered saline (PBS). Single-cell suspensions were fixed using 70% ethanol for 2 h, and subsequently permeabilized with PBS containing 1 mg/ml propidium iodide (Sigma-Aldrich), 0.1% Triton X-100 (Sigma-Aldrich) and 2 μg DNase-free RNase (Sigma-Aldrich) at room temperature. Flow cytometry was done with a FACS Calibur analyzer (Becton Dickinson, Mountain, View, CA), capturing 10,000 events for each sample. Results were analyzed with ModFit LT™ software (Verity Software House, Topsham, ME).
Western blot analysis
Cell lysates were subjected to polyacrylamide gel electrophoresis and blotted onto Immobilion polyvinylidene difluoride membranes (Millipore, Bedford, MA). Antibodies were purchased from Cell Signaling Technology (Beverly, MA), Abcam Inc. (Cambridge, MA), Sigma Aldrich, GenScript (Piscataway, NJ) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and specific proteins were detected by the enhanced chemiluminescence system (GE Health Care, Piscataway, NJ).
Flow cytometric analyses for CSC marker
24 h following Quinomycin treatment, cells were subjected to direct immunofluorescence staining followed by flow cytometric analyses. Briefly, the cells were harvested and suspended in PBS containing 0.5% BSA for 10 minutes at room temperature followed by the addition of 10 μl phycoerythrin conjugated DCLK1 antibody (Abcam Inc, Cambridge, MA). The samples were analyzed using a FACS Calibur analyzer (Becton Dickinson, Mountain, View, CA), capturing 10,000 events for each sample. Results were analyzed with ModFit LT™ software (Verity Software House, Topsham, ME).
Pancreatosphere assay
Cells were cultured in RPMI 1640 supplemented with 20 ng/ml bFGF 10 mL per 500 mL of 50X B27 supplement, EGF 20 ng/ml (all from Life Technologies) at low densities (3000 cells/mL) in 6 well low adhesion plates. Cells were treated with Quinomycin. After 5 days, the number and size of pancreatospheres were determined using Celigo (Cyntellect Inc., San Diego, CA). For second and third passages, cells were grown in the absence of these compounds.
Plasmids and transfections
MiaPaCa-2 and PanC-1 cells were transfected with plasmid EF.hICN1.CMV.GFP encoding the Notch-1 intracellular domain (NICD) or the empty vector EF.v-CMV.GFP (Addgene Inc, Cambridge, MA), and subsequently treated with 5 nM Quinomycin for 24 h and harvested for western blot analysis. Cell proliferation and apoptosis were detected using hexoaminadase assay and Apo-one Homogeneous Caspase-3/7 Assay kit, respectively. In addition, pancreatospheres assays were also performed.
MiaPaCa-2 xenograft tumors in mice
Five-week-old male athymic nude mice, purchased from Charles River Laboratory were utilized for in vivo experiments. They were maintained with water and standard mouse chow ad libidum and used in protocols approved by the University’s Animal Studies Committee. Animals were injected with 1 × 106 MiaPaCa-2 cells in the left and right flank and allowed to form a xenograft. One week following implantation, and after observing the presence of a palpable tumor, Quinomycin (20 μg/kg body weight) was administered intraperitoneally daily for 21 d. Tumors were measured weekly. At the end of treatment the animals were euthanized, and the tumors were removed, weighed and use for histology (hematoxylin & eosin), immunohistochemistry, and gene expression studies.
Immunohistochemistry
Paraffin embedded tissues were cut to 4 μm sections, deparaffinized and blocked with Avidin/Biotin for 20 min. The slides were incubated with primary antibodies for overnight at 4°C. Next the slides were treated with a broad spectrum secondary antibody (Invitrogen) and HRP-conjugate for one hour and then developed with DAB (Invitrogen). Finally, the slides were counterstained with hematoxylin. The slides were examined in Nikon Eclipse Ti microscope under a 40X objective.
Statistical analysis
All values are expressed as the mean ± SEM. Data was analyzed using an unpaired 2-tailed t test. A P value of less than 0.05 was considered statistically significant.
Author contributions
Conception and design: D. Subramaniam. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S. Ponnurangam, D. Subramaniam. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Ponnurangam, P. Dandawate, S. Anant, D. Subramaniam. Writing, review, and/or revision of the manuscript: S. Anant, A. Dhar, OW. Tawfik, S. Umar, SJ. Weir, A. Sugumar, RA. Jensen, SB. Padhye, A. Balakrishnan, D. Subramaniam. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S. Anant, RR. Parab, PD. Mishra, P. Ranadive, R. Sharma, G. Mahajan, A. Balakrishnan. Study supervision: S. Anant, D. Subramaniam. Other (performed experiments): S. Ponnurangam, D. Subramaniam.
GRANT SUPPORT
This work was supported by Thomas O’Sullivan Foundation, a pilot grant from the NCI-designated University of Kansas Cancer Center (P30CA168524–01; DS), and NIH Grants CA182872 and CA190291 SA). S. Anant is an Eminent Scientist of the Kansas Biosciences Authority. The Flow Cytometry Core Laboratory is sponsored, in part, by the NIH COBRE program of the NCRR P20 RR016443 and The University of Kansas Cancer Center P30CA168524–01 grants.
ACKNOWLEDGMENTS
We thank Ms. Dannita R. Youngblood for her secretarial help. We also thank members of the Anant laboratory for their discussion during the course of this study.
CONFLICTS OF INTEREST
No potential conflicts of interest are disclosed.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015; 65:5–29.
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer research. 2014; 74:2913–2921.
3. Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. Journal of clinical oncology. 1997; 15:2403–2413.
4. Burris H, Storniolo AM. Assessing clinical benefit in the treatment of pancreas cancer: gemcitabine compared to 5-fluorouracil. Eur J Cancer. 1997; 33:S18–22.
5. Foster BJ, Clagett-Carr K, Shoemaker DD, Suffness M, Plowman J, Trissel LA, Grieshaber CK, Leyland-Jones B. Echinomycin: the first bifunctional intercalating agent in clinical trials. Investigational new drugs. 1985; 3:403–410.
6. Park JY, Ryang YS, Shim KY, Lee JI, Kim HS, Kim YH, Kim SK. Molecular signaling cascade in DNA bisintercalator, echinomycin-induced apoptosis of HT-29 cells: evidence of the apoptotic process via activation of the cytochrome c-ERK-caspase-3 pathway. The international journal of biochemistry & cell biology. 2006; 38:244–254.
7. Park JY, Chang JH, Bae KS, Lee KH, Choi SJ, Park JY, Ryang YS, Kim SK. NF-kappaB-dependency and consequent regulation of IL-8 in echinomycin-induced apoptosis of HT-29 colon cancer cells. Cell biology international. 2008; 32:1207–1214.
8. Wang Y, Liu Y, Tang F, Bernot KM, Schore R, Marcucci G, Caligiuri MA, Zheng P, Liu Y. Echinomycin protects mice against relapsed acute myeloid leukemia without adverse effect on hematopoietic stem cells. Blood. 2014; 124:1127–1135.
9. Chang AY, Kim K, Boucher H, Bonomi P, Stewart JA, Karp DD, Blum RH. A randomized phase II trial of echinomycin, trimetrexate, and cisplatin plus etoposide in patients with metastatic nonsmall cell lung carcinoma: an Eastern Cooperative Oncology Group Study (E1587). Cancer. 1998; 82:292–300.
10. Wadler S, Tenteromano L, Cazenave L, Sparano JA, Greenwald ES, Rozenblit A, Kaleya R, Wiernik PH. Phase II trial of echinomycin in patients with advanced or recurrent colorectal cancer. Cancer chemotherapy and pharmacology. 1994; 34:266–269.
11. Brown TD, Goodman PJ, Fleming TR, Taylor SA, Macdonald JS. Phase II trial of echinomycin in advanced colorectal cancer. A Southwest Oncology Group study. Investigational new drugs. 1991; 9:113–114.
12. Shevrin DH, Lad TE, Guinan P, Kilton LJ, Greenburg A, Johnson P, Blough RR, Hoyer H. Phase II trial of echinomycin in advanced hormone-resistant prostate cancer. An Illinois Cancer Council study. Investigational new drugs. 1994; 12:65–66.
13. Marshall ME, Wolf MK, Crawford ED, Taylor S, Blumenstein B, Flanigan R, Meyers FJ, Hynes HE, Barlogie B, Eisenberger M. Phase II trial of echinomycin for the treatment of advanced renal cell carcinoma. A Southwest Oncology Group study. Investigational new drugs. 1993; 11:207–209.
14. Muss HB, Blessing JA, DuBeshter B. Echinomycin in recurrent and metastatic endometrial carcinoma. A phase II trial of the Gynecologic Oncology Group. American journal of clinical oncology. 1993; 16:492–493.
15. Schilsky RL, Faraggi D, Korzun A, Vogelzang N, Ellerton J, Wood W, Henderson IC. Phase II study of echinomycin in patients with advanced breast cancer: a report of Cancer and Leukemia Group B protocol 8641. Investigational new drugs. 1991; 9:269–272.
16. Hakes T, Markman M, Phillips M. A phase II trial of echinomycin in metastatic cervix carcinoma. Investigational new drugs. 1990; 8:311–312.
17. Muss HB, Blessing JA, Baker VV, Barnhill DR, Adelson MD. Echinomycin (NSC 526417) in advanced ovarian cancer. A phase II trial of the Gynecologic Oncology Group. American journal of clinical oncology. 1990; 13:299–301.
18. Gradishar WJ, Vogelzang NJ, Kilton LJ, Leibach SJ, Rademaker AW, French S, Benson AB. A phase II clinical trial of echinomycin in metastatic soft tissue sarcoma. An Illinois Cancer Center Study. Investigational new drugs. 1995; 13:171–174.
19. Taylor SA, Metch B, Balcerzak SP, Hanson KH. Phase II trial of echinomycin in advanced soft tissue sarcomas. A Southwest Oncology Group study. Investigational new drugs. 1990; 8:381–383.
20. Yonekura S, Itoh M, Okuhashi Y, Takahashi Y, Ono A, Nara N, Tohda S. Effects of the HIF1 inhibitor, echinomycin, on growth and NOTCH signalling in leukaemia cells. Anticancer research. 2013; 33:3099–3103.
21. Koch U, Radtke F. Notch and cancer: a double-edged sword. Cellular and molecular life sciences. 2007; 64:2746–2762.
22. Miele L, Golde T, Osborne B. Notch signaling in cancer. Current molecular medicine. 2006; 6:905–918.
23. Ma J, Xia J, Miele L, Sarkar FH, Wang Z. Notch Signaling Pathway in Pancreatic Cancer Progression. Pancreatic disorders & therapy. 2013; 3:1000114.
24. Arumugam TV, Chan SL, Jo DG, Yilmaz G, Tang SC, Cheng A, Gleichmann M, Okun E, Dixit VD, Chigurupati S, Mughal MR, Ouyang X, Miele L, et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nature medicine. 2006; 12:621–623.
25. Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L, Dziubinski ML, Simeone DM. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PloS one. 2014; 9:e91983.
26. Mohammed A, Janakiram NB, Madka V, Brewer M, Ritchie RL, Lightfoot S, Kumar G, Sadeghi M, Patlolla JM, Yamada HY, Cruz-Monserrate Z, May R, Houchen CW, et al. Targeting pancreatitis blocks tumor-initiating stem cells and pancreatic cancer progression. Oncotarget. 2015; 6:15524–15539. doi:10.18632/oncotarget.3499.
27. Plentz R, Park JS, Rhim AD, Abravanel D, Hezel AF, Sharma SV, Gurumurthy S, Deshpande V, Kenific C, Settleman J, Majumder PK, Stanger BZ, Bardeesy N. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2009; 136:1741–1749 e1746.
28. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer research. 2006; 66:1883–1890; discussion 1895–1886.
29. Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clinical cancer research. 2008; 14:5646–5648.
30. Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C, Yao Q, Li M. Overcoming drug resistance in pancreatic cancer. Expert opinion on therapeutic targets. 2011; 15:817–828.
31. Ohara Y, Oda T, Sugano M, Hashimoto S, Enomoto T, Yamada K, Akashi Y, Miyamoto R, Kobayashi A, Fukunaga K, Morishita Y, Ohkohchi N. Histological and prognostic importance of CD44(+) /CD24(+) /EpCAM(+) expression in clinical pancreatic cancer. Cancer science. 2013; 104:1127–1134.
32. Rasheed Z, Wang Q and Matsui W. Isolation of stem cells from human pancreatic cancer xenografts. Journal of visualized experiments. 2010:2169.
33. Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, Kopetz S, McConkey DJ, Evans DB, Gallick GE. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PloS one. 2011; 6:e20636.
34. Sureban SM, May R, Ramalingam S, Subramaniam D, Natarajan G, Anant S, Houchen CW. Selective blockade of DCAMKL-1 results in tumor growth arrest by a Let-7a MicroRNA-dependent mechanism. Gastroenterology. 2009; 137:649–659, 659 e641–642.
35. Sureban SM, May R, Lightfoot SA, Hoskins AB, Lerner M, Brackett DJ, Postier RG, Ramanujam R, Mohammed A, Rao CV, Wyche JH, Anant S, Houchen CW. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer research. 2011; 71:2328–2338.
36. Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, Isomura A, Kawada K, Sakai Y, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nature genetics. 2013; 45:98–103.
37. Metcalfe C, de Sauvage FJ. A tumor-specific stem cell. Nature genetics. 2013; 45:7–9.
38. Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, Zhang H, Pasricha PJ, Bardeesy N, Matsui W, Maitra A, Leach SD. DCLK1 Marks a Morphologically Distinct Subpopulation of Cells with Stem Cell Properties in Pre-invasive Pancreatic Cancer. Gastroenterology. 2014; 146:245–56.
39. Subramaniam D, Nicholes ND, Dhar A, Umar S, Awasthi V, Welch DR, Jensen RA, Anant S. 3,5-bis(2,4-difluorobenzylidene)-4-piperidone, a novel compound that affects pancreatic cancer growth and angiogenesis. Molecular cancer therapeutics. 2011; 10:2146–2156.
40. Boatright KM, Salvesen GS. Mechanisms of caspase activation. Current opinion in cell biology. 2003; 15:725–731.
41. Weiss GR, Hersh M, Kuhn JG, Ludden TM, von Hoff DD, Kisner DL, Pirtle TE. A phase I and pharmacokinetic comparison of hepatic arterial and peripheral vein infusions of bisantrene for liver cancer. Cancer chemotherapy and pharmacology. 1985; 15:144–148.
42. Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Molecular cancer. 2007; 6:24.
43. Cohen B, Shimizu M, Izrailit J, Ng NF, Buchman Y, Pan JG, Dering J, Reedijk M. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast cancer research and treatment. 2010; 123:113–124.
44. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, Li Y, Wolfe MS, Shachaf C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes & development. 2006; 20:2096–2109.
45. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O’Neil J, Neuberg D, Weng AP, Aster JC, Sigaux F, Soulier J, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:18261–18266.
46. Saez R, Craig JB, Kuhn JG, Weiss GR, Koeller J, Phillips J, Havlin K, Harman G, Hardy J, Melink TJ, et al. Phase I clinical investigation of amonafide. Journal of clinical oncology. 1989; 7:1351–1358.
47. Lai KW, Koh KX, Loh M, Tada K, Subramaniam MM, Lim XY, Vaithilingam A, Salto-Tellez M, Iacopetta B, Ito Y, Soong R. MicroRNA-130b regulates the tumour suppressor RUNX3 in gastric cancer. Eur J Cancer. 2010; 46:1456–1463.
48. Kuhn JG, Von Hoff DD, Hersh M, Melink T, Clark GM, Weiss GR, Coltman CA. Phase I trial of echinomycin (NSC 526417), a bifunctional intercalating agent, administered by 24-hour continuous infusion. European journal of cancer & clinical oncology. 1989; 25:797–803.
49. Lee KM, Yasuda H, Hollingsworth MA, Ouellette MM. Notch 2-positive progenitors with the intrinsic ability to give rise to pancreatic ductal cells. Laboratory investigation; a journal of technical methods and pathology. 2005; 85:1003–1012.
50. Mann CD, Bastianpillai C, Neal CP, Masood MM, Jones DJ, Teichert F, Singh R, Karpova E, Berry DP, Manson MM. Notch3 and HEY-1 as prognostic biomarkers in pancreatic adenocarcinoma. PloS one. 2012; 7:e51119.