The cover for issue 81 of Oncotarget features Figure 1, "Molecular Mechanisms of Autophagy," by Servante, et al.
On one face, especially in the early phases of cancer formation, autophagy can act as a cellular housekeeper to eliminate damaged organelles and recycle macromolecules, thus acting as tumor suppressor. The researchers present an overview of the relevance of autophagy in central nervous system tumors including how its chemical modulation can serve as a useful adjunct to chemotherapy, and use this knowledge to consider how targeting of autophagy may be relevant in pediatric brain tumors.
Dr. Juliette Servante from the School of Life Sciences, at the University of Nottingham, Medical School, in Nottingham, NG7 2UH, UK said, "Central nervous system brain tumors are both the leading cause of cancer-related death in childhood/adolescence and the most common form of solid Tumor in this age group."
Central nervous system brain tumors are both the leading cause of cancer-related death in childhood/adolescence and the most common form of solid Tumor in this age group. The mainstay of treatment of pediatric brain tumors is surgery with optional adjuvant radiotherapy or chemotherapy determined through patient age, Tumor type, degree of surgical resection, location and grade. Autophagy may also affect cell survival as aberrant proteins, if left, could build up within the cell and have the potential to affect signalling and transport mechanisms.
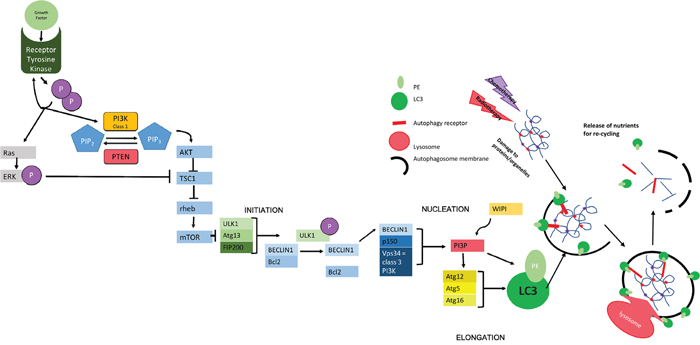
Figure 1: Molecular Mechanisms of Autophagy. The initiation of autophagy is controlled through a series of complexes involving a group of evolutionally conserved proteins known as "autophagy related proteins" (ATGs) which eventually lead to the production of scaffold protein LC3-II; essential for autophagosome function. The initial complex involved incorporates ULK1/2 (uncoordinated 51-like kinase 1/2), ATG13 (a regulator of ULK1 auto-induction) and FIP200 (also a regulator) [38] and its assembly results in auto-phosphorylation of ATG13/ULK1. A subsequent conformational change in this preliminary complex allows the formation of further complexes [39]. The second complex formed at the site of autophagosome construction involves Beclin 1, which has been identified as a Tumor suppressor [40]. Beclin 1 interacts with the anti-apoptotic regulator Bcl2 [41, 42]. This coupling is broken under situations of starvation which allows Beclin 1 to associate with VPS34 (a class 3 PI3K) and p150 (also known as VPS15) to produce PI3P. PI3P interacts with one of several WIPI proteins (WD40 repeat protein interacting with phospho-inositides) and the WIPI protein subtype determines the rate of autophagy [39, 43, 44]. In the next step of autophagy, stimulation of ATG12 allows the formation of a complex that helps in the conversion of LC3-I to LC3-II via the addition of phosphatidylethanolamine (PE) and in the positioning of this modified protein on the developing autophagosome [45] where it acts as a scaffold protein [46]. The specificity of autophagy comes from the involvement ofautophagy receptors, such as SQSTM1/p62, that can simultaneously bind to the autophagosomal membrane (via LC3) and to ubiquitin modifications used to mark autophagic targets [47]. The growing autophagosome encircles both the receptor and its target for recycling as well as other cellular waste, forming a double-membrane vesicle that is able to fuse to a lysosome either directly or via fusion with an endosome derivative (multi-vesicular body, MVB [15, 48]). Fusion with the lysosome allows the release of digestive enzymes into the autophagosome with consequent catabolism of proteins and organelles resulting in the release of amino acids for recycling [49]. The chief inhibitor of autophagy, mTOR works to inhibit the initial ULK1-ATG13-FIP200 complex. mTOR is a protein kinase - active when energy supply is sufficient [19, 43] - that hyperphosphorylates ATG13 and prevents auto-phosphorylation of ULK1, thereby inhibiting further steps [50, 51]. mTOR works downstream of growth factors and is also controlled by feedback of both cellular energy levels and protein availability. This includes the monitoring of amino acid levels in lysosomes using v-ATPase (vacuolar-type H+ ATPase) in the lysosomal membrane. Where levels of amino acids are sufficient, the binding of growth factors to tyrosine kinase receptors leads to receptor autophosphorylation and consequent activation of both PI3K and Ras. Class 1 PI3K aids the phosphorylation of PIP2-PIP3., thus triggering AKT to inhibit the formation of a complex between TSC1 and TSC2. The phosphorylation of ERK by Ras also inhibits this interaction. The TSC1-2 complex normally acts to inactivate the GTPase rheb. When active, rheb upregulates mTOR activity. Therefore, action of AKT indirectly up-regulates mTOR, meaning that autophagy is inactive [52]. This is reversed during periods of starvation where nutrients are less abundant, meaning that mTOR becomes inactive and autophagy occurs at an enhanced rate. PTEN (phosphatase/tensin homolog on chromosome 10) is a phosphatase acting on lipids to cause the conversion of PIP3 back to PIP2, thus increasing levels of cellular autophagy via reduced mTOR activity [53].
Autophagy modulation has also been considered as a treatment strategy for brain tumors both in adults and children and although its exact role in pediatric CNS neoplasms is not yet known, build-up of autophagosomes is enhanced in tumors such as gangliogliomas; in treatment with chemotherapy agent temozolomide; and in association with cell death.
Current evidence for the potential of a combinatorial treatment strategy comes mostly from studies of adult tumors; this will be explored with consideration of the distinctions between adult and childhood brain tumors.
The Juliette Servante research team concluded, "Disease-associated genetic mutation can have an effect on baseline cellular autophagy rates as demonstrated in V600E mutation of BRAF and this could make autophagy modulation particularly useful in affected cells."
Full text - https://doi.org/10.18632/oncotarget.26186
Correspondence to - Juliette Servante - [email protected]
Keywords - autophagosome, autophagy, childhood brain tumors, chloroquine, mTOR



