
INTRODUCTION
HCC is one of the most common malignancies worldwide [1]. Currently, there is no effective systemic chemotherapy for patients with advanced HCC because HCC is relatively chemotherapy-resistant and exhibits high rates of recurrence. Cancer stem cells (CSCs) are generally considered to be key contributors to tumor initiation, maintenance, chemoresistance, radioresistance, recurrence, and metastasis. CD133+ CSCs from lung cancer are in enrichment after cisplatin treatment and have stable cisplatin-resistant phenotype in vitro [2]. Thus, the efficient identification and isolation of CSCs from cancer cell lines or tumor tissues is necessary and helpful for the development of anti-cancer strategies targeting CSCs. Multiple of cell surface markers such as CD13, CD44, DLK1, ALDH1, CD133 have been used to isolate and identify CSCs in solid tumors [3-10], in which CD133 has been identified as a marker for enrichment of CSCs in HCC. Compared with CD133- cells, CD133+ cells exhibit significantly higher proliferation potential as well as a greater capacity for colony formation and tumor initiation in vivo [5-10]. CD133+ cells are typically reactivated in HCC, and the expression of CD133 increases in higher-stage tumors. This enhancement of CD133 expression typically indicates poor patient prognosis [11]. However, little is known regarding the characteristics and molecular regulation of CD133 and CD133+ cancer stem-like cells in HCC.
Ikaros is a member of the Kruppel family of zinc finger DNA-binding protein [12], which is the crucial regulator of normal hematopoiesis, and has been identified as a tumor suppressor associated with the development of leukemia [13, 14, 15]. Loss of Ikaros function is thought to be essential to the development of lymphoid leukemia, including adult B-cell acute lymphoblastic leukemia (B-ALL), acute myeloid leukemia and infant T-cell ALL [13, 16, 17]. Ikaros mutation or polymorphism is a typical genetic feature in human B-ALL [18]. In general, Ikaros binds to promoter regions and regulates target gene expression by recruiting promoter regions into pericentromeric heterochromatin. As a result, its target genes are suppressed via chromatin remodeling. The expression of Hes1 and Deltex are repressed by the direct binding of Ikaros to their promoters [19, 20]. Ikaros also plays important role in the development of nervous system, and the cerebral cortex and hypothalamic–pituitary somatotroph development [21, 22]. The essential function of Ikaros in leukemia is obvious; however, the role of Ikaros in solid tumors remains unclear, particularly in hepatocellular carcinoma (HCC).
In this study, we showed that Ikaros interacted with the transcription repressor CtBP as a complex and inhibited CD133 expression via direct binding to the CD133 P1 promoter in HCC. Moreover, biological activation of CD133+ cancer stem-like cells was regulated by Ikaros in HCC. We confirmed that ETS1 up-regulated the expression of Ikaros. In addition, patients with higher Ikaros expression had longer survival. Thus, our results suggest that reactivation of Ikaros could be a novel strategy for treatment of HCC.
RESULTS
P1 promoter is the main 5`-UTR pattern of CD133 in HCC
In our previous study, we showed that CD133 was used to isolate and identify CSCs in HCC and CD133+ HCC cells exhibited the biological characteristics of cancer stem cell in HCC [6, 7]. To understand the regulatory mechanism of CD133 expression, we examined the CD133 promoter. Five alternative promoters (P1, P2, P3, P4, and P5) were identified in the 5’-UTR of CD133 (Supplementary Figure S1A) [23]. Reporter assays revealed that the P1 promoter exhibited the highest activity of the five pattern promoters of CD133 in Hep3B, Huh7 and PLC/PRF5 (Figure 1A), which have higher expressions of CD133 mRNA and protein (Supplementary Figure S1B and C), and RT-PCR further confirmed that the P1 promoter was the principal promoter of CD133 (Figure 1B).
We successively deleted the sequences of P1 promoter and investigated the activity of these promoters. The promoter activity was increased when -260/-150-bp sequences were deleted (Figure 1C). We concluded that this region might contain transcription repressor binding sites. We further analyzed the transcription factor binding sites in the P1 promoter and multiples of transcription factors sites were found in this region (Figure 1D). Some of these transcription factors were able to down-regulate the expression of CD133, such as c-ETS1, Ikaros and Nkx3.1 (Supplementary Figure S1D). Taken together, these results showed that the P1 promoter was the key promoter of CD133 in HCC.
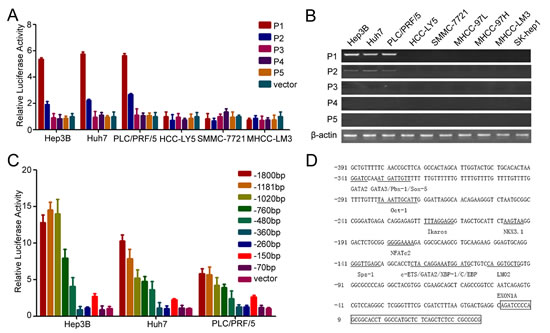
Figure 1: P1 promoter is the key promoter of CD133 in HCC. (A) The promoter activity of P1, P2, P3, P4, and P5 in HCC cells. (B) RT-PCR analysis of CD133 expression pattern for 5`-UTR containing exons 1A (P1), 1B (P2), 1C (P3), 1D (P4), and 1E (P5) in HCC cells. (C) The promoter activity of the deleted P1 promoter in Hep3B, Huh7, and PLC/PRF/5 cells. (D) Analysis of CD133 P1 promoter. Transcription factors were predicted using TFSEARCH (http://mbs.cbrc.jp/research/db/TFSEARCH.html).
Ikaros inhibits CD133 expression by directly binding to the CD133 promoter
One Ikaros binding site was observed at -212/-221-bp region in the CD133 P1 promoter. Ikaros is a crucial regular in the differentiation of hematopoietic stem cells and has been identified as a tumor suppressor associated with the development of leukemia [13, 14, 24, 25]. To identify whether Ikaros also played an important role in CD133 expression, we examined the P1 promoter activity when Ikaros was overexpressed. Report assays showed that the P1 promoter activity was inhibited when Ikaros was enforced to express in HCC cells (Figure 2A). ChIP assays and DNA pull-down assays confirmed that Ikaros directly bound to the CD133 P1 promoter in HCC cells (Figure 2B and C). However, CD133 promoter activity was unchanged in response to Ikaros overexpression when the Ikaros binding site in the CD133 promoter was mutated (Supplementary Figure S2A). The 157 phenylalanine (F), 162 arginine (R), and 184 arginine (R) have been confirmed to be necessary for Ikaros DNA binding activity and subcellular localization [26]. Therefore, we constructed three mutants in which these three critical amino acids were mutated to alanine (A). Report assays showed that P1 promoter activity had no change when these mutant Ikaros were overexpressed (Supplementary Figure S2B).
To further determine whether Ikaros could down-regulate CD133 expression, CD133 expression was detected after ectopic overexpression or knockdown of Ikaros. Overexpressed Ikaros decreased the expression of CD133, while silenced Ikaros increased CD133 expression (Figure 2D). The mRNA and protein levels of Ikaros and CD133 were negatively correlated in nine HCC cell lines using qPCR and western blot (Supplementary Figure S1B and C). To better understand the correlation between Ikaros and CD133, immunohistochemical staining (IHC) analysis was performed in 102 human primary HCC samples. IHC analysis showed that Ikaros protein levels inversely correlated with CD133 expression in HCC tissues (Figure 2E and F). Together, these results suggested that Ikaros inhibited the expression of CD133 via direct binding to the CD133 P1 promoter in HCC.
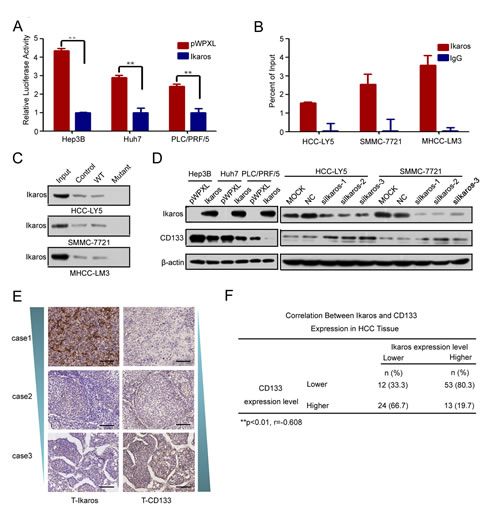
Figure 2: Ikaros represses CD133 expression in HCC. (A) The promoter activity of P1 after the transfection of Ikaros into Hep3B, Huh7, and PLC/PRF/5 (PLC) cells. **, p<0.01. (B) Binding of Ikaros on the CD133 gene promoter was assayed by chromatin immunoprecipitation (ChIP). ChIP was performed using antibodies to Ikaros in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. A negative control with irrelevant antibody (IgG) was included for comparison. qPCR was performed on the bound fractions. (C) DNA pull-down assay to detect Ikaros binding to the P1 promoter using either wild-type (WT) or Ikaros mutant oligonucleotide sequences from the CD133 promoter in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. (D) Immunoblot analysis of CD133 expression when Ikaros was overexpressed or silenced. (E) IHC analysis of Ikaros and CD133 in HCC samples. Representative images are shown. Scale bar, 100 μm. (F) The analysis of the correlation between Ikaros and CD133 in HCC tissue. **, p<0.01; r=-0.608; n=102. Data represent mean ± SD of three independent experiments with technical triplicates for each, and statistical analysis was performed using Student’s t test.
CtBP could interact with Ikaros and regulate CD133 expression in HCC cells
Usually, Ikaros interacts with co-repressors as complexes to regulate target genes. The co-immunoprecipitation (Co-IP) assay was performed to identify the co-repressors of Ikaros in HCC cells. Approximately, 40-50 kD proteins were precipitated from the three HCC cell lines (Figure 3A). We suspected that CtBP, with a molecular weight of approximately 48 kD, interacted with Ikaros as a co-repressor [27]. To determine whether Ikaros interacts with CtBP in HCC, we analyzed precipitated samples using western blot. CtBP was identified in anti-Ikaros precipitated samples and Ikaros was also detected in anti-CtBP precipitated fractions (Figure 3B). Next, we investigated the subcellular location of Ikaros and CtBP using immunofluorescence staining. Results demonstrated that these proteins were co-localized in HCC cells (Figure 3C). ChIP assays showed CtBP was able to bind to CD133 P1 promoter, which further confirmed the interaction between CtBP and Ikaros (Figure 3D). Uniformly, the CD133 promoter activity was inhibited by overexpression of CtBP (Figure 3E). Taken together, these findings confirmed that CtBP interacted with Ikaros and regulated CD133 expression in HCC.
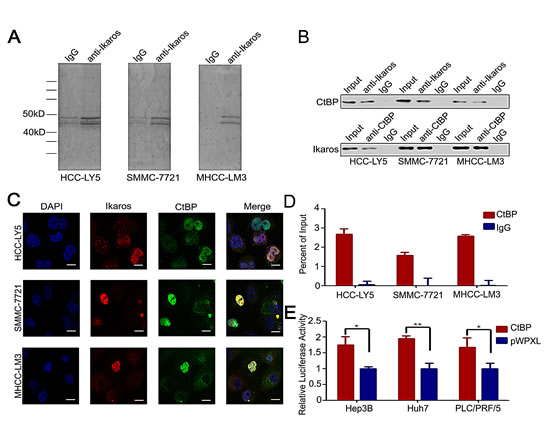
Figure 3: CtBP interacts with Ikaros as a transcription repressor complex. (A) Co-immunoprecipitation assay to detect Ikaros interaction proteins in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. (B) Immunoblot analysis of co-immunoprecipitated mixture. (C) Confocal imaging analysis of CtBP and Ikaros colocalization in the nucleus. Red indicates Ikaros; green indicates CtBP; and blue indicates DAPI. Scale bar, 10 μm. (D) Binding of CtBP on the CD133 gene promoter was assayed by ChIP, which was performed using antibodies to CtBP in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. A negative control with irrelevant antibody (IgG) was included for comparison. qPCR was performed on the bound fractions. (E) The promoter activity of P1 after the transfection of CtBP into Hep3B, Huh7, and PLC/PRF/5 cells. **, p<0.01; *, p<0.05. Data represent mean ± SD of three independent experiments with technical triplicates for each, and statistical analysis was performed using Student’s t test.
ETS1 up-regulates Ikaros expression via the MAPKs pathway
To detect which pathway participates in the regulation of Ikaros and CD133 expression, we analyzed the Ikaros promoter. One binding site for the transcription factor ETS1 was found in the Ikaros promoter at the -69/-59-bp region (Figure 4A). Reporter assays showed that Ikaros promoter activity was significantly enhanced in response to ETS1 overexpression (Figure 4A). Ohnishi et al showed that the ETS family transcription factors regulate CD133 via direct binding of CD133 to the P5 promoter in colon cancer cells [28]. One ETS1 binding site was also predicted in the CD133 P1 promoter. However, ChIP assays revealed that ETS1 did not bind to this site (Supplementary Figure S3A).
ChIP assay and DNA pull-down assay further confirmed that ETS1 directly bound to the Ikaros promoter (Figure 4B and C). When ETS1 was silenced, we did not detect the binding between ETS1 and Ikaros promoter (Supplementary Figure S3B). Ikaros expression was upregulated by overexpressing ETS1 and down-regulated by ETS1 knockdown, while CD133 expression exhibited the opposite changes compared with Ikaros (Figure 4D and E). ETS1 activity was inhibited via phosphorylated by Erk1/2 [29, 30]. In addition, specific inhibitors PD98059 and U0126 were utilized to block Erk1/2 activity. The expression of phosphorylated Erk1/2 and ETS1, Ikaros, and CD133 also correspondingly changed after treatment with these inhibitors (Figure 4F). Taken together, these results indicated that the MAPKs/ETS1 pathway regulates Ikaros and CD133 expression in HCC.
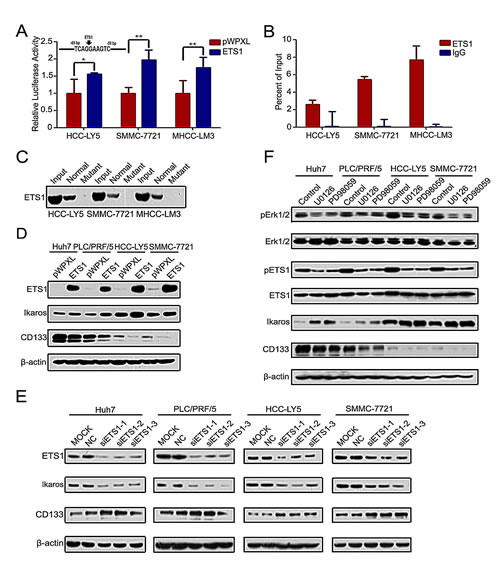
Figure 4: MAPKs/ETS1 pathway regulates the expression of Ikaros and CD133. (A) The promoter activity of Ikaros responding to ETS1 overexpressing in HCC-Y5, SMMC-7721, and MHCC-LM3 cells. **, p<0.01; *, p<0.05. The Ikaros promoter was analyzed using TFSEARCH (http://mbs.cbrc.jp/research/db/TFSEARCH.html). (B) Binding of ETS1 on the Ikaros gene promoter was assayed by ChIP which was performed using antibodies to ETS1 in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. A negative control with irrelevant antibody (IgG) was included for comparison. qPCR was performed on the input and bound fractions. (C) DNA pull-down assay to detect ETS1 binding to the Ikaros promoter in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. (D and E) Immunoblot analysis of Ikaros and CD133 expression when ETS1 overexpression or knockdown. (F) Immunoblot analysis of the expression of Erk1/2, pErk1/2, ETS1, pETS1, Ikaros, and CD133 when specific MAPKs inhibitors (U0126, PD98059) were added to Huh7, PLC/PRF/5, HCC-LY5, and SMMC-7721 cells. Data represent mean ± SD of three independent experiments with technical triplicates for each, and statistical analysis was performed using Student’s t test.
Ikaros inhibits the tumorigenic and self-renewal capacity of CD133+ HCC cells
Next, we determined the function of Ikaros in HCC. Cell-proliferation assay revealed that forced Ikaros expression remarkably inhibited the cell growth of HCC cells in vitro (Supplementary Figure S4A). We found that Ikaros had more effect to HCC cells with high-level expression of CD133. Colony-formation assay using sorted CD133+/- cells from HCC-LY5, SMMC-7721 and MHCC-LM3 cells also showed that the suppression of growth by Ikaros overexpression was more effective in CD133+ cells than CD133- cells (Supplementary Figure S4B). The self-renewal capacity of Huh7 and CD133+ PLC/PRF/5 cells was significantly inhibited by transfection with Ikaros (Figure 5A). When HCC cells transfected with Ikaros were treated with Cisplatin, the IC50 value was decreased. These cells were also more susceptible to Doxorubicin and Sorafenib (Supplementary Figure S4C). Next, CD133+ or CD133- cells transfected with Ikaros or control from SMMC-7721 or HCC-LY5 were subcutaneously injected into non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice. Ikaros significantly inhibited the tumorigenicity of CD133+ cells (Figure 5B). Western blot showed that the expression of CD133 from xenograft mouse models was inhibited by Ikaros in vivo (Supplementary Figure S4D).
Ikaros not only repressed CD133 expression, but also decreased the proportion of CD133+ HCC cells upon forced Ikaros expression (Huh7 cells for around 28.1%; PLC/PRF/5 cells for around 83.2%) (Figure 5C). In addition, the proportion of CD133+ HCC cells were increased when Ikaros was silenced (HCC-LY5 cells for approximately 34 times; SMMC-7721 cells for approximately 72 times) (Figure 5D). We further examined differentiated molecular marker Cytokeratin 18 (CK18) and Cytokeratin 19 (CK19) in Ikaros-transfected HCC cells. CK18 and CK 19 were increased and decreased, respectively, when Ikaros was overexpressed. Moreover, CK18 and CK19 had opposite changes after Ikaros was knocked down (Figure 5E). These data confirmed that Ikaros could efficiently inhibit tumorigenicity and the self-renewal capacity of CD133+ CSCs in HCC.
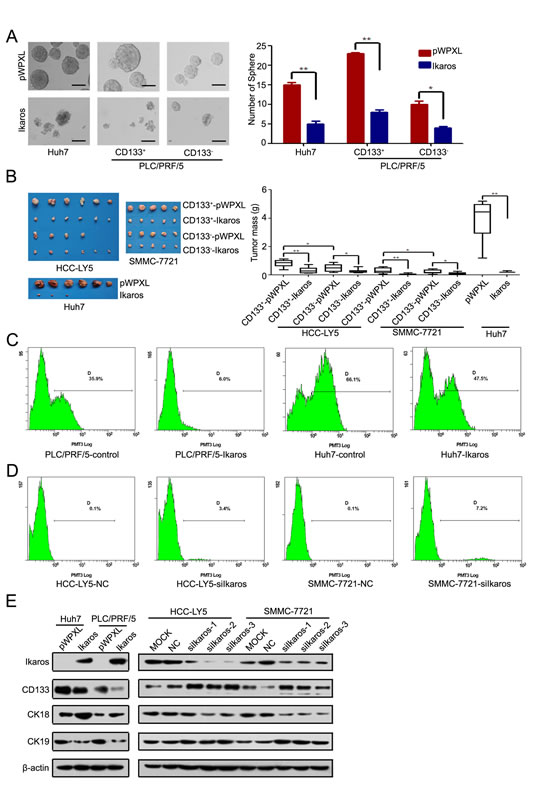
Figure 5: Ikaros inhibits the tumor growth and self-renewal of CD133+ HCC cells. (A) A representative image of a CSC sphere in self-renewal assays (left panel). The mean values of Huh7, CD133+/- PLC/PRF/5 cells sphere from three independent experiments are shown in the right panel. (Scale bar, 100 μm) (B) The size of tumors from NOD/SCTD mice injected with Ikaros-overexpressing or control cells (left panel). The weight of tumors from NOD/SCTD mice injected with Ikaros-overexpressing or control cells in the right panels. **, p<0.01; *, p<0.05. (C) Flow cytometric analysis of the percentage of CD133+ cells when Ikaros was overexpressed in Huh7 and PLC/PRF/5 cells. (D) Flow cytometric analysis of the percentage of CD133+ cells when Ikaros was knockdown in HCC-LY5 and SMMC-7721 cells. (E) Immunoblot analysis of expression of CD133, CK18, and CK19 when Ikaros was enforced to express or silenced. Data represent mean ± SD of three independent experiments with technical triplicates for each, and statistical analysis was performed using Student’s t test.
High Ikaros levels indicate longer survival in HCC patients
We analyzed Ikaros expression in 102 pairs of human primary HCC and matched noncancerous liver specimens using IHC. The IHC analysis demonstrated that Ikaros expression was at low levels in HCC samples (Figure 6A), and 17.8% (18/102) of human primary HCC specimens with Ikaros protein staining was higher than paired nontumorous liver specimens. Western blot showed that the protein levels of Ikaros were down-regulated in primary HCC tissues (Figure 6B).
According to the IHC results, the expression intensity of Ikaros protein was scored as 0, 1 for weak and strong immunostaining, respectively. The results showed that Ikaros expression was negatively associated with histological grade of HCC and tumor size (Table 1). However, there was no correlation between Ikaros expression and other clinicopathological indexes, including age, gender, serum alpha-fetoprotein (AFP), intrahepatic metastasis, hepatitis B surface antigen (HBsAg) levels, and with or without cirrhosis (Table 1). Further, overall survival analysis revealed that the high expression of Ikaros was closely associated with good outcomes in HCC patients (Figure 6C).
Table 1: Relationship between Ikaros expression and clinicopathological features in HCC tissues
|
Clinicopathological Features |
Ikaros Expression (Cancer) |
|||
Score 0 |
Score 1 |
|||
Case |
Case (%) |
Case (%) |
p Value |
|
Age (years) |
||||
<60 |
76 |
28 (77.8) |
48(72.7) |
0.619 |
≥60 |
26 |
8(22.2) |
18(27.3) |
|
Gender |
||||
male |
84 |
27(75) |
57(76.4) |
0.15 |
female |
18 |
9(25) |
9(13.6) |
|
AFP |
||||
≤20 |
31 |
11(30.6) |
20(30.3) |
0.517 |
>20 |
71 |
25(69.4) |
46(69.7) |
|
HBV |
||||
negative |
19 |
10(27.8) |
9(13.6) |
0.067 |
positive |
79 |
24(72.2) |
55(76.4) |
|
Cirrhosis |
||||
absent |
27 |
11(30.6) |
16(24.2) |
0.49 |
present |
75 |
25(69.4) |
50(75.8) |
|
Intrahepatic metastasis |
||||
absent |
74 |
29(80.6) |
45(68.2) |
0.181 |
present |
28 |
7(19.4) |
21(31.8) |
|
Histological grade |
||||
I, II |
57 |
10(27.8) |
47(71.2) |
0.035* |
III, IV |
45 |
26(72.2) |
19(29.8) |
|
Tumor size |
||||
≤5cm |
55 |
9(25.7) |
46(69.7) |
0.043* |
>5cm |
46 |
26(74.5) |
20(30.3) |
|
P value represents the probability from a Chi-square test for different immunohistochemical scores of Ikaros in HCC tissues.
*, p<0.05.
We then tested relative molecular in 7 pairs of HCC and their matched adjacent liver tissue samples (Figure 6D). Western blot showed that phosphorylation of Erk1/2 and ETS1 was increased, and Ikaros expression decreased and CD133 expression increased in HCC tissue. Overall, we proposed that Ikaros interacting with CtBP played roles as transcription repressors in inhibiting CD133 expression via binding to P1 promoter, and MAPKs pathways indirectly regulated the expression of Ikaros and CD133 via ETS1 (Figure 6E).
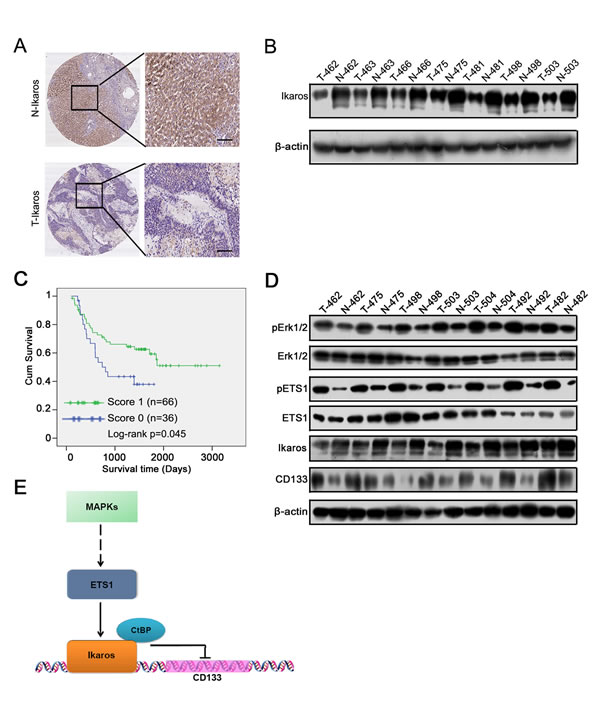
Figure 6: The clinical significance of Ikaros in HCC. (A) The IHC analysis of Ikaros in HCC samples. Scale bar, 100 μm. (B) Immunoblot analysis of Ikaros expression in HCC samples. (C) The overall survival analysis of HCC patients with Ikaros expression. *, p<0.05; n=102; Log-rank p=0.045. (D) Immunoblot analysis of the expression of Erk1/2, ETS1, Ikaros, and CD133 in HCC samples. (E) The function model of Ikaros in HCC. MAPK pathway regulates the activity of ETS1 via phosphorylation of ETS1. ETS1 is able to up-regulate Ikaros expression, and Ikaros interacting with CtBP inhibits CD133 expression via directly binding to CD133 P1 promoter.
DISCUSSION
Although Ikaros plays an indispensable role in the development of leukemia, its role in solid tumors remains unknown. Yang et al examined the relationship between Ikaros expression and prognosis in 13 types of cancers and found that the relationship varied between cancers, even between the same cancers obtained from different databases [31]. In the current study, we confirmed for the first time that Ikaros plays an important role in HCC. We determined that Ikaros functions as a transcription repressor and inhibits CD133 expression in HCC.
In our study and other previous studies, CD133 was identified as a marker of CSCs in HCC [5, 6, 7]. Overexpression of CD133 in cancers is closely related to the poor prognosis associated [32]. Although several patterns of CD133 regulation have been reported, its mechanism still remains unclear. CD133 expression was regulated by Sp1/myc [33]. CD133 and CD133+ CSCs was found to be regulated by miR-142-3p in HCC [34], and Lupeol inhibited liver tumor-initiating cells and decreased CD133 expression through phosphatase and tensin homolog modulation [35]. CD133 expression is enhanced under hypoxic conditions via HIF-1α/mTOR signaling in glioblastoma [31, 36]. Furthermore, epigenetic modification was another important factor for the regulation of CD133 expression in many tumors. TGFβ has been shown to elevate CD133 expression via demethylation of the CD133 P1 promoter by inhibiting the expression of DNMT1 and DNMT3β [37] and Promoter methylation decreased CD133 expression in ovarian cancer and glioblastoma [33, 38].
In this study, we determined that Ikaros suppresses CD133 expression by directly binding to the CD133 P1 promoter. We found that Ikaros induced the differentiation of CD133+ cancer stem-like cells. Thus, we concluded that inhibition of the stemness property of CSCs was one functional role of Ikaros in HCC. Ikaros protein was partially localized to the cytoplasm of HCC samples and HCC cells (Figure 6A and Supplementary S5A and B). Wild-type Ikaros localized in the nucleus after forced Ikaros expression in HCC cells (Supplementary Figure S6A). This result indicated that the subcellular localization of Ikaros in HCC was important for its function. In addition, 157 phenylalanine, 162 arginine, and 184 arginine are necessary for Ikaros DNA binding activity and subcellular localization [26]. Mutant Ikaros could not localize to the nucleus and could not regulate CD133 expression when these three amino acids were mutated to alanine (Supplementary Figure S6B and C). However, part of the Ikaros was found to be subcellular localized in the cytoplasm of HCC as assessed by IHC. Altered localization of Ikaros may be difficult to act as a transcription factor in HCC cells, which requires confirmation in future studies.
In present study, we found that pETS1 was higher in HCC samples than their matched noncancerous liver specimens, and there is no difference in ETS1 expression. Although Ito et al reported that ETS1 was over-expressed in HCC [41]. Cowley et al reported that the p-ETS1 is the inhibitory form of ETS1 activity [30], therefore, we consider that phosphorylation modification is critical for ETS1 function and its activity in HCC samples. We also found that HCC samples Ito et al detected are HCC patients with hepatitis virus C (HCV), however, the HCC samples we used are HCC patients with hepatitis virus B (HBV). This different pathogens may result in the discrepancy of ETS1 expression between our data and that of Ito et al.
In this study, we confirmed that CtBP could interact with Ikaros as a complex, and repressed CD133 protein expression in HCC. CtBP, as a transcription repressor, plays important roles in multiples of cancers. CtBP1 interacted with Ikaros in pituitary tumor cells, and modulated their survival in response to hypoxia [42]. CtBP also by binding with p19Arf can inhibit the invasion of hepatocellular carcinoma cells [43] and decrease the sensitivity of breast cancer cells to mechanistically diverse cancer chemotherapeutic agents via p53-dependent and -independent roles [44]. In colon carcinomas, high levels of Zeb1 and CtBP were correlated with low levels of E-cadherin [45]. CtBP also drived the EMT, stem cell pathways and genome instability in breast cancer [46]. Usually, CtBP interacts with other transcription factors regulated the expression of down-stream genes. CtBP and PRDM16 co-control the brown and white fat-selective gene programs [47]. And in leukemic transformation of hematopoietic cells, Evi-1 interacted with CtBP [48] and repressed the transforming growth factor beta signaling [49]. Importantly, CtBP contributed to the activities of oncogenes Hdm2, Znf217, and multiple drug resistance genes MDR1 [50, 51, 52]. CtBP interacted with other transcription factors as complex, inhibited down-stream genes expression. In addition, CtBP might participate in multiples of biological process and interacted with other corepressors in HCC. In present study, Ikaros was one partner we determined in HCC. CtBP-Ikaros complex functioned as a transcription repression complex and inhibited CD133 expression. Other function of CtBP is needed to be explored in future.
In summary, we provide evidence that Ikaros acts as a transcription repressor in HCC, and our results indicate that Ikaros may be a potential target for anticancer therapy of HCC by enhancing its activity.
MATERIALS AND METHODS
Cell lines
Hep3B, PLC/PRF/5, and 293T cells were all obtained from the American Type Culture Collection (Manassas, VA, USA). Huh7 cells were obtained from the Riken Cell Bank (Tsukuba, Japan). SMMC-7721 cells were provided by the Cell Bank of the Institute of Biochemistry and Cell Biology, China Academy of Sciences (Shanghai, China). MHCC-LM3 cells were provided by the Liver Cancer Institute of Zhongshan Hospital, Fudan University (Shanghai, China). HCC-LY5 cells were established from a human primary HCC tissue in our lab.
Cell culture
All cell lines used in this study were cultured in DMEM (Sigma-Aldrich) supplemented with 10% bovine serum (Hyclone), penicillin (500 U/ml), and streptomycin (500 μg/ml) and were incubated in 5% CO2 at 37°C. For the differentiation-inducing assay of CSCs in vitro, HCC cells were plated in six-well plates (NUNC, USA) in a monolayer culture with serum-free chemically defined medium (CDM). The CDM consisted of DMEDM/F12 supplemented with 0.5 × B27 supplements, 10 ng /ml basic fibroblast growth factor (bFGF) (Millipore), 10 ng epithelial growth factor (EGF) (Millipore), 500 U/ml penicillin, and 500 μg /ml streptomycin.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay was performed in HCC-LY5, SMMC-7721, and MHCC-LM3 cells. The cells were cross-linked with 10% paraformaldehyde for 10 min at 37°C, and crosslinking was reversed with 1 M glycine for 5 min at 37°C. After washing with 1× PBS buffer, the cells were harvested in T-PER Tissue Protein Extraction Reagent (Thermo Scientific) for 5 min on ice and centrifuged at 2,000 g for 5 min. The precipitants were suspended in nuclei lysis buffer, and the DNA was shredded into fragments of 1,000 base pairs by sonication. Antibodies against Ikaros, CtBP and ETS1 (Santa Cruz Biotechnology) using protein G agarose beads (Sigma-Aldrich) were added and incubated overnight at 4°C. After reversing the crosslinks, the DNA was isolated and used for polymerase chain reaction (PCR) analysis. Primers for quantitative PCR (qPCR) are listed in Supplementary Table 1
DNA pull-down assay
For DNA pull-down assays, 100 μg of nuclear protein per sample was incubated with 1 μg of biotinylated double-stranded oligonucleotides for 30 min at 37°C. Subsequently, DNA-protein complexes were incubated with streptavidin-agarose beads (Invitrogen) for 2 h at 37°C. After washing, the complexes were subjected to western blotting analyses. Sequences for DNA pull-down are provided as follows: Ikaros-normal:TCAGCTTTTGGGAATCTCCTGTCA; Ikaros-control: GC TTTTGGGAATCTCCTGTCA; Ikaros-normal: GAGAGTTTTTAGGAGGGTAGCTGC; Ikaros-mutant: GAGAGTTTTTAGTAGCTGC; ETS1-normal: CGAGTGAG CAACTTCAGGAATCATTGGAAGAAG; ETS1-mutant: CGAGTGAGCAACTTT TCCGCATCATTGTGAAGAAAG.
Co-immunoprecipitation
Co-immunoprecipitation assays were performed using HCC-LY5, SMMC-7721, and MHCC-LM3 cells. After washing with 1× PBS buffer, the cells were harvested in T-PER Tissue Protein Extraction Reagent (Thermo Scientific) for 5 min on ice and were centrifuged at 2,000 g for 5 min. The precipitants were suspended in RIPA (Santa Cruz Biotechnology) lysis buffer for 50 min on ice and centrifuged at 5,000 g for 15 min. Antibodies against Ikaros and CtBP (Santa Cruz Biotechnology) with protein G agarose beads (Sigma-Aldrich) were added and incubated overnight at 4°C. After washing, the complexes were subjected to western blot analysis.
Luciferase reporter assay
Cells were plated in 96-well culture plates for 24 h and were transfected with the appropriate constructs. Renilla and firefly luciferase activity were measured according to the manufacturer’s instructions (Promega).
Quantitative real-time PCR
RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol and was reverse-transcribed into cDNA using PrimeScriptTM RT Reagent Kit (TaKaRa). Primers for quantitative RT-PCR are provided in Supplementary Table 1.
Western blotting
Western blotting was performed as described by the SuperSignal West Femto Maximum Sensitivity Substrate Kit (Pierce). The primary and HRP-conjugated secondary antibodies are listed in Supplementary Table 2.
Plasmid constructs, lentivirus production, and cell transfection
Full-length human Ikaros (Origene) was subcloned into pWPXL (Addgene) at the BamH I and EcoR II sites. The CD133 full-length and deletion reporter constructs were generated and cloned into a pGL3-enhancer vector (Promega) at the Kpn I and Hind III sites. Three Ikaros single DNA binding sites mutants (K137A, R162A, and R184A) were generated using the megaprimer PCR method as previously described [39]. Virus packaging and cell transfection were performed as previously described [40]. Primers for cloning are provided in Supplementary Table 3.
RNA interference-based gene knockdown experiments
Small-interfering RNA (siRNA) oligos targeting Ikaros, ETS1 and a negative control (Cat. No. B01001) were synthesized and annealed by GenePharma (Shanghai, China). Three fragments were designed to target the corresponding gene transcripts, and the silencing effects of the sequences were validated by western blotting analyses. Sequences for siRNA are provided as follows: siIkaros-1: AAGACCTGTGCAAGATAGGAT; siIkaros-2: GUCGUGGCCAGUAAUGUUATT; siIkaros-3: GACGCACUCCGUUGGUAAATT; siETS1-1: ACUUGCUACCAUCC CGUAC; siETS1-2: GGACAAGCCUG UCAUUCCU; siETS1-3: GCAGCAACUUG AAUUUGCUCACCAA.
Magnetic-activated cell sorting
HCC-LY5, SMMC-7721, and MHCC-97L cells were magnetically isolated from CD133+ and CD133- cells with corresponding antibodies using the Easysteo PE Selection Kit (StemCell Technologies) according to the manufacturer’s instructions.
Immunofluorescent confocal imaging
Cells were seeded onto glass slides for 24 h, fixed in 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100 for 15 min. The slides were then incubated with primary antibody in blocking solution overnight at 4°C in a humidified chamber. The glass slides were then washed three times in PBS and incubated in Alexa 594-conjugated secondary antibody and 4’6-diamino-2-phenylindole (DAPI) in blocking solution for 40 min at 37°C in a humidified chamber. Images were obtained with a confocal laser microscope (IX-70, Olympus 40M, Olympus, London, UK).
Immunohistochemistry
Paraffin-embedded tissue array sections (5μm in thickness) were prepared and immunodetections were detected by immunofluorescence according to the procedures described previously [6]. The results were visualized and photographed under an Axioskop 2 microscope (Carl Zeiss, Oberkochen, Germany) with a DP70 CCD system (Olympus, Tokyo, Japan).
Plate colony formation assay
Two thousand cells were seeded into six-well culture plates and fixed seven days later using 10% formaldehyde for 30 min at 37°C. The cells were stained using GIEMSA (Sigma-Aldrich) for 30 min at 37°C. After washing, the cells cell colonies were quantified.
MTT
Three thousand cells were seeded into 96-well culture plates and incubated for 24 h. Next, 100 μL MTT (5 mg /ml) was added to each well. Cells were incubated for 4 h at 37°C. The media was removed, and 100 μL DMSO was added in the dark for 2 h at 37°C. The OD was recorded at an absorbance of 570 nm.
Fluorescence activated cell sorting
Huh7, PLC/PRF/5, HCC-LY5, and SMMC-7721 cells were incubated with a PE-conjugated CD133/1(AC133) antibody (Miltenyi Biotec, Germany) and sorted into CD133+ and CD133- cell subpopulations on an Epics Altra flow cytometer (Beckman Coulter, USA)
Drugs and drug-resistance test
Cisplatin (Sigma-Aldrich), Doxorubicin (Sigma-Aldrich), Sorafenib (Biochemparterner) were diluted to final concentrations with corresponding culture medium. After treated with drugs for 48 h, 100 μL MTT (5 mg /ml) was added. Cells were incubated for 4 h at 37°C. Removed media and added 100 μL DMSO in dark for 2 h at 37°C. OD was recorded absorbance at 570 nm.
Tumor xenograft models
Six to eight-week-old BALB/c (nu/nu) mice were randomly divided into groups and inoculated with the suspended HCC cells (5×106 Huh7 cells, 5×103 CD133+ or CD133- HCC cells were inoculated into the mice). At the end of the experimental period, the animals were sacrificed. Immediately after killing, xenograft tumors were weighed and fixed in neutral buffered formalin. Fixed tumor tissues were analyzed using western blotting analyses.
Patients
One hundred and two human HCC tissue samples were obtained from patients who underwent surgical treatment at the First Affiliated Hospital of Zhejiang University (Hangzhou, China), the Qidong Liver Cancer Institute (Qidong, China) or the Guangxi Cancer Institute (Nanning, China). The 102 HCC patients included 84 males and 18 females (mean age: 52.21 years, ranging from 30 to 76 years). None of the patients received chemotherapy or other treatment before surgery. All procedures were performed under consensus agreements and in accordance with the China Ethical Review Committee. All tissue samples were fixed in 4% phosphate-buffered neutral formalin for at least 72 hr and routinely embedded in paraffin.
Statistical analysis
Statistical analyses were performed using SPSS 13.0 software. The results were presented as the mean ± SD and compared using Student’s t test. Survival analysis was performed using the Kaplan-Meier method. Statistical computations were performed using GraphPad Prism version 5.0. p<0.05 was considered significant. *p < 0.05; **p<0.01.
ACKNOWLEDGMENTS
All authors have no financial, professional or personal conflicts to disclose.
This work was supported in part by grants from the National Key Program for Basic Research of China (973) (2015CB553905), National Natural Science Foundation of China (81472726, 81272438, 81301859, 81372192), Key Discipline and Specialty Foundation of Shanghai Municipal Commission of Health and Family Planning and the National Key Sci-Tech Special Project of China (2013ZX10002-011), the SKLORG Research foundation (91-13-02, 91-14-09).
REFERENCES
1. Sukowati CH, Rosso N, Croce LS, Tiribelli C. Hepatic cancer stem cells and drug resistance: Relevance in targeted therapies for hepatocellular carcinoma. World J Hepatol. 2010; 2: 114-126.
2. Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009; 106: 16281-16286.
3. Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, Barnard GF, Doki Y, Mori M. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010; 120: 3326–3339.
4. Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006; 25: 1696-1708.
5. Xu X, Liu RF, Zhang X, Huang LY, Chen F, Fei QL, Han ZG. DLK1 as a potential target against cancer stem/progenitor cells of hepatocellular carcinoma. Mol Cancer Ther. 2012; 11: 629-638.
6. Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, Yang S, Zheng S, Gu J. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007; 120: 1444-1450.
7. Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, Li J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010; 126: 2067-2078.
8. Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006; 351: 820-824.
9. Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007; 132: 2542-2556.
10. Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, Ng IO, Man K, To KF, Lai PB, Lo CM, Guan XY, Chan KW. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012; 55: 807-820.
11. Song W, Li H, Tao K, Li R, Song Z, Zhao Q, Zhang F, Dou K. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract. 2008; 62: 1212-1218
12. Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science. 1992; 258: 808-812.
13. Yagi T, Hibi S, Takanashi M, Kano G, Tabata Y, Imamura T, Inaba T, Morimoto A, Todo S, Imashuku S. High frequency of Ikaros isoform 6 expression in acute myelomonocytic and monocyt leukemias: implications for up-regulation of the antiapoptotic protein Bcl-XL in leukemogenesis. Blood. 2002; 99: 1350-1355.
14. Jäger R, Gisslinger H, Passamonti F, Rumi E, Berg T, Gisslinger B, Pietra D, Harutyunyan A, Klampfl T, Olcaydu D, Cazzola M,Kralovics R. Deletions of the transcription factor Ikaros in myeloproliferative neoplasms. Leukemia. 2010; 24: 1290-1298.
15. Singh H, Medina KL, Pongubala JMR. Contingent gene regulatory networks and B cell fate specification. Proc Natl Acad Sci USA. 2005; 102: 4949-4953.
16. Nakase K, Ishimaru F, Avitahl N, Dansako H, Matsuo K, Fujii K, Sezaki N, Nakayama H, Yano T, Fukuda S, Imajoh K, Takeuchi M,Miyata A, et al. Dominant negative isoform of the Ikaros gene in patients with adult B-cell acute lymphoblastic leukemia. Cancer Res. 2000; 60: 4062-4065.
17. Nakayama H, Ishimaru F, Avitahl N, Sezaki N, Fujii N, Nakase K, Ninomiya Y, Harashima A, Minowada J, Tsuchiyama J, Imajoh K,Tsubota T, Fukuda S, et al. Decreases in Ikaros activity correlate with blast crisis in patients with chronic myelogenous leukemia. Cancer Res. 1999; 59: 3931-3934.
18. Ezzat S, Yu S, Asa SL. Ikaros isoforms in human pituitary tumors: distinct localization, histone acetylation, and activation of the 5’ fibroblast growth factor receptor-4 promoter. Am J Pathol. 2003; 163: 1177-1184.
19. Dumortier A, Jeannet R, Kirstetter P, Kleinmann E, Sellars M, dos Santos NR, Thibault C, Barths J, Ghysdael J, Punt JA, Kastner P,Chan S. Notch activation is an early and critical event during T-Cell leukemogenesis in Ikaros-deficient mice. Mol Cell Biol. 2006; 26: 209-220.
20. Kathrein KL, Chari S, Winandy S. Ikaros directly represses the notch target gene Hes1 in a leukemia T cell line: implications for CD4 regulation. J Biol Chem. 2008; 283: 10476-10484.
21. Alsiö JM, Tarchini B, Cayouette M, Livesey FJ. Ikaros promotes early-born neuronal fates in the cerebral cortex. Proc Natl Acad Sci USA. 2013; 110: 716–725.
22. Ezzat S, Mader R, Fischer S, Yu S, Ackerley C, Asa SL. An essential role for the hematopoietic transcription factor Ikaros in hypothalamic–pituitary-mediated somatic growth. Proc Natl Acad Sci USA. 2006; 103: 2214-2219.
23. Shmelkov SV, Jun L, St Clair R, McGarrigle D, Derderian CA, Usenko JK, Costa C, Zhang F, Guo X, Rafii S. Alternative promoters regulate transcription of the gene that encodes stem cell surface protein AC133. Blood. 2004; 103: 2055-2061.
24. Nichogiannopoulou A, Trevisan M, Neben S, Friedrich C. Georgopoulos K. Defects in hemopoietic stem cell activity in Ikaros mutant mice. J Exp Med. 1999; 190: 1201-1214.
25. Lopez RA, Schoetz S, DeAngelis K, O’Neill D, Bank A. Multiple hematopoietic defects and delayed globin switching in Ikaros null mice. Proc Natl Acad Sci USA. 2002; 99: 602-7.
26. Cobb BS, Morales-Alcelay S, Kleiger G, Brown KE, Fisher AG, Smale ST. Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev. 2000; 14: 2146-2160.
27. Koipally J, Georgopoulos K. Ikaros interactions with CtBP reveal a repression mechanism that is independent of histone deacetylase activity. J Biol Chem. 2000; 275: 19594-19602.
28. Ohnishi S, Maehara O, Nakagawa K, Kameya A, Otaki K, Fujita H, Higashi R, Takagi K, Asaka M, Sakamoto N, Kobayashi M, Takeda H. Hypoxia-Inducible Factors Activate CD133 Promoter through ETS Family Transcription Factors. PLoS One. 2013; 8: e66255.
29. Grund EM, Spyropoulos DD, Watson DK, Muise-Helmericks RC. Interleukins 2 and 15 regulate Ets1 expression via ERK1/2 and MNK1 in human natural killer cells. J Biol Chem. 2005; 280: 4772-4778.
30. Cowley DO, Graves BJ. Phosphorylation represses Ets-1 DNA binding by reinforcing autoinhibition. Genes Dev. 2000; 14: 366-376.
31. Yang L, Luo Y, Wei J. Integrative genomic analyses on Ikaros and its expression related to solid cancer prognosis. Oncol Rep. 2010; 24: 571-577.
32. McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res. 2009; 7: 489-497.
33. Gopisetty G, Xu J, Sampath D, Colman H, Puduvalli VK. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by Sp1/myc and promoter methylation. Oncogene. 2013; 32: 3119-3129
34. Chai S, Tong M, Ng KY, Kwan PS, Chan YP, Fung TM, Lee TK, Wong N, Xie D, Yuan YF, Guan XY, Ma S. Regulatory role of miR-142-3p on the functional hepatic cancer stem cell marker CD133. Oncotarget. 2014 (Epub ahead of print).
35. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. Lupeol targets liver tumor-initiating cells through phosphatase and tensin homolog modulation. Hepatology. 2011; 53: 160-170.
36. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, Pollack IF, Park DM. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene. 2009; 28: 3949-3959.
37. You H, Ding W, Rountree CB. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology. 2010; 51: 1635-1644.
38. Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, Huang Z, Bentley RC, Mori S, Fujii S, Marks JR, Berchuck A,Murphy SK. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2009; 28: 209-218.
39. Barik S. Site-directed mutagenesis in vitro by megaprimer PCR. Methods in Mol biology. 1996; 57: 203-215.
40. Hu C, Li H, Li J, Zhu Z, Yin S, Hao X, Yao M, Zheng S, Gu J. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation by Akt signaling. Carcinogenesis. 2008; 29: 2289-2297.
41. Ito Y, Miyoshi E, Takeda T, Sakon M, Noda K, Tsujimoto M, Monden M, Taniguchi N, Matsuura N. Expression and Possible Role of ets-1 in Hepatocellular Carcinoma. Am J Clin Pathol. 2000; 114:719-725.
42. Dorman K, Shen Z, Yang C, Ezzat S, Asa SL. CtBP1 interacts with Ikaros and modulates pituitary tumor cell survival and response to hypoxia. Mol Endocrinol. 2012; 26: 447-457.
43. Chen YW, Paliwal S, Draheim K, Grossman SR, Lewis BC. p19Arf inhibits the invasion of hepatocellular carcinoma cells by binding to C-terminal binding protein. Cancer Res. 2008; 68: 476-82.
44. Birts CN, Harding R, Soosaipillai G, Halder T, Azim-Araghi A, Darley M, Cutress RI, Bateman AC, Blaydes JP. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol Cell. 2010; 103: 1-19.
45. Peña C, García JM, García V, Silva J, Domínguez G, Rodríguez R, Maximiano C, García de Herreros A, Muñoz A, Bonilla F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int J Cancer. 2006; 119: 2098–2104.
46. Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S, Baek S, Hunter K, Yang H, Lee M, Zvosec C, Khramtsova G, Cheng F, Perou CM, Miller CR, Raab R,Olopade OI, Gardner K. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun. 2013; 4: 1449.
47. Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008; 22: 1397-409.
48. Nitta E, Izutsu K, Yamaguchi Y, Imai Y, Ogawa S, Chiba S, Kurokawa M, Hirai H. Oligomerization of Evi-1 regulated by the PR domain contributes to recruitment of corepressor CtBP. Oncogene. 2005; 24: 6165-6173.
49. Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood. 2001; 97: 2815-2822.
50. Mirnezami AH, Campbell SJ, Darley M, Primrose JN, Johnson PW, Blaydes JP. Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. Curr Biol 2003; 13: 1234–1239.
51. Quinlan KG, Verger A, Yaswen P, Crossley M. Amplification of zinc finger gene 217 (ZNF217) and cancer: when good fingers go bad. Biochim Biophys Acta. 2007; 1775: 333–340
52. Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z,Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR,Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 2008; 40: 310–315.