The cover for issue 60 of Oncotarget features Figure 8, "Modeling of palbociclib resistance in KRAS-mutant NSCLC cells and effects of MAP kinase pathway inhibition," by Haines, et al.
The research demonstrates that KRAS-mutant NSCLC cell lines are initially sensitive to the CDK4/6 inhibitor palbociclib, but readily acquire resistance associated with increased expression of CDK6, D-type cyclins and cyclin E. Resistant cells also demonstrated increased ERK1/2 activity and sensitivity to MEK and ERK inhibitors. Moreover, MEK inhibition reduced the expression and activity of cell cycle proteins mediating palbociclib resistance.
Dr. Geoffrey I. Shapiro from the Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA and the Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, USA said, "KRAS mutations occur in approximately 15-30% of non-small cell lung cancers."
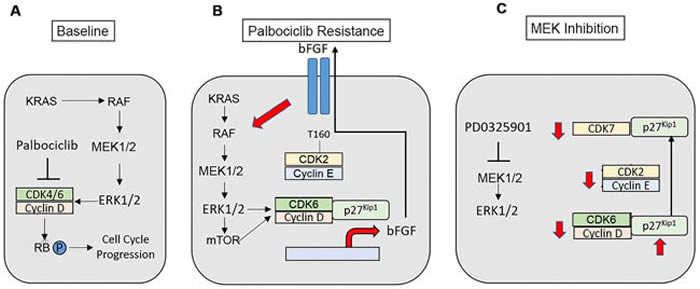
Figure 8: Modeling of palbociclib resistance in KRAS-mutant NSCLC cells and effects of MAP kinase pathway inhibition. A. KRAS-mutant NSCLCs signal via the MAP kinase pathway to stimulate cell cycle progression. B. A bFGF autocrine/paracrine loop propagates and maintains palbociclib resistance in NSCLC. Increased signals through MEK-ERK1/2-mTOR enhance the expression, assembly and activity of cyclin D-CDK6, which promotes palbociclib-resistant proliferation and growth and enhances the transcription of the FGFR ligand bFGF. bFGF is secreted, where its activates its receptor to further enhance MEK-ERK1/2 signaling within the individual resistant cell as well as in neighboring cells. Increased MAP kinase activity in the palbociclib-resistant state also leads to increased cyclin E-CDK2 activity. C. MAP kinase pathway inhibition is expected to disrupt the autocrine loop, reducing the expression of D-cyclins and CDK6. MEK inhibition causes increased expression of p27Kip1, as well as a redistribution of p27Kip1 from CDK4/6 to CDK7 with maintenance of the association of p27Kip1 with CDK2. These events block the activating phosphorylation of CDK2 mediated by CAK, and directly block CDK2 activity. Therefore, MAP kinase inhibition reduces the activity of all G1 CDK complexes in palbociclib-resistant cells.
ERK1/2 activation, downstream of activated KRAS and MEK, is well described for its role in promoting cell cycle progression and proliferation by enhancing the expression of D-cyclins that are critical for promoting the transition from G1 to S phase. In lung cancer cell lines and genetically-engineered mouse models, depletion, genetic ablation or inhibition of CDK4/6 has demonstrated synthetic lethality in KRAS-mutant, but not wild-type backgrounds, suggesting the therapeutic relevance of selective CDK4/6 inhibition.
Moreover, CDK4/6 inhibition has been reported to synergize with MEK inhibition in a variety of cancers driven by mutant RAS, including NSCLC. Here, the research team demonstrates that the initial efficacy of palbociclib in both in vitro and in vivo models of KRAS-mutant NSCLC is complicated by the rapid onset of acquired resistance, mediated by increased expression of CDK6, cyclin D1, cyclin D3 and cyclin E.
Additionally, they established the importance of increased ERK1/2 activity in palbociclib-resistant cells that mediates D-cyclin and CDK6 expression; ERK activity is controlled upstream in part by FGFR1 and exerts its effects through mTOR activation.
The Geoffrey I. Shapiro research team concluded, "The present study highlights a novel mechanism of palbociclib resistance in KRAS-mutant NSCLC that stems from the ERK-dependent induction of CDK6 and cyclins D1 and D3, as well as increased expression of cyclin E. Increased expression of G1 cyclin-CDK complexes results in part from increased FGFR1 mediated-signaling that is transferable and maintained via bFGF secretion and that causes activation and dependence on a MEK-ERK-mTOR pathway."
Full text - https://doi.org/10.18632/oncotarget.25803
Correspondence to - Geoffrey I. Shapiro - [email protected]
Keywords - CDK4/6 inhibitor, MEK inhibitor, FGFR1, KRAS-mutant non-small cell lung cancer, drug resistance



