
Introduction
Thyroid cancer is the most common endocrine malignancy with complex carcinogenesis mechanisms. The incidence of thyroid cancer increased over the past 4 decades [1, 2]. Thyroid cancer is the second most common cancer in women next to breast cancer and the fifth most common cancer in all gender in Saudi Arabia [3]. Differentiated thyroid cancer (DTC) accounts for ~90% of all thyroid cancers and is comprised of papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC) [4].
Major advances in DNA sequencing technologies revealed various molecular abnormalities in coding [4, 5] and non-coding genes [6] of several signaling pathways in thyroid cancer. Somatic genetic alterations in these pathways are mostly mutually exclusive in the course of malignant transformation with varying rates among different populations. DTCs frequently (~70%) harbor activating somatic alterations in the genes of mitogen-activated protein kinase (MAPK) signaling pathway [7, 8]. The most prevalent genetic alterations are the hotspot point mutation BRAFV600E [9, 10], RAS single point mutations, and fusions in RET/PTC [11], NTRK1/3 and PAX8-PPARγ [8, 12]. A low frequency of mutations has also been reported in the receptor tyrosine kinases (RTKs) including EGFR [13] and members of the phosphatidylinositol-3 kinase (PI3K) pathway such as PIK3CA, PTEN and AKT1 [14–16]. Previous studies also reported some point mutations in the genes of metabolic and regulatory pathways which include IDH1 [17] and TERT. Somatic alterations of these genes are strongly associated with distinct clinicopathological features of the tumors [18, 19]. Next-generation whole genome sequencing (WGS) of PTCs revealed additional somatic driver alterations that include EIF1AX, PPM1D, and CHEK2 [4]. Although similar histology is observed in both pediatric and adult cases, the genetic profile and the frequency of the commonly identified genetic mutations in adult DTCs are different from those in the pediatric DTCs [20] suggesting significant differences in the pathogenesis between adult and pediatric DTCs. Poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancers (ATC) are aggressive subtypes of thyroid cancer [7, 8]. In addition to the frequent occurrence of the oncogenic mutations of BRAFV600E and NRAS, mutations in the other critical genes such as TERT, TP53, EIF1AX, PIK3CA, PTEN, ALK, and mTOR mutations were frequently detected in PDTC and ATC [5, 7, 8, 21, 22, 23].
Lysophosphatidic acid receptor 4 (LPAR4) is a 41.895 kDa protein molecule encoded by 370 amino acids (except stop codon) which are translated from 1110 nucleotides. This gene is located on the X chromosome (Xq21.1). LPAR4 is also variably called as G-protein coupled receptor 23 (GPCR23), purinergic receptor 9, P2Y purinoceptor 9, P2Y5-like receptor, LPA receptor 4, and GPR23. It is one of the six subtypes (LPAR1-6) of G protein-coupled transmembrane receptors (GPCR) that is activated by ubiquitous bioactive ligand named lysophosphatidic acid (LPA), an extracellular biological lipid that is framed by a glycerol, a fatty acid, and a phosphate. LPARs are classified into two families, the endothelial cell differentiation gene (Edg) family (LPAR1-3) and the non-Edg purinergic receptor family (LPAR4-6) [24]. The LPAR4 has three vital functions at the molecular level which includes G protein-coupled receptor activity, binding to lysophosphatidic acid, and lysophosphatidic acid receptor activity. It is mainly involved in the signaling pathways of G protein-coupled receptor, positive regulation of cytosolic calcium ion concentration involved in phospholipase C-activating G protein-coupled signaling, and positive regulation of Rho protein signal. LPA induced LPAR-mediated PI3K/AKT signaling via Gαi/o subunits is implicated in various biological functions including proliferation, motility, invasion, and tumorigenicity and it confers chemotherapy-resistance in certain cancers [25].
Recently, whole exome sequencing of a Chinese population with DTCs identified frequent somatic mutations in the genes of the GPCR-mediated PI3K pathway [26]. However, to date, this axis was not analyzed in detail in other populations. Furthermore, the roles of this axis were not studied in pure pediatric and adult cases. Here, we studied mutations in the major genes (LPAR4, PIK3CA, and PTEN) of the GPCR-mediated PI3K pathway in a large series of pediatric and adult DTCs, and in aggressive thyroid cancer (PDTC and ATC) samples (n = 323) from a Saudi Arab population. We also analyzed the most comprehensive TCGA study data of DTCs (n = 507) and aggressive thyroid cancers (PDTCs and ATCs) (n = 117) to determine if there are differences in the rates of these mutations among different populations. Further, to understand the prevalence and importance of genetic mutations of the GPCR-mediated PI3K pathway in other types of human cancers, we comprehensively analyzed the largest cohort of TCGA data from pan-cancer cell lines (n = 1080) and pan-tumor samples (n = 9020). This study on thyroid cancer in the pediatric and adult population along with the detailed analyses of international data provides a comprehensive data on the GPCR-mediated PI3K pathway not only in pediatric, adult DTCs and aggressive thyroid cancers (PDTC and ATC) but also in other human solid tumors.
Results
GPCR-mediated PI3K pathway genes (LPAR4, PIK3CA, and PTEN) are mutated both in pediatric and adult DTCs with similar low prevalence
Overall, GPCR-mediated PI3K pathway genes harbored mutations in 3.37% (3/89) and 2.45% (5/204) of pediatric and adult DTCs, respectively. PIK3CA mutations occurred in 2.24% (2/89) of pediatric DTC (both cases were conventional PTCs) and in 1.5% of (3/204) adult DTC [two in follicular variant PTC (FV-PTC) and one in tall cell variant (TPTC)]. PTEN mutations were present in1.1% (1/89) of pediatric conventional PTC and 1% (2/204) adult DTC (1 conventional PTC and 1 follicular variant PTC). The former one harbored a novel mutation (Q214H). Overall, in both pediatric and adult DTCs, PIK3CA mutations were found in 1.7% (5/293) and the PTEN mutations were found in 1% (3/293). We did not identify LPAR4 gene mutations in any of the samples analyzed. MNG samples had no mutation in any of the genes analyzed. We also found no mutation in the LPAR4, PIK3CA and PTEN genes of the 13 cases of PDTCs and ATCs (aggressive thyroid cancers). All the mutation positive samples were further analyzed for hotspot and non-hotspot mutations of commonly mutated genes in DTC which include BRAFV600E and TERT promoter mutations. We found 2 samples concomitantly harboring PIK3CA and BRAF mutations and one of the two BRAF mutations was a non-hotspot frameshift mutation (1798inTAC) and the other one was the common BRAFV600E mutation. Similarly, two samples with PTEN mutations coexisted with BRAFV600E hotspot mutation. However, we did not find TERT promoter mutations in any of the sequenced samples (Figure 1 and 3, Tables 1 and 2).
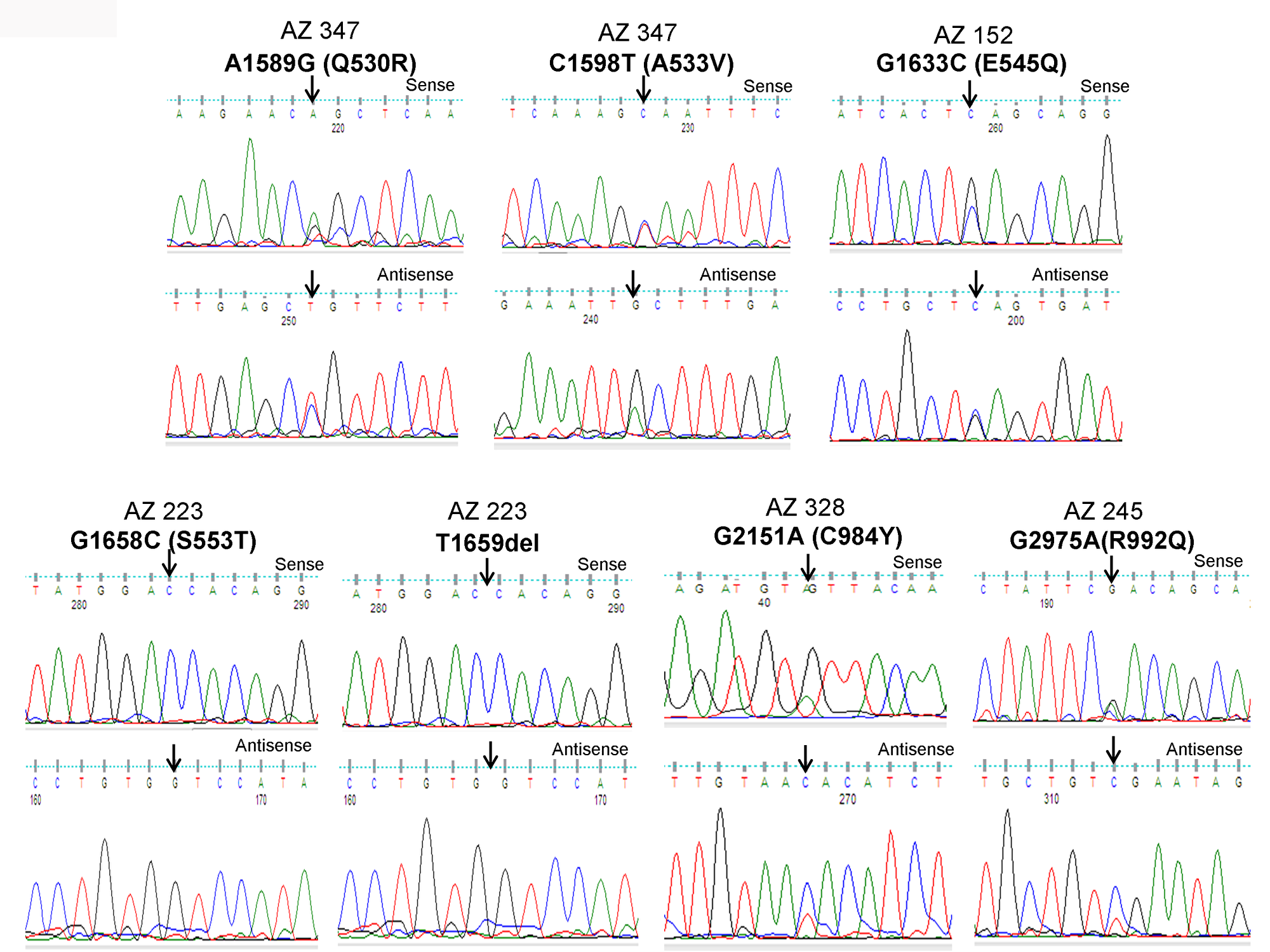
Figure 1: Identification of PIK3CA mutations in pediatric and adult DTCs. The illustration shows the sequencing electropherogram of the PIK3CA gene in various samples. The sequence shown on top and bottom indicates the sense and antisense strand, respectively. Altered nucleotide positions and its related amino acid (within bracket) are shown on top of each corresponding sequence. Mutated nucleotides are indicated by arrows. Nucleotide number refers to the position within the coding sequence of the PIK3CA gene, where nucleotide position 1 indicates the first nucleotide of the translation initiation codon. All samples were sequenced in 2 repeated experiments with independent PCR by sense and antisense primers.
Table 1: Summary of mutations identified in LPAR4, PIK3CA and PTEN genes of GPCR-mediated PI3K pathway in pediatric and adult thyroid cancer
| Samples | Tumor subtype | Number of Samples | LPAR4 | PIK3CA | PTEN |
|---|---|---|---|---|---|
| MNG | 17 | 0/17 (0%) | 0/17 (0%) | 0/17 (0%) | |
| Pediatric DTC | CPTC | 72 | 0/72 (0%) | 2/72 (2.7%) | 1/72 (1.4%) |
| FV-PTC | 7 | 0/7 (0%) | 0/7 (0%) | 0/7 (0%) | |
| TPTC | 2 | 0/2 (0%) | 0/2 (0%) | 0/2 (0%) | |
| DSC | 3 | 0/3 (0%) | 0/3 (0%) | 0/3 (0%) | |
| FTC | 3 | 0/3 (0%) | 0/3 (0%) | 0/3 (0%) | |
| HCC | 2 | 0/2 (0%) | 0/2 (0%) | 0/2 (0%) | |
| 89 | 0/89 (0%) | 2/89 (2.24%) | 1/89 (1.1%) | ||
| Overall | 3/89 (3.37%) | ||||
| Adult DTC | CPTC | 114 | 0/114 (0%) | 0/114 (0%) | 1/114 (0.9%) |
| FV-PTC | 55 | 0/55 (0%) | 2/55 (3.6%) | 1/55 (1.8%) | |
| TPTC | 29 | 0/29 (0%) | 1/29 (3.5%) | 0/29 (0%) | |
| DSC | 3 | 0/3 (0%) | 0/3 (0%) | 0/3 (0%) | |
| HCC | 3 | 0/3 (0%) | 0/3 (0%) | 0/3 (0%) | |
| 204 | 0/204 (0%) | 3/204 (1.5%) | 2/204 (1%) | ||
| Overall | 5/204 (2.45%) | ||||
| Total DTCs | 293 | 0/293 (0%) | 5/293 (1.7%) | 3/293 (1%) | |
| Aggressive cancer | PDTC | 10 | 0/10 (0%) | 0/10 (0%) | 0/10 (0%) |
| ATC | 3 | 0/3 (0%) | 0/3 (0%) | 0/3 (0%) | |
| 13 | 0/13 (0%) | 0/13 (0%) | 0/13 (0%) | ||
| 0/13 (0%) |
Mutations are identified in important domains and predict potential function
As illustrated in Figure 2A and 2B, PIK3CA mutations occur in both exons 9 and 20 coding helical and kinase domains of the p110α, respectively. Five out of seven mutations accumulated in the helical domains while the other two were in the kinase domain of PIK3CA. Of the 5 helical domain mutations, only one mutation was found in the helical domain hotspot at codon E545 (E545Q) and other mutations were away from the helical domain hotspot. Six (Q530R, A533V, E545Q, S553T, C984Y, and R992Q) out of these seven PIK3CA mutations were missense single point mutations and one (T1659del) had a single nucleotide deletion (Table 2). To predict the interacting network of the PIK3CA, we used STRING network analysis tool and found that PIK3CA is an important key player in the signaling network as it closely interacts with other oncogenic drivers such as PIK3R1, HRAS, NRAS, AKT, etc (Figure 2C). These results suggest that the mutations are likely to be driver mutations in DTCs. Figure 3A–3C shows the PTEN mutations identified in DTCs and of 3 mutations, one occurred in the phosphatase domain, and other two in the C2 domain. To predict the PTEN interaction network, we constructed a PTEN interactome that revealed a strong interaction of PTEN with PIK3CA/B, AKT2/1, P53, PDGFR, etc. This result shows that PTEN is an important member of this network and mutations in the PTEN would likely impair its normal function suggesting that mutations identified are likely loss of function mutations (Figure 3D).
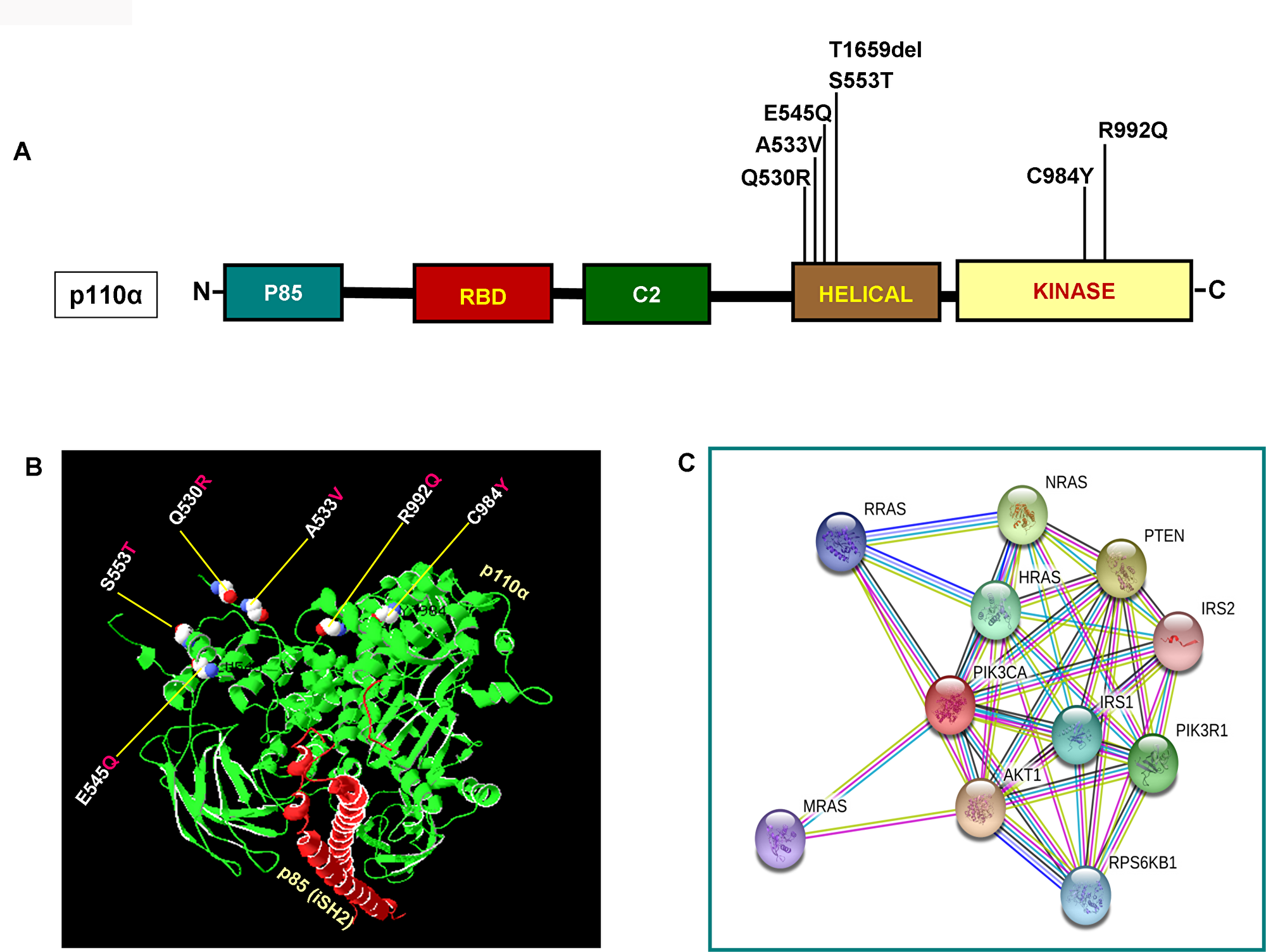
Figure 2: Functional domains of p110α and its interactome. (A) Schematic diagram of p110α protein. Shown are various somatic mutations identified in the PIK3CA gene of pediatric and adult DTCs and located in various corresponding domains of p110α. (B) Structure of p110α with DTCs associated mutations identified in this study. Shows ribbon diagram of native modeled structure of p110α /niSH2 (p85) as a heterodimer and its PDB ID is 2RDO. Mutated amino acid residues in DTCs were plotted in p110α native protein structure using SWISS PDB viewer. All the mutated residues are shown in a sphere shape, p110α is shown in green color and the niSH2 domain of p85α is shown in red color. (C) Interactome shows the interacting network of PIK3CA (p110α). The network nodes represent proteins and the red-colored node indicates the query protein and first shell of interactors. Bond in light blue and purple color shows known interactions from the curated database and experimentally determined, respectively. Bond in green, red and dark blue shows predicted interactions from gene neighborhood, gene-fusions, and gene co-occurrence, respectively. Black indicates co-expression while parrot green and pale blue shows text mining and protein homolog, respectively.
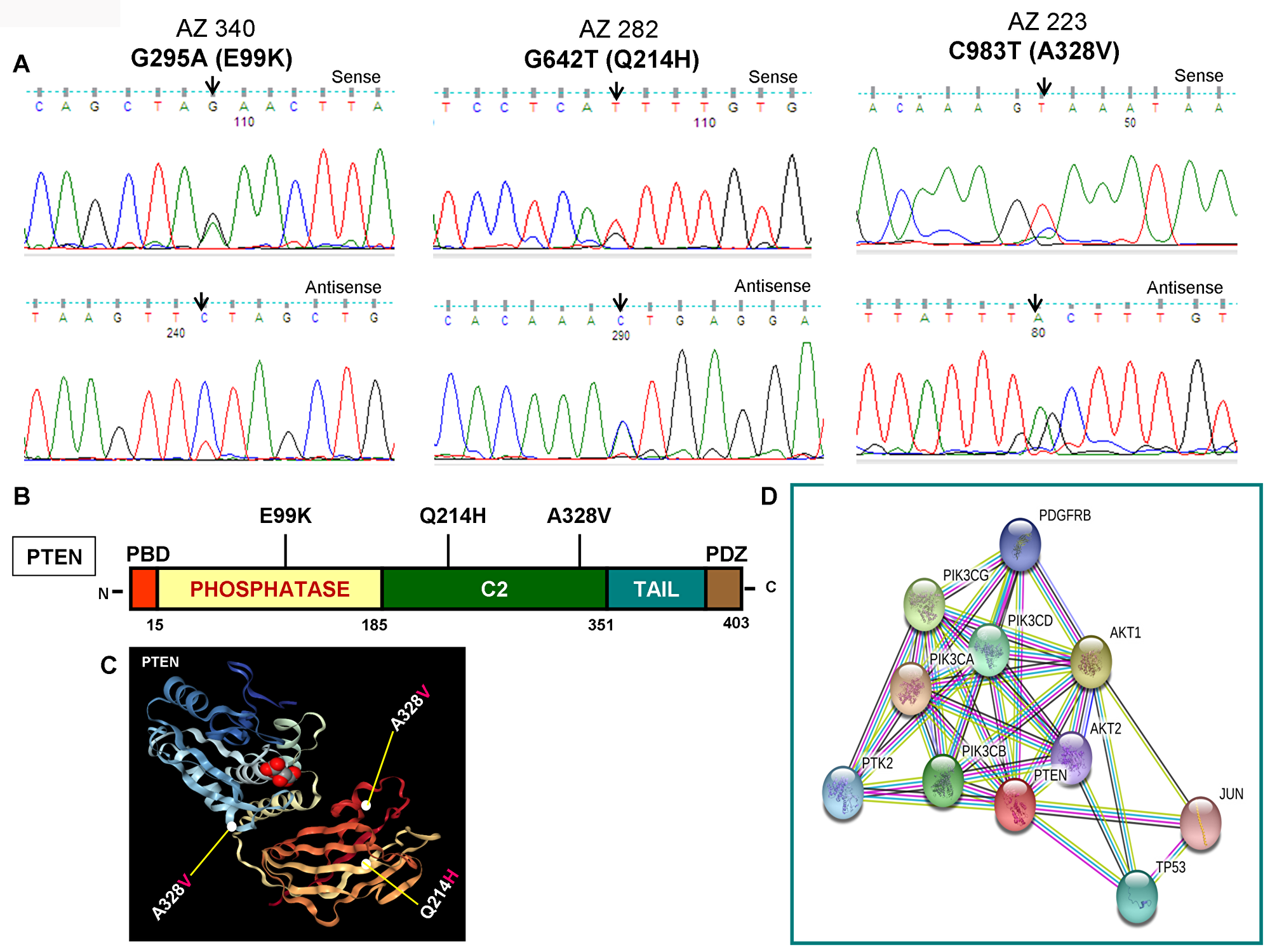
Figure 3: Identification of PTEN mutations, corresponding domains, and interactome of PTEN. (A) Chromatopherograms of the sequences of PTEN mutated samples. Sequences shown on the top and bottom indicate the sense and antisense strand, respectively of a sample. Altered nucleotide positions and its related amino acid (within bracket) are shown on top of each corresponding sequence. Arrows indicate the mutated nucleotides. Nucleotide number refers to the position within the coding nucleotides of the PIK3CA gene, where nucleotide position 1 shows the first nucleotide of the translation initiation codon. (B) Schematic diagram of PTEN protein. Marked-amino acids are various somatic mutations identified in the PTEN gene of pediatric and adult DTCs and located in various corresponding domains of PTEN. (C) A native modeled 3D structure of PTEN with pediatric and adult DTCs-associated mutations identified in this study. Ribbon diagram shows native modeled structure of PTEN protein and its PDB ID is 1D5R. Mutated amino acid residues in thyroid cancer are plotted in PTEN native protein structure using WebGL viewer as explained in materials and methods. All the mutated residues are marked in a sphere shape. (D) Interactome shows the interacting network of PTEN protein. The network nodes represent proteins and the red-colored node indicates the query protein and first shell of interactors. The bond represents protein-protein associations. Color of the bond and the type of associations are as mentioned above in Figure 2D.
Table 2: Somatic mutations of GPCR-mediated PI3K pathway genes: PIK3CA and PTEN mutations identified in pediatric and adult DTCs
| Tumor No | Group | Sex | Age | Histology | Gene | Exon | Nucleotide | Codon | Amino acid | Mutation | Status | BRAF | TERT | COS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AZ347 | Pediatric | F | 13 | CPTC | PIK3CA | 09 | A1589G | CAG-CGG | Q530R | Missense | Heterozygous | - | - | + |
| AZ347 | Pediatric | F | 13 | CPTC | PIK3CA | 09 | C1598T | GCA-GTA | A533V | Missense | Heterozygous | - | - | + |
| AZ152 | Adult | F | 44 | FV-PTC | PIK3CA | 09 | G1633C | GAG-CAG | E545Q | Missense | Heterozygous | 1798inTAC | - | + |
| AZ223 | Adult | F | 42 | FV-PTC | PIK3CA | 09 | G1658C | AGT-ACT | S553T | Missense | Heterozygous | - | - | + |
| AZ223 | Adult | F | 42 | FV-PTC | PIK3CA | 09 | T1659del | - | - | Frameshift | Homozygous | - | - | + |
| AZ328 | Pediatric | F | 13 | CPTC | PIK3CA | 20 | G2951A | TGT-TAT | C984Y | Missense | Heterozygous | - | - | + |
| AZ245 | Adult | F | 62 | TCPTC | PIK3CA | 20 | G2975C | CGA-CAA | R992Q | Missense | Heterozygous | V600E | - | + |
| AZ340 | Pediatric | F | 18 | CPTC | PTEN | 05 | G295A | GAA-AAA | E99K | Missense | Heterozygous | V600E | - | + |
| AZ282 | Adult | F | 42 | CPTC | PTEN | 07 | G642T | CAG-CAT | Q214H | Missense | Heterozygous | V600E | - | Novel |
| AZ223 | Adult | F | 42 | FV-PTC | PTEN | 08 | C983T | GCA-GTA | A328V | Missense | Heterozygous | - | - | + |
Comprehensive TCGA data analyses of PTCs show a relatively low incidence of mutation in GPCR-mediated PI3K pathway genes
As we found a low frequency of mutations in the GPCR-mediated PI3K pathway in the pediatric and adult DTCs of Saudi Arabian origin, we attempted to analyze the TCGA data of DTCs to determine whether the different cohort analyzed in the TCGA show a different prevalence of mutations in this pathway. As seen in Figure 4A OncoPrint, 0.2% (1/507) of cases had a somatic missense mutation in LPAR4; 0.4% (2/507) of the cases had missense mutations in PIK3CA and 1.2 % (6/507) cases harbored PTEN mutations consisting mainly of missense mutations and deep deletions. Overall, the GPCR-mediated pathway genes showed mutations in only 1.8% (9/507) of PTCs (Figure 4A–4C). This reflects low prevalence of mutations in this pathway regardless of the ethnic population and age group (pediatrics and adults) (Figure 4B–4E; Table 3).
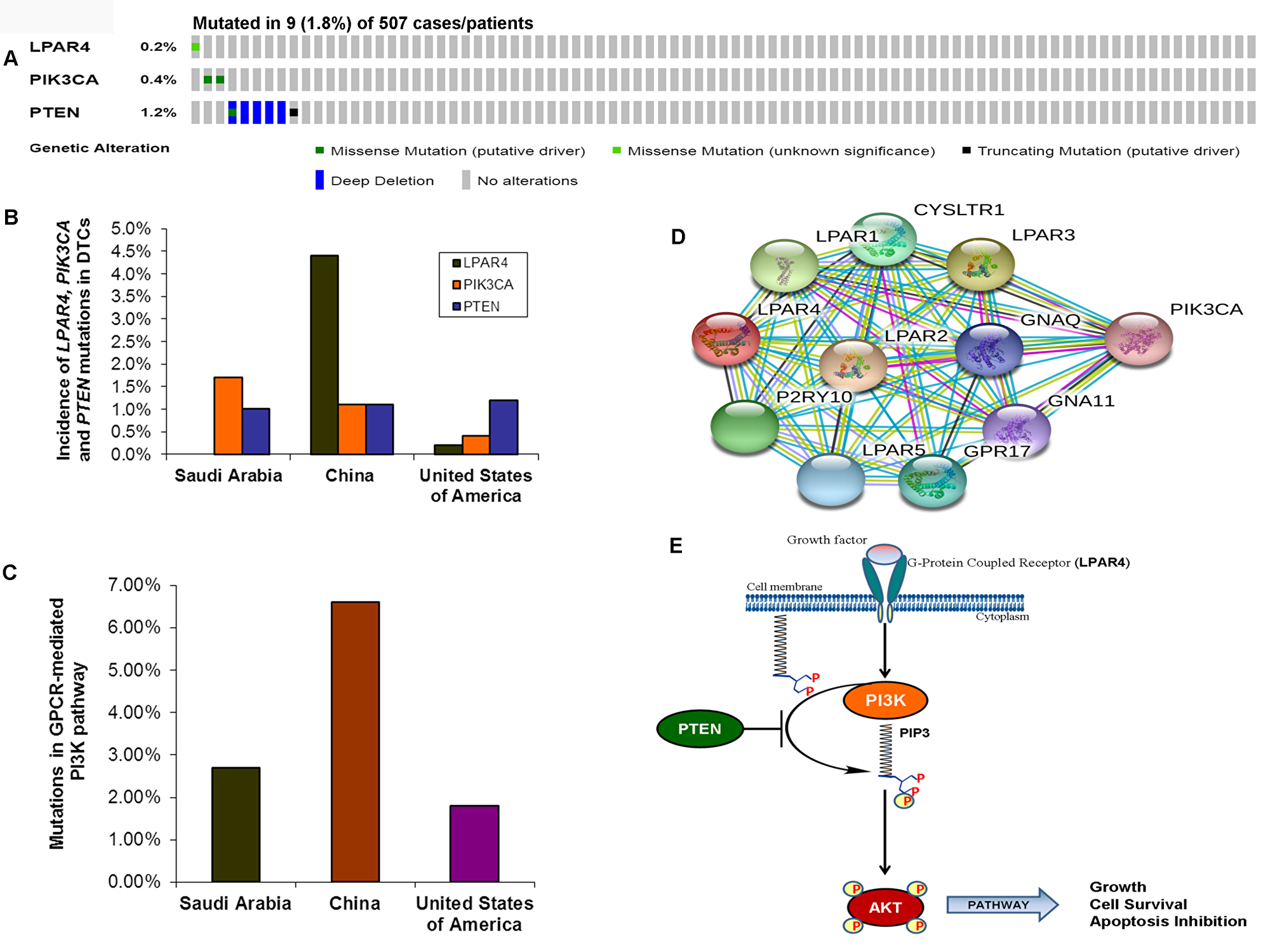
Figure 4: Prevalence of GPCR-mediated PI3K pathway mutations in DTCs. (A) OncoPrint of GPCR-mediated PI3K pathway- Thyroid cancer TCGA. The OncoPrint summarizes genomic alterations in LPAR4, PIK3CA and PTEN genes across the TCGA sample set. Each horizontal row indicates a gene and each vertical column shows a tumor sample. Green bars show nonsynonymous mutations and blue bars shows homozygous deletions. (B) Prevalence of LPAR4, PIK3CA, and PTEN in DTCs of various studies. The bar shows each LPAR4, PIK3CA and PTEN mutations in Sanger sequencing in the present study, and next-generation sequencing studies from China and USA (TCGA). (C) Mutations in the GPCR-mediated PI3K pathway. Bar indicates the combined frequency of mutation in the GPCR-mediated PI3K pathway in DTCs of the present study and other studies. (D) Interactome shows the interacting network of LPAR4 protein. The network nodes represent proteins and the red-colored node indicates the query protein and first shell of interactors. Bond represents protein-protein associations. Bond color and the type of associations are as mentioned above in Figure 3D. (E) Schematic diagram of the GPCR (LPAR4)-mediated PI3K signaling pathway. Growth factor activates the LPAR4 and activated LPAR4 triggers potential downstream signaling members such as PIK3CA, PTEN, and AKT, and the active PI3K pathway leads to growth, cell survival and inhibition of apoptosis.
Table 3: Mutations of GPCR-mediated PI3K pathway genes in differentiated thyroid cancer of various ethnic populations
| Genes | Saudi Arabia (Sanger sequencing)* | China (Whole Exome Seq.)1 | United States of America, TCGA (Whole Genome seq.)2 |
|---|---|---|---|
| [Mutations/Samples (Percentage)] | [Mutations/Samples (Percentage)] | [Mutations/Samples (Percentage)] | |
| LPAR4 | 0/293 (0%) | 4/91 (4.4%) | 1/507 (0.2%) |
| PIK3CA | 5/293 (1.7%) | 1/91 (1.1%) | 2/507 (0.4%) |
| PTEN | 3/293 (1%) | 1/91 (1.1%) | 6/507 (1.18%) |
| Overall | 8/293 (2.7%) | 6/91 (6.6%) | 9/507 (1.8%) |
TCGA data analyses of PDTCs and ATCs (aggressive thyroid cancers) show a relatively high incidence of mutations in GPCR-mediated PI3K pathway genes
In our current study, analysis of the GPCR-mediated PI3K pathway in the aggressive phenotypes of thyroid cancer (PDTC and ATC) showed no mutation. However, we had a limited number of cases with these aggressive forms (13 cases only). We, therefore, analyzed the TCGA data of PDTCs and ATCs to determine whether the results we obtained corroborates with the TCGA data of the different ethnic populations. As illustrated in Figure 5A and 5B, this pathway harbors mutations in 30% (10/33) and 6% (5/84) of ATC and PDTC, respectively. As seen in Figure 5C–5F, each PIK3CA and PTEN genes harbored somatic mutations in 7% (8/117) of aggressive thyroid cancers (PDTCs and ATCs) while no mutation was found in LPAR4 gene. Overall the GPCR-mediated pathway genes mutations were detected in 13% (15/117) of aggressive thyroid cancers (PDTCs and ATCs). These results suggest that the mutational prevalence of this pathway gene may be different among various ethnic cohorts. Furthermore, our analyses of this pathway genes revealed that the presence of PIK3CA mutation significantly predicts a poor overall survival (p = 0.0289) in patients with aggressive phenotypes suggesting a vital role of PIK3CA mutation in PDTCs and ATCs (Figure 5G).
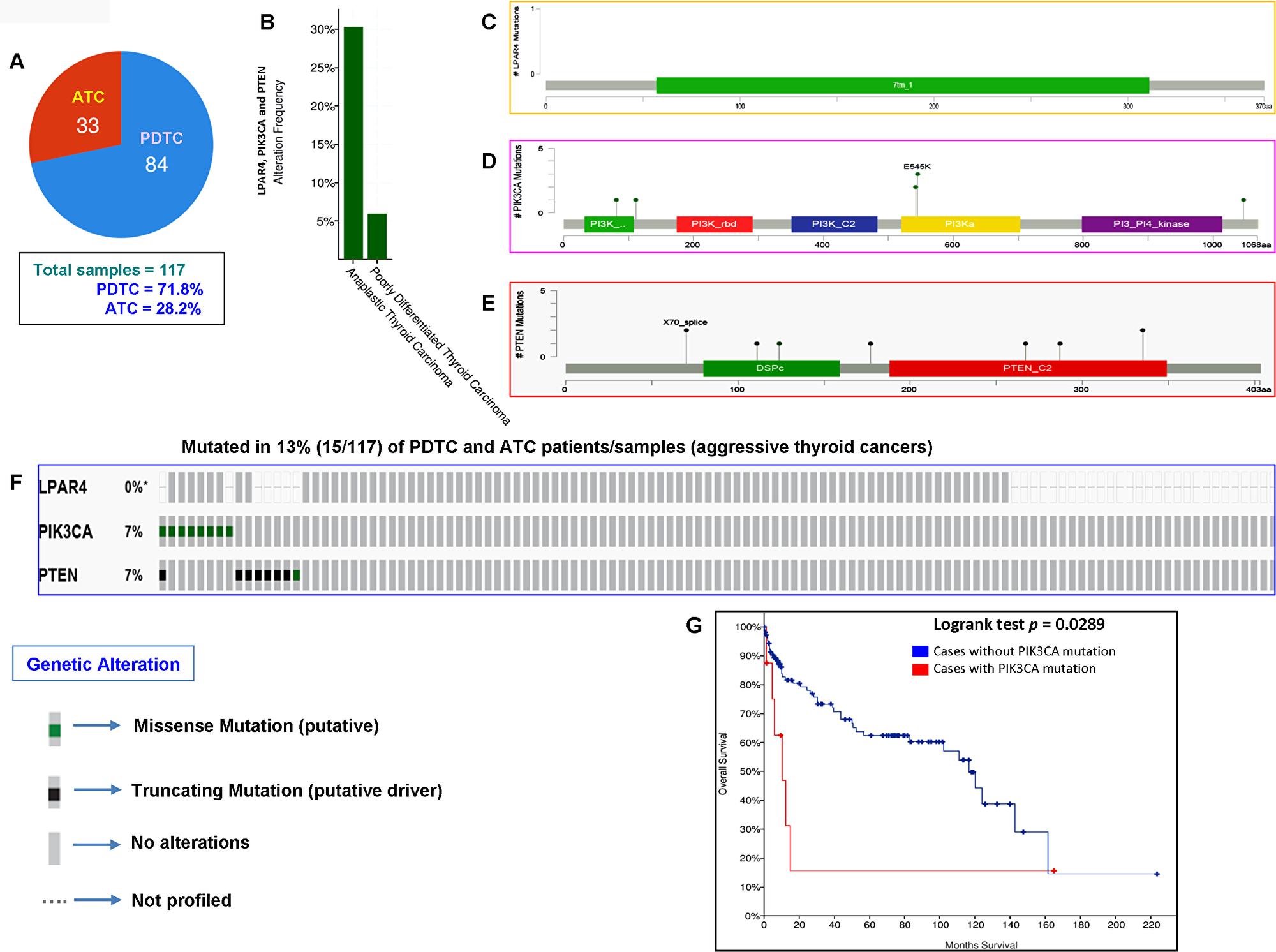
Figure 5: Prevalence of GPCR-mediated PI3K pathway mutations in PDTC and ATC (aggressive thyroid cancers). (A) Pie chart of aggressive thyroid cancers (PDTC and ATC). The chart shows the type of tumors, the number of samples in each type and total number of samples studied. (B) The histogram indicates the mutation frequency of GPCR-mediated PI3K pathway genes in thyroid cancers. The bar shows the overall frequency of LPAR4, PIK3CA and PTEN mutations in 6% and 30% of PDTC and ATC patients, respectively. (C–E) Mutation tab. The schematic diagrams indicate GPCR-mediated PI3K members (from top) LPAR4, PIK3CA (p110α) and PTEN protein domains and position of a particular mutation. The length of the line connected between mutation annotation and protein directly correspond to the number of mutated samples. The most frequent mutation is indicated in the diagram. (F) The OncoPrint tab. Tab shows the LPAR4, PIK3CA and PTEN mutations across the PDTC and ATC (MSKCC cohort). Each row represents a particular gene of GPCR-mediated PI3K pathway and each column shows a tumor sample. The green squares plotted on the columns are non-synonymous mutations. (G) Survival curve. The diagram is the Kaplan–Meier plot of overall survival of aggressive thyroid cancer-bearing patients (PDTC and ATC) absence or presence of PIK3CA mutations in blue and red color, respectively.
Construction of mutational landscape and mutated genes-mediated interactome reveals a complex multi-protein interaction network in aggressive thyroid cancers (PDTC and ATC)
Unlike DTCs, the PDTCs and ATCs are rare but aggressive thyroid cancers [7, 8]. Therefore, to find whether there is any difference in the mutational landscape and mutated genes-mediated protein-protein interaction network pattern between DTC and aggressive thyroid cancer (PDTC and ATC), we constructed and analyzed the mutational landscape and mutated genes-mediated interactome for DTC and aggressive thyroid cancers. In the mutational landscape, our analysis of TCGA data of DTCs showed 3600 mutated genes. We found 186 mutated genes in the TCGA data of aggressive thyroid cancers (PDTC and ATC). As illustrated in Figure 6A and 6B, we used the top 188 mutated genes for building the mutational landscape and protein-protein interaction network that covered all the mutated genes with a mutational prevalence of ≥ 0.70% in DTC and ≥ 0.80% in aggressive thyroid cancers. Projection of a top 50 mutated genes in DTC and aggressive thyroid cancers capped all the mutated genes with a mutational prevalence of ≥1% and 2.6%, respectively. Interestingly, compared with the DTC-specific interactome (3 major clusters include BRAF, NRAS, and P53), the aggressive thyroid cancers-mediated interactome reveals very complex (>13 major clusters by vital protein molecules such as NRAS, P53, AKT3, BRAF, TERT, PTEN, PIK3CA, EGFR, RAC1, CTNNB1, EBP300, CREBBP, MAPK1, etc.) and multi-node interactions in each protein molecule. Overall, it also shows the involvement of more number of mutated protein molecules and formation of multiple highly clustered interaction networks (Figure 6C and 6D. This interaction network truly reflects the complexity and advanced disease pattern of aggressive thyroid cancer phenotypes, PDTC and ATC.
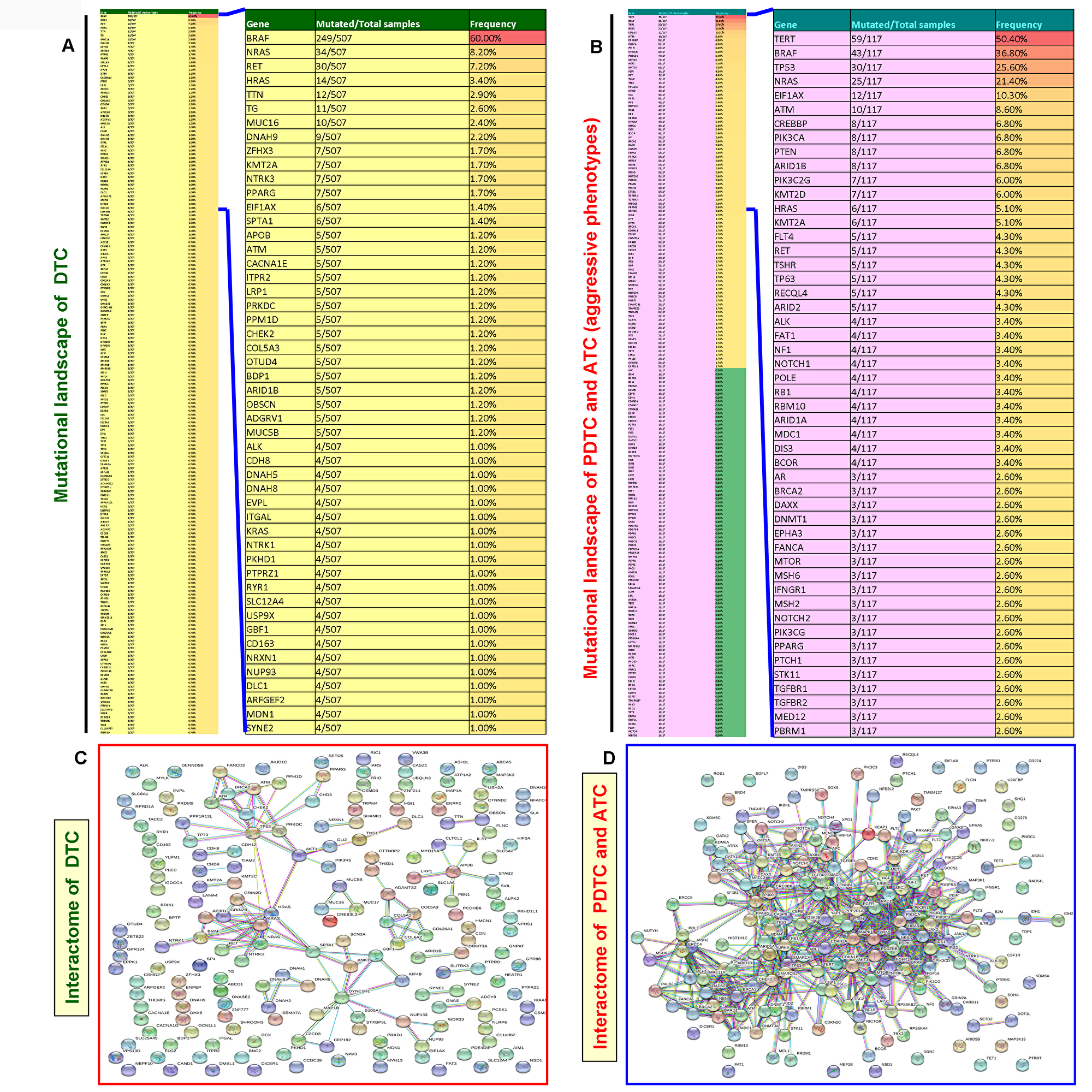
Figure 6: Mutated gene-mediated interactome of DTC and aggressive thyroid cancers (PDTC and ATC). (A) Mutational landscapes of DTC. The thin column shows the top 188 mutated genes in DTC and extracted after analyses of TCGA data of DTC with a gene mutational prevalence cutoff of ≥ 0.70% in cBioPortal. The projected wide column indicates the top 50 mutated genes in DTC. (B) Mutational landscapes of aggressive thyroid cancers (PDTC and ATC). The thin column shows 186 genes which include all the mutated genes in PDTC and ATC. These genes were extracted as explained in Figure 6A. The projected wide column indicates the top 50 mutated genes in aggressive thyroid cancers (PDTC and ATC). (C and D) The interaction network (C and D) show the mutated gene-mediated interactome of DTC and aggressive thyroid cancers (PDTC and ATC), respectively. The interactome of DTC and aggressive thyroid cancers (PDTC and ATC) was constructed by STRING v10 after extracting the full set of mutated genes indicated in the mutational landscape in (A and B), respectively.
Analyses of pan-cancer cell lines in TCGA reveal highly frequent mutations of GPCR-mediated PI3K pathway genes
To assess the rates of GPCR-mediated PI3K pathway and whether mutations in this pathway play role in other human cancers, we initially analyzed the pan-cancer cell lines sequenced data from TCGA. From the OncoPrint (Figure 7A–7F), 3.3% (2/60) of cases had a somatic mutation in LPAR4 gene and all the alterations were missense mutations. A high rate of 50% (1/2) LPAR4 mutations was found in prostate cancer cell line but only two cell lines were reported. This was followed by colorectal cancer cell lines [14.29% (1/7)]. Overall, 10.3% (111/1080) of cell lines had somatic mutations in the PIK3CA gene. Colon and ovarian cancer cell lines had a high frequency of the PIK3CA mutation of 28.57% (2/7) followed by invasive breast carcinoma [20% (1/5)]. Overall, 11.2% (121/1080) cell lines had somatic mutations in the PTEN gene. The PTEN showed mutations in 20% (1/5) of breast cancer, 16.67% (1/6) of brain and leukemia followed by 14.29% (1/7) of colon cancer cell lines. Overall this pathway contributed to 24.8% of cases of pan-cancer cell lines (Figure 7G). As shown in Figure 7H, this pathway genes had frequent mutations [57.14% (4/7)] in colon cancer cell lines followed by 50% (2/4), 40% (2/5), 28.57% (2/7) of prostate, breast, ovarian cancer cell lines, respectively. These results suggest that the pathway may be frequently mutated in solid tumors.
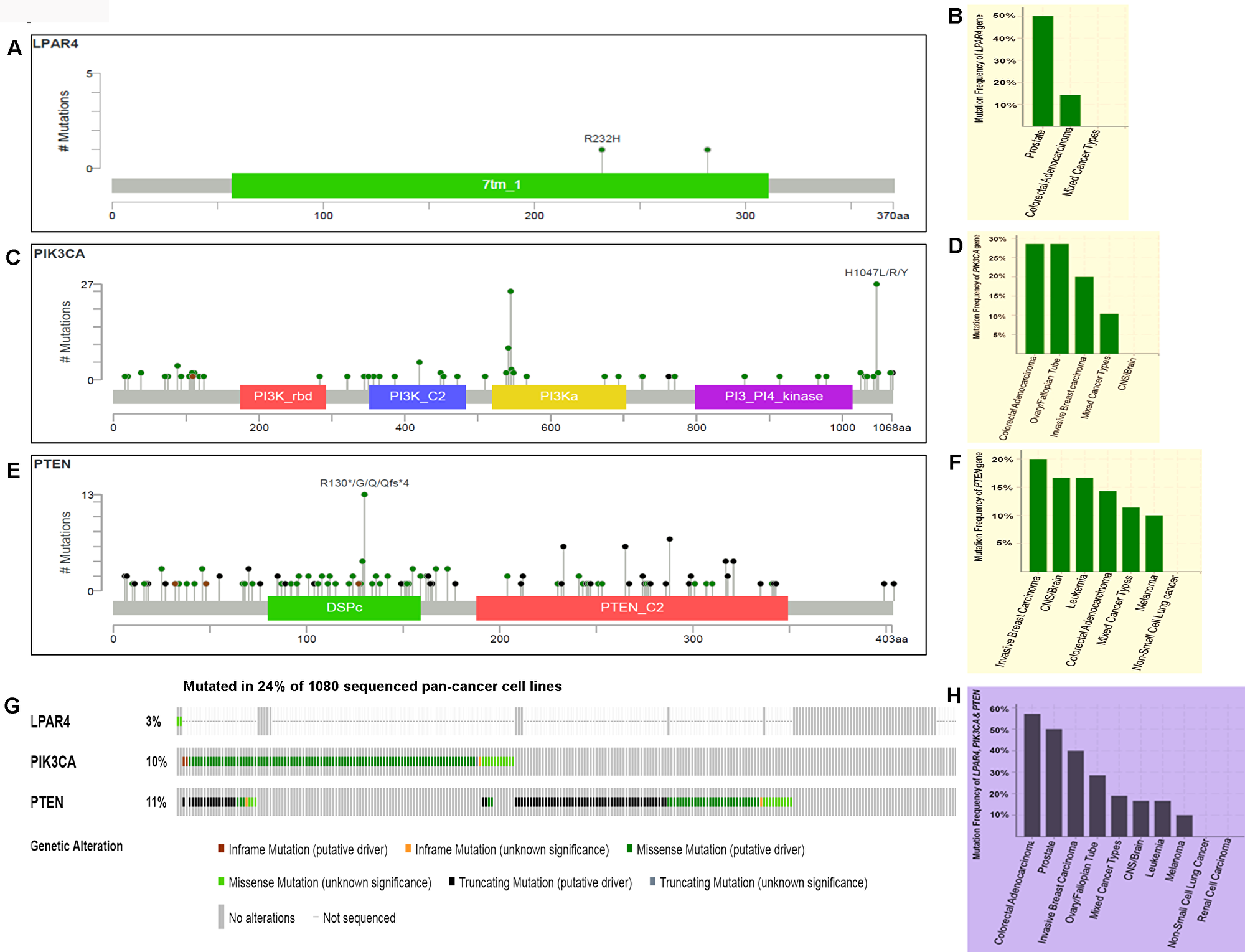
Figure 7: Prevalence of somatic mutations in genes of GPCR-mediated PI3K pathway in the pan-human cancer cell lines. (A) Schematic diagram of LPAR4 protein. The diagram shows the domains and the positions of somatic mutations. The length of the line connecting the indicated mutation to the protein represents the number of samples which is positive for the mutation. The labeled mutation in the diagram represents the most recurrent mutation. (B) The frequency of LPAR4 mutation. Bars indicate the frequency of LPAR4 gene mutations across the human cancer cell lines. Only positive and a few negative cases are shown in the figure. (C) Schematic diagram of p110α protein (PIK3CA). The diagram shows various domains and the positions of somatic mutations as stated above in Figure 7A. (D) The frequency of PIK3CA (p110α) mutation. Bars indicate the frequency of somatic mutations of the PIK3CA gene across the human cancer cell lines as explained in Figure 7B. (E) Schematic diagram of PTEN protein. The diagram shows the domains and the positions of somatic mutations as stated above in Figure 7A. (F) The frequency of PTEN mutation. Bars indicate the frequency of somatic mutations of the PTEN gene across the human cancer cell lines as explained in Figure 7B. (G) OncoPrint of GPCR-mediated PI3K pathway of pan-human cancer cell lines. The OncoPrint bar summarizes genomic alterations in LPAR4, PIK3CA and PTEN genes across the TCGA sample set of 1080 human pan-cancer cell lines. Each horizontal row indicates a gene and each vertical column shows a tumor sample. Green bars show nonsynonymous mutations, blue bars show homozygous deletions, black shows truncating mutations, no color indicates the absence of mutation and gap shows not sequenced. (H) Frequency of GPCR-mediated PI3K pathway genes (combined) across the type of human cancer cell lines. Highest and the lowest frequency of these pathway mutations were observed in colorectal adenocarcinoma and melanoma, respectively.
TCGA data analyses display frequent mutations of GPCR-mediated PI3K pathway genes in human solid cancers
To confirm the high frequency of GPCR-mediated pathway mutations found in human cancer cell lines, we analyzed the pan-cancer sequencing data from TCGA. As seen in Figure 8A and 8G, LPAR4 showed 0.8% (75/9020) mutation in pan-human solid cancers. Lung adenocarcinoma revealed 3.18% (21/660), the highest frequency of mutation in this gene followed by 2.89% (14/484) in lung squamous cell carcinoma and 2.56% (1/39) cutaneous melanoma (Figure 8B). PIK3CA had mutations in 18% (1625/9020) of tumors (Figure 8C and 8G) and it was frequently mutated in breast mixed ductal and lobular carcinoma 51.72% (45/87) followed by 48.94% of breast invasive lobular (69/141) and 40.47 % of ductal carcinoma (607/1500), respectively Figure 8D. PTEN showed a 6% (551/9020) of mutations in pan-human solid cancers (Figure 8E and 8G). As illustrated in Figure 8F, PTEN harbored high frequent mutations in uterine endometrioid carcinoma [51.79% (159/307)] next to cervical endometrioid carcinoma [33.33% (1/3)] and lung squamous cell carcinoma 11.57% (56/484)]. Overall this gene set was somatically mutated in 24.8% (Figure 8G) of solid cancers. This pathway’s mutations were frequently observed in 58.96% (181/307) of uterine endometrioid carcinoma, 52.87% (46/87) of breast mixed ductal and lobular carcinoma, 51.77% (73/141) of breast invasive lobular carcinoma (Figure 8H). These results show that this pathway is frequently mutated in human pan-tumor samples and could be a potentially important therapeutic target.
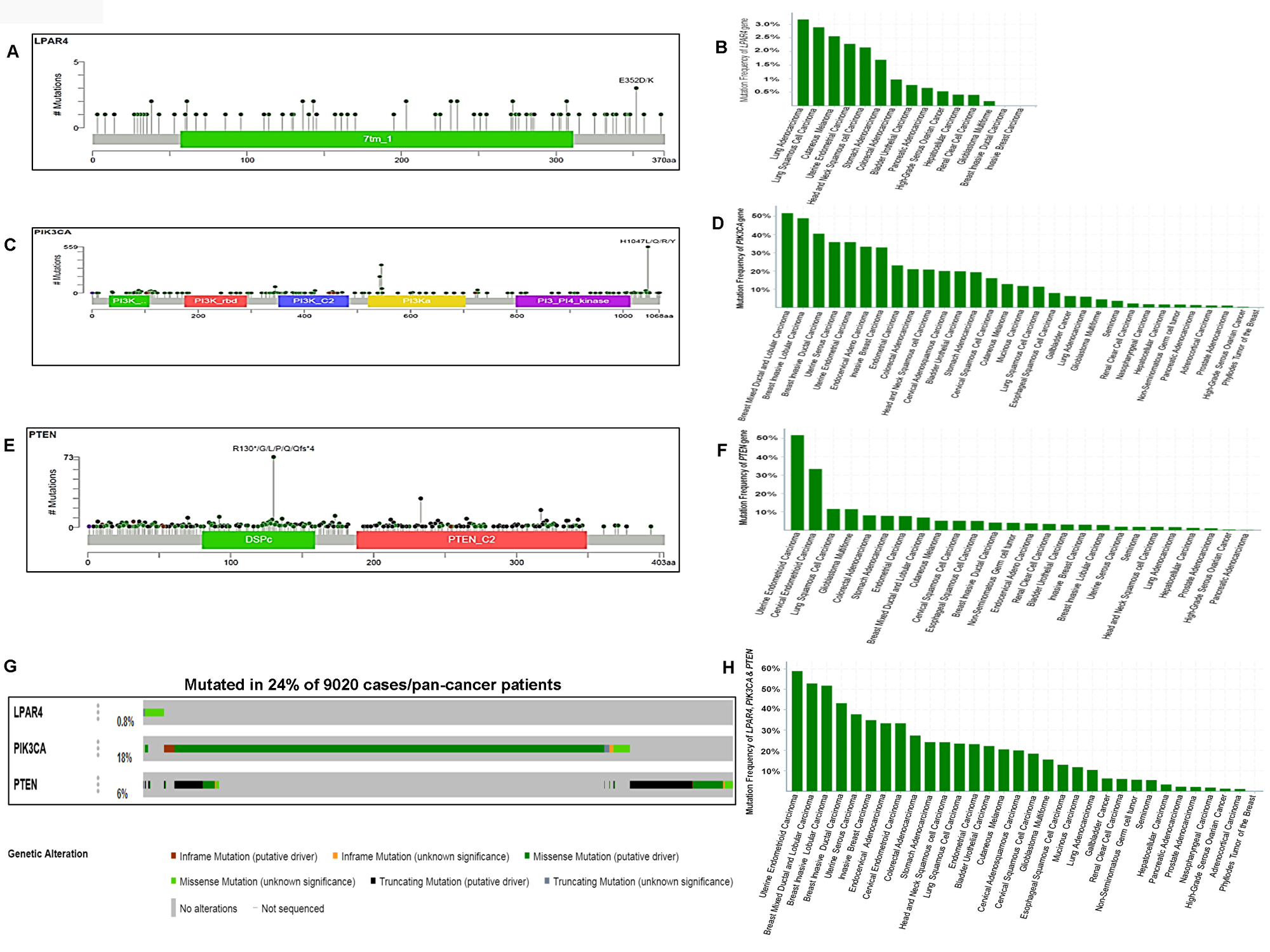
Figure 8: Somatic mutations of the GPCR-mediated PI3K pathway in pan-human cancers (solid tumors) and their association with patient survival. (A) Schematic diagram of LPAR4 protein. The diagram shows the domain and the positions of somatic mutations. The length of the line connecting the indicated mutation to the protein represents the number of samples which is positive for the mutation. The labeled mutation in the diagram represents the most recurrent mutation. (B) The frequency of LPAR4 mutation in pan-human cancers (solid). The histogram shows the somatic mutation frequencies of the LPAR4 gene mutations across cross cancer studies. (C) Schematic diagram of p110α protein (PIK3CA). The diagram shows various domains and the positions of somatic mutations identified in pan-human cancer (solid) as stated above in Figure 8A. (D) The frequency of PIK3CA mutation in pan-human cancers (solid). The histogram shows the somatic mutation frequencies of the PIK3CA gene cancer studies. (E) Schematic diagram of PTEN protein. The diagram shows various domains and the positions of somatic mutations identified in pan-human cancer (solid) as stated above in Figure 8A. (F) The frequency of PTEN mutation in pan-human cancers (solid). The histogram shows the somatic mutation frequencies of the PTEN gene across cancer studies. (G) OncoPrint of GPCR-mediated PI3K pathway of pan-human cancer. The OncoPrint bar summarizes genomic alterations in LPAR4, PIK3CA and PTEN genes across the TCGA sample set of 9020 pan-cancer samples. Each horizontal row indicates a gene and each vertical column shows a tumor sample. Green bars show nonsynonymous mutations, blue bars show homozygous deletions, black shows truncating mutations, no color indicates the absence of mutation and gap shows not sequenced. (H) Frequency of GPCR-mediated PI3K pathway gene mutations (combined) across the type of pan-human cancer. The highest frequency of these pathway mutations was observed in uterine endometrioid carcinoma and various types of breast carcinoma.
GPCR-mediated PI3K pathway genes significantly co-occur in human solid cancers
To determine any relationship between oncogenic activation and loss of tumor suppressor function in the GPCR-mediated PI3K pathway, mutations in the LPAR4, PIK3CA, and PTEN mutations were analyzed in relation to each other both in pan-cancer cell lines and pan-solid tumor samples (Table 4). Our analyses demonstrated that PIK3CA and PTEN mutations co-occurred significantly in pan-cancer cell lines (p = <0.001). However, LPAR4 vs PIK3CA and LPAR4 vs PTEN were not related in either mutually exclusive or a co-occurring manner (p = 0.805 and p = 0.788, respectively) in pan-cancer cell lines. To see the consistency of this relationship, we also analyzed the co-occurrence of mutations in these genes in pan-tumor cases. This showed that PIK3CA and PTEN mutations significantly co-occurred (p = <0.001). In addition, LPAR4 and PTEN also exhibited a significant co-occurrence (p = 0.003). However, LPAR4 and PIK3CA did not show any significant co-occurrence and/or mutual exclusivity (p = 0.499). These results suggest a significantly combined role of PIK3CA with PTEN and LPAR4 with PTEN in the human solid tumors. We also performed individual network analysis and found that PIK3CA and PTEN could strongly interact and this was consistent with our data as it co-occurred significantly in pan-cancer cell lines and tumors. LPAR4 could interact strongly with various PI3K pathway genes including PIK3CA except for PTEN (Figure 4D).
Table 4: Co-occurrence and mutual exclusivity of somatic mutations in GPCR-mediated PI3K pathway genes in pan-cancer
| Samples | Gene A | Gene B | Neither | A Not B | B Not A | Both | Log Odds Ratio | p-Value | Tendency | Significance |
|---|---|---|---|---|---|---|---|---|---|---|
| Cancer cell lines | PIK3CA | PTEN | 876 | 83 | 93 | 28 | 1.156 | <0.001 | Co-occurrence | Significant |
| LPAR4 | PIK3CA | 967 | 2 | 111 | 0 | <-3 | 0.805 | Mutual exclusivity | No | |
| LPAR4 | PTEN | 957 | 2 | 121 | 0 | <-3 | 0.788 | Mutual exclusivity | No | |
| Pan-cancers | PIK3CA | PTEN | 7058 | 1452 | 359 | 171 | 0.853 | <0.001 | Co-occurrence | Significant |
| LPAR4 | PTEN | 8430 | 60 | 519 | 11 | 1.091 | 0.003 | Co-occurrence | Significant | |
| LPAR4 | PIK3CA | 7358 | 59 | 1591 | 12 | –0.061 | 0.499 | Mutual exclusivity | No |
DISCUSSION
PI3K pathway genes are more frequently altered in high-grade thyroid cancers particularly in FTCs, PDTCs, and ATCs than in the well-differentiated types. [5, 16, 7, 8, 21–23]. Previous studies focused mostly on PIK3CA and/or PTEN of the PI3K pathway while several other studies examined individual genes of the PI3K pathway in thyroid cancers mainly FTCs and ATCs but have not studied the complete axis [27, 14, 15, 28]. Recently, WES of a Chinese population with PTCs revealed frequent mutations in the GPCR-mediated PI3K pathway and identified mutations of LPAR4 as novel PTC driver alterations [26]. LPAR4 is implicated in cell proliferation, migration, invasion, and tumorigenesis [24, 25]. However, to date, knowledge on the somatic mutations of LPAR4 in DTCs remains unknown except in the Chinese population [26]. LPAR4 mutations were not studied in pure pediatric and adult DTC groups, and aggressive thyroid cancers (PDTC and ATC). Furthermore, the prevalence of genetic alterations of this axis has not been explored in DTC, PDTC, and ATC from Saudi Arabia, a population of unique genetic makeup with high rates of thyroid cancer and consanguinity. We therefore specifically studied genetic alterations of the LPAR4-PIK3CA-PTEN of the PI3K pathway axis in pediatric and adult DTCs, PDTCs and ATCs derived from Saudi population. To determine if there is any difference in the rates of these mutations between different ethnic populations, we also analyzed this axis in the TCGA data of PTCs and aggressive thyroid cancers (PDTC and ATC). We also analyzed this axis in human pan-cancer cell lines and other solid tumors to understand the relative significance of the PI3K pathway in human tumorigenesis.
Our study revealed GPCR-mediated PI3K pathway mutations in 3.37% and 2.45% of pediatric and adult DTCs, respectively. In addition, pediatric and adult DTCs showed no significant differences in the prevalence of these mutations. Similarly, low frequent (4.8%) mutations have also been found in this axis (3.6%, PIK3CA and 1.2%, PTEN) in DTC from a Greek population [29]. However, a relatively high frequency (6.6%) of this pathway-mutations was found in a Chinese population with DTC [26] suggesting that ethnic differences, sample selection, and sequencing method are likely to influence the prevalence of mutations in this pathway. Moreover, consistent with previous studies [28–30], our study also suggests that PI3K pathway alterations are quite rare events and may indicate that the GPCR-mediated PI3K pathway genes are mutated only in a small fraction of DTCs. Although somatic mutations of PIK3CA and PTEN genes have previously been documented in sporadic thyroid cancers, they were mostly observed in high-grade tumors of FTCs and ATCs [31, 14, 30, 16].
In this study, of 293 DTC cases, 5 harbored 7 different PIK3CA mutations as two cases had concomitant mutations (Q530R; A533V and S553T; T1659del). Out of the 7 PIK3CA mutations, 5 and 2 were identified in the helical and kinase domain, respectively. Most of the mutations identified were localized in non-hotspot codons of the helical and kinase domains with the exception of a helical domain mutation (E545Q). Further, we identified PTEN mutations in 3 of 293 cases. All the PIK3CA and PTEN mutations identified in this study were localized to various functional domains. PI3K pathway mutations are reported to be mutually exclusive in human cancers including thyroid cancer. However, only one case had concomitant PIK3CA and PTEN mutation suggesting a weaker oncogenic potential of PIK3CA and requirement of a loss of tumor suppressor-like PTEN. Recent deep sequencing of DTCs (PTCs) identified PIK3CA and PTEN mutations in samples with distant metastasis [32] suggesting that the DTCs with these mutations are likely to progress into aggressive poorly differentiated thyroid cancer (PDTCs). Further, PIK3CA/PTEN mutation-positive samples were negative for the most commonly identified TERT promoter mutations. Nonetheless, of the 5 PIK3CA mutation positive cases, 2 had concomitant BRAF mutation. BRAF mutation in combination with a gain-of-function PIK3CA or loss-of-function PTEN/TP 53 mutation scenario has only been seen in the ATCs. A recent study demonstrated that BRAF mutant induced PTC was indolent and could not lead to end-stage disease. Similarly, PIK3CA mutant was unable to transform thyrocytes on its own but when co-expressed with BRAF mutant, that lead to the development of lethal ATC in mice suggesting that tumors bearing both BRAF and PIK3CA mutations are likely to progress into PDTCs and lethal ATCs [33]. On the other hand, thyroid cancer patients with PIK3CA mutations were shown to have a distinct pathological profile as PIK3CA mutations are likely to arise during the tumor progression [34]. Furthermore, 2 of 3 PTEN mutation-positive cases had BRAF mutation. It has been shown that BRAFV600E and PTEN loss facilitates the progression of thyroid cancers when the fibroblast-mediated collagen remodeling takes place within the tumor microenvironment suggesting a possibility of synergism in tumor progression [35].
In addition, we analyzed TCGA, a large series of DTCs (n=507) from a completely different ethnic background [4]. Consistent with our data, we observed a low prevalence (1.8%) of GPCR-mediated PI3K pathway gene mutations (0.2% of LPAR4, 0.4% of PIK3CA and 1.18% of PTEN). The prevalence of both our study (2.7%) and TCGA (1.8%) is relatively lower compared to that reported from the study from Chinese cases (6.6%) (Table 3) [26]. Although the low prevalence of GPCR-mediated PI3K pathway mutations is identified both in this study and TCGA data, they are located in the important functional domains including helical and kinase domains and interact with the important key players in the signaling network, and hence the mutations are likely to be oncogenic/tumor progression drivers.
The PDTCs and ATCs are rare but aggressive phenotypes of thyroid cancers particularly the ATC which is deadly cancer with less than 4 months median survival from the initial diagnosis [7, 8]. Analyses of these aggressive thyroid cancers in our study revealed no mutation in the GPCR-mediated PI3K pathway (LPAR4-PIK3CA-PTEN) genes. In contrast, analyses of TCGA data of aggressive thyroid cancers (PDTC and ATC) showed an overall 13% mutation rate in this pathway. Interestingly, this pathway had mutations in 30% of ATCs but only in 6% of PDTC. PIK3CA was found to be mutated in 7% of cases and PTEN mutations were also found in 7% while mutation was not detected in LPAR4. To assess whether these relatively frequent mutations have any prognostic value, we performed survival analysis in the genes of GPCR-mediated PI3K pathway in aggressive thyroid cancer (PDTC and ATC). Patients bearing PIK3CA mutations showed a significant poor overall survival (p = 0.0289) compared with people bearing wild-type PIK3CA suggesting the important role of this gene in aggressive thyroid cancer. Although the PI3K pathway is frequently genetically altered in PDTC and ATC [31, 14, 30, 16], the sample size is limited in our study and that could explain the absence of mutations in this pathway in the 13 cases of PDTC and ATC we studied. Alternatively, ethnic differences might be the reason for this apparent difference in the rate of mutations in PDTC and ATC in our study compared to TCGA data. Moreover, construction of mutational landscape and mutated gene-derived protein-protein interaction network (interactome) prominently reflected the simple disease pattern of DTC and complexity of the advanced disease (PDTC and ATC) and may assist in guiding the right molecular therapeutic target.
As GPCR-mediated PI3K pathway axis has never been analyzed in other solid malignancies, we mined pan-cancer cell lines (n = 1080) and tumor (n = 9020) from TCGA data. Unlike the low prevalence observed in DTCs and thyroid TCGA, both pan-cancer cell lines (24.8%) and tumors (24.8%) had high mutation frequency which is even higher than (6.6%) that reported in DTC from a Chinese population [26]. LPAR4 mutations in pan-tumors (0.8%) were comparable to that of DTCs though pan-cancer cell lines had a slightly higher prevalence (3.3%). LPAR4 mutations have previously been reported in 16% (1/6) only in human colon cancer cell lines [36] and recently in PTCs [26]. However, PIK3CA and PTEN showed more frequent mutation rates in pan-cancer cell lines and tumors and were very consistent with the previous reports [37, 38, 16] suggesting that more proven contribution of this pathway gene in the pathogenesis of solid pan-tumors.
Co-occurrence and mutual exclusivity inferred by a statistical analysis could show a rough relationship between different genes and they could serve as a platform for drug discovery. We, therefore, analyzed LPAR4, PIK3CA and PTEN genes in TCGA data of pan-cancer cell lines and tumors. This showed that PIK3CA and PTEN could significantly co-occur in pan-cancer cell lines (p = <0.001) and tumors (p = <0.001). As the interactome could reveal the interaction among the different genes, we performed network analysis and found that PIK3CA and PTEN could strongly interact and this was consistent with our data as it co-occurred significantly in pan-cancer cell lines and tumors. It has been widely demonstrated that PIK3CA and PTEN mutations were mutually exclusive; suggesting that tumorigenic signaling through the PI3K pathway could occur either with activation of PIK3CA/inactivation of PTEN [38, 39]. In contrast, it has been reported that coexistence of PIK3CA/PTEN mutations at high frequency (26%) in endometrial carcinoma. Further, PIK3CA mutations were more frequent in tumors with PTEN mutations (46%) compared with those without PTEN mutations (24%) [40] and this concomitant mutations (1.3%) have also been reported from Chinese breast cancers [41]. Further, an oncogenic PIK3CA mutation coupled with PTEN loss was shown to initiate ovarian tumors in mice [42]. This unique genetic representation indicates that the combination of PIK3CA/PTEN alterations might play an important synergistic role in tumorigenesis of these cancers. Moreover, LPAR4 and PTEN could co-occur in pan-tumor samples (p = 0.003). Nevertheless, network analysis revealed that LPAR4 could interact strongly with various PI3K pathway genes including PIK3CA except for PTEN. In contrast, LPAR4 was rarely analyzed for mutation except for a report from Chinese PTCs [26]. We did not find any significant mutual exclusivity among LPAR4, PIK3CA, and PTEN. Though data mining studies are more informative, they cannot draw a definite conclusion on the initiating event for the malignancy and only the prospective experimental studies could confirm that hypothesis.
In conclusion, data from our study and TCGA data of thyroid showed a similar low prevalence of mutations in GPCR-mediated PI3K pathway genes. Pediatric and adult cases showed no significant differences in mutational rates in this pathway. These results suggest that this pathway contributes to the pathogenesis of a smaller portion of DTCs but may have a major role in the progression of tumors harboring mutations in this pathway, leading to more aggressiveness and metastasis leading to PDTC and ATC. In addition, TCGA data analyses reveal that GPCR-mediated PI3K pathway genes are frequently mutated and they are likely playing a key role in the pathogenesis of PDTC and ATC and other human solid malignancies.
Materials and Methods
Tumor tissue samples and their histopathological features
Tumor samples were collected from the Department of Pathology, King Faisal Specialist Hospital and Research Centre (KFSH&RC), Riyadh, and the study was approved by the Institutional Review Board (IRB) (RAC-2130015). We used a total of 323 samples which includes 17 multinodular goiters (MNGs), 89 pediatric, 204 adult DTCs and 13 aggressive thyroid cancers. Pediatric DTCs includes 72 (81%) conventional papillary thyroid cancer (CPTC), 7 (7.9%) follicular variant-papillary thyroid cancer (FV-PTC), 2 (2.2%) tall cell variant PTC (TPTC), 3 (3.4%) diffuse sclerosing type papillary thyroid cancer (DSC), 3 (3.4%) follicular thyroid cancer (FTC) and 2 (2.2%) Hürthle Cell Cancer (HCC). The pediatric DTCs include all patients with the age at diagnosis of ≤ 18 years. Adult DTCs includes 114 (55.9%) CPTC, 55 (27%) FV-PTC, 29 (14.2%) TPTC, 3 (1.5%) HCC and 3 (1.5%) DSC. All the aggressive thyroid cancers were adults and consist of 10 (77%) poorly differentiated thyroid cancer (PDTC) and 3 (23%) anaplastic thyroid cancer (ATC).
Genomic DNA extraction
Slices of ~10-micron thickness of tumor samples were dissected from formalin fixed and paraffin embedded tissue (FFPE) after histological diagnosis was confirmed by an experienced endocrine pathologist (H.A). Genomic DNA was extracted as previously described [43] using the Gentra Puregene DNA extraction kit (Qiagen, Valencia, CA, USA).
PCR amplification and sequencing
We PCR amplified exon 1 of the LPAR4 gene using a forward primer with a 5’ M13 tag (LPAR4FW- 5’-GTA AAA CGA CGG CCA GTC TCT TCG CAA GCC TGC TAC-3’and a reverse primer with a 5’ M13 tag (LPAR4RV-5’-CAG GAA ACA GCT ATG ACC GCA AGG CAC AAG GTG ATT GG-3’ and the PCR conditions were as follows: initial denaturation at 94º C for 2 min and 35 cycles consisting denaturation at 94º C for 30 sec, annealing at 60º C for 30 sec with extension at 72º C for 45 sec followed by a 7 min final extension at 72º C. Primers for exons 9 & 20 of PIK3CA gene and exons 5, 6, 7 and 8 of PTEN gene was as reported previously [14]. All the mutation-positive cases were further screened for the common mutations in exon 15 of BRAF and TERT promoter using primers as described earlier [10, 18]. We limited our search to these exons as mutations were mostly harbored in these exons. The PCR amplicons were confirmed on 2.5% agarose gel and the successfully-amplified samples were subjected to direct sequencing using the Big Dye Terminator v3.1 cycle sequencing kit and by ABI PRISM 3730X1 genetic analyzer (Applied Biosystems). Identified mutations were confirmed in both forward and reverse directions by an independent PCR amplification and sequencing. The sequencing results were read against the GeneBank Accession No: LPAR4 (NM_005296.2), PIK3CA (NM_006218.3), PTEN (NM_000314.6), BRAF (NM_004333.4) and TERT (AF098956.1).
3D structure
The p110α protein in complex with p85 (niSH2) was downloaded from https://www.rcsb.org/ using the PDB ID: 2RDO. Amino acid residues of p110α (PIK3CA) somatic mutations identified in pediatric/adult DTCs were plotted in p110α native protein structure using SWISS PDB viewer as indicated previously [35]. The structure of PTEN was downloaded using the PDB ID: 1D5R as mentioned above. PTEN mutations identified in pediatric/adult DTCs were viewed and plotted using NGL viewer, a WebGL based 3D viewer powered by MMTF (Macromolecular Transmission Format) as described before [44].
Construction of interactive signaling network
Protein interacting network (interactome) for LPAR4, PIK3CA and PTEN protein molecules were constructed using a pre-computed database for the exploration and analysis of protein-protein interactions named STRING v10 (Search Tool for the Retrieval of Interacting Genes/Proteins) database [45]. All the protein-protein interaction networks were built with an interaction score of the highest confidence (0.900). The minimum required interaction score is 0.150, i.e., considered as low confidence.
Construction of mutational landscape and mutated gene-mediated interactome
The mutational landscape was constructed by analyzing the respective DTC (TCGA) and aggressive thyroid cancers (PDTC and ATC) from the TCGA data (MSKCC) of the respective phenotype in cBioPortal. Mutated gene-based interactome (protein-protein interaction network) was built by STRING v10 [45]. Initially, the data was filtered and used only the genetically altered genes (only the genes with mutation and deletion) in related phenotype and all the genes with copy number variations (CNV) were excluded from our further analyses. Our analyses showed 3600 mutated genes in DTC. We therefore filtered the mutated genes using gene with ≥ 0.70% of mutation as a cutoff and that extracted 188 genes and used all these genes for constructing DTC interactome and out of these genes, top 50 were projected for better visibility. Similarly, in PDTCs and ATCs, we used the same cutoff of 188 which covered all the 186 mutated genes in PDTC and ATC and used them for constructing interactome and top 50 mutated genes were projected.
TCGA data analyses for the mutation in the GPCR-mediated PI3K pathway (LPAR4, PIK3CA, and PTEN) genes
Differentiated thyroid cancer (DTC): TCGA data analyses
Data consisting of 507 DTCs from TCGA (Papillary Thyroid Carcinoma, TCGA, Cell 2014) were analyzed for this study and only mutations and deletions were included. Copy number variations (CNV) were excluded from this study.
Aggressive thyroid cancer: poorly-differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC) - TCGA data analyses
TCGA data of 117 aggressive thyroid cancer samples that include 84 PDTCs and 33 ATCs (Poorly-Differentiated and Anaplastic Thyroid Cancers, MSKCC, JCI 2016) were analyzed in this study. Copy number variations (CNV) were excluded in this study while only the mutations and deletions were included.
Pan-human cancer cell line: TCGA data analyses
In total, 1080 pan-cancer cell lines from 2 different studies, Cancer Cell Line Encyclopedia (Novartis/Broad, Nature 2012) and NCI-60 Cell Lines (NCI, Cancer Res. 2012) were analyzed.
Pan-human solid cancer sample: TCGA data analyses
A total of 9020 pan-solid cancer samples (derived from 9017 patients) from 20 different studies were analyzed which include Adrenocortical carcinoma (TCGA, Provisional), Bladder Urothelial carcinoma (TCGA, Nature 2014), Breast Cancer (METABRIC, Nature 2012 & Nat Commun 2016), Cervical Squamous Cell carcinoma and Endocervical Adenocarcinoma (TCGA), Colorectal Adenocarcinoma (DFCI, cell Reports), Esophageal Squamous Cell Carcinoma (UCLA, Nat Genet 2014), Gallbladder Carcinoma (Shanghai, Nat Gene 2014), Genomic Hallmarks of Prostate adenocarcinoma (CPC-GENE, Nature 2017), Genomic Profile of Patients with Advanced germ Cell Tumors (MSK, JCO 2016), Glioblastoma (TCGA, Cell 2013), Head and Neck Squamous Cell carcinoma (TCGA, Nature 2015), Hepatocellular Carcinomas (Inserm, Nat Genet 2014), Kidney Renal Clear cell Carcinoma (TCGA, Nature 2013), Nasopharyngeal Carcinoma (Singapore, Nat Gene 2014), Ovarian Serous Cystadenocarcinoma (TCGA, Nature 2011), Pan-Lung Cancer (TCGA, Nat Genet 2016), Pancreatic Adenocarcinoma (QCMG, Nature 2016), Stomach Adenocarcinoma (TCGA, Nature 2014), Uterine Corpus Endometrial Carcinoma (TCGA, Nature 2013) and WES of pretreatment melanoma tumors (UCLA, Cell 2016) and excludes thyroid TCGA DTC data. All the data mentioned above were derived and various analyses were performed in the cBioPortal for Cancer Genomics (www.cbioportal.org).
Statistical analyses
Mutual exclusivity/co-occurrence analysis of mutations in pan-cancer cell lines and pan-solid tumor samples were performed by calculating the odds ratio followed by Fisher’s exact test. Kaplan–Meier plots with a log-rank test were used to calculate the overall and disease-free survival of pan-solid tumors (except thyroid cancer) with at least one mutation or without mutation in the query gene. All the analyses were performed with the tools available in the cBioPortal for Cancer Genomics (www.cbioportal.org). A p-value < 0.05 was considered as statistically significant.
ACKNOWLEDGMENTS
This work was supported by King Abdulaziz City for Science and Technology, Riyadh, Saudi Arabia (12-BIO2952-20).
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
References
1. Pellegriti G, Frasca F, Regalbuto C, Squatrito S, Vigneri R. Worldwide increasing incidence of thyroid cancer: update on epidemiology and risk factors. J Cancer Epidemiol. 2013; 2013:965212. https://doi.org/10.1155/2013/965212. [PubMed].
2. Pstrąg N, Ziemnicka K, Bluyssen H, Wesoły J. Thyroid cancers of follicular origin in a genomic light: in-depth overview of common and unique molecular marker candidates. Mol Cancer. 2018; 17:116. https://doi.org/10.1186/s12943-018-0866-1. [PubMed].
3. Alzahrani AS, Alomar H, Alzahrani N. Thyroid Cancer in Saudi Arabia: A Histopathological and Outcome Study. Int J Endocrinol. 2017; 2017:8423147. https://doi.org/10.1155/2017/8423147. [PubMed].
4. Agrawal N, Akbani R, Aksoy BA, Ally A, Arachchi H, Asa SL, Auman JT, Balasundaram M, Balu S, Baylin SB, Behera M, Bernard B, Beroukhim R, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cancer Genome Atlas Research Network. Cell. 2014; 159:676–90. https://doi.org/10.1016/j.cell.2014.09.050.
5. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, Schultz N, Berger MF, Sander C, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest. 2016; 126:1052–66. https://doi.org/10.1172/JCI85271. [PubMed].
6. Murugan AK, Munirajan AK, Alzahrani AS. Long noncoding RNAs: emerging players in thyroid cancer pathogenesis. Endocr Relat Cancer. 2018; 25:R59–82. https://doi.org/10.1530/ERC-17-0188. [PubMed].
7. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013; 13:184–99. https://doi.org/10.1038/nrc3431. [PubMed].
8. Fagin JA, Wells SA Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med. 2016; 375:2307. https://doi.org/10.1056/NEJMra1501993. [PubMed].
9. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005; 12:245–62. [PubMed].
10. Murugan AK, Qasem E, Al-Hindi H, Shi Y, Alzahrani AS. Classical V600E and other non-hotspot BRAF mutations in adult differentiated thyroid cancer. J Transl Med. 2016; 14:204. https://doi.org/10.1186/s12967-016-0958-x. [PubMed].
11. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990; 60:557–63. https://doi.org/10.1016/0092-8674(90)90659-3. [PubMed].
12. Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am J Surg Pathol. 2002; 26:1016–23. https://doi.org/10.1097/00000478-200208000-00006. [PubMed].
13. Murugan AK, Dong J, Xie J, Xing M. Uncommon GNAQ, MMP8, AKT3, EGFR, and PIK3R1 mutations in thyroid cancers. Endocr Pathol. 2011; 22:97–102. https://doi.org/10.1007/s12022-011-9155-x. [PubMed].
14. Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, Wang Y, Trink A, El-Naggar AK, Tallini G, Vasko V, Xing M. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007; 13:1161–70. https://doi.org/10.1158/1078-0432.CCR-06-1125. [PubMed].
15. Abubaker J, Jehan Z, Bavi P, Sultana M, Al-Harbi S, Ibrahim M, Al-Nuaim A, Ahmed M, Amin T, Al-Fehaily M, Al-Sanea O, Al-Dayel F, Uddin S, Al-Kuraya KS. Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J Clin Endocrinol Metab. 2008; 93:611–18. https://doi.org/10.1210/jc.2007-1717. [PubMed].
16. Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010; 20:697–706. https://doi.org/10.1089/thy.2010.1646. [PubMed].
17. Murugan AK, Bojdani E, Xing M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 2010; 393:555–59. https://doi.org/10.1016/j.bbrc.2010.02.095. [PubMed].
18. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, Sun H, El-Naggar AK, Xing M. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2013; 20:603–10. https://doi.org/10.1530/ERC-13-0210. [PubMed].
19. Qasem E, Murugan AK, Al-Hindi H, Xing M, Almohanna M, Alswailem M, Alzahrani AS. TERT promoter mutations in thyroid cancer: a report from a Middle Eastern population. Endocr Relat Cancer. 2015; 22:901–08. https://doi.org/10.1530/ERC-15-0396. [PubMed].
20. Alzahrani AS, Qasem E, Murugan AK, Al-Hindi HN, AlKhafaji D, Almohanna M, Xing M, Alhomaidah D, AlSwailem M. Uncommon TERT Promoter Mutations in Pediatric Thyroid Cancer. Thyroid. 2016; 26:235–41. https://doi.org/10.1089/thy.2015.0510. [PubMed].
21. Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011; 71:4403–11. https://doi.org/10.1158/0008-5472.CAN-10-4041. [PubMed].
22. Murugan AK, Liu R, Xing M. Identification and characterization of two novel oncogenic mTOR mutations. Oncogene. March 2019. https://doi.org/10.1038/s41388-019-0787-5. [Epub Ahead of Print] [PubMed].
23. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, Rubinstein JC, Choi M, Kiss N, Nelson-Williams C, Mane S, Rimm DL, Prasad ML, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet. 2015; 24:2318–29. https://doi.org/10.1093/hmg/ddu749. [PubMed].
24. Matayoshi S, Chiba S, Lin Y, Arakaki K, Matsumoto H, Nakanishi T, Suzuki M, Kato S. Lysophosphatidic acid receptor 4 signaling potentially modulates malignant behavior in human head and neck squamous cell carcinoma cells. Int J Oncol. 2013; 42:1560–68. https://doi.org/10.3892/ijo.2013.1849. [PubMed].
25. Takahashi K, Fukushima K, Onishi Y, Inui K, Node Y, Fukushima N, Honoki K, Tsujiuchi T. Lysophosphatidic acid (LPA) signaling via LPA4 and LPA6 negatively regulates cell motile activities of colon cancer cells. Biochem Biophys Res Commun. 2017; 483:652–57. https://doi.org/10.1016/j.bbrc.2016.12.088. [PubMed].
26. Pan W, Zhou L, Ge M, Zhang B, Yang X, Xiong X, Fu G, Zhang J, Nie X, Li H, Tang X, Wei J, Shao M, et al. Whole exome sequencing identifies lncRNA GAS8-AS1 and LPAR4 as novel papillary thyroid carcinoma driver alternations. Hum Mol Genet. 2016; 25:1875–84. https://doi.org/10.1093/hmg/ddw056. [PubMed].
27. García-Rostán G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, Herrero A, Fusco A, Cameselle-Teijeiro J, Santoro M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005; 65:10199–207. https://doi.org/10.1158/0008-5472.CAN-04-4259. [PubMed].
28. Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008; 93:278–84. https://doi.org/10.1210/jc.2007-1076. [PubMed].
29. Sozopoulos E, Litsiou H, Voutsinas G, Mitsiades N, Anagnostakis N, Tseva T, Patsouris E, Tseleni-Balafouta S. Mutational and immunohistochemical study of the PI3K/Akt pathway in papillary thyroid carcinoma in Greece. Endocr Pathol. 2010; 21:90–100. https://doi.org/10.1007/s12022-010-9112-0. [PubMed].
30. Wang Y, Hou P, Yu H, Wang W, Ji M, Zhao S, Yan S, Sun X, Liu D, Shi B, Zhu G, Condouris S, Xing M. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J Clin Endocrinol Metab. 2007; 92:2387–90. https://doi.org/10.1210/jc.2006-2019. [PubMed].
31. Dahia PL, Marsh DJ, Zheng Z, Zedenius J, Komminoth P, Frisk T, Wallin G, Parsons R, Longy M, Larsson C, Eng C. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997; 57:4710–13. [PubMed].
32. Gandolfi G, de Biase D, Sancisi V, Ragazzi M, Acquaviva G, Pession A, Piana S, Tallini G, Ciarrocchi A. Deep sequencing of KIT, MET, PIK3CA, and PTEN hotspots in papillary thyroid carcinomas with distant metastases. Endocr Relat Cancer. 2014; 21:L23–26. https://doi.org/10.1530/ERC-14-0361. [PubMed].
33. Charles RP, Silva J, Iezza G, Phillips WA, McMahon M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol Cancer Res. 2014; 12:979–86. https://doi.org/10.1158/1541-7786.MCR-14-0158-T. [PubMed].
34. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, Janakiraman M, Solit D, Knauf JA, Tuttle RM, Ghossein RA, Fagin JA. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009; 69:4885–93. https://doi.org/10.1158/0008-5472.CAN-09-0727. [PubMed].
35. Jolly LA, Novitskiy S, Owens P, Massoll N, Cheng N, Fang W, Moses HL, Franco AT. Fibroblast-Mediated Collagen Remodeling Within the Tumor Microenvironment Facilitates Progression of Thyroid Cancers Driven by BrafV600E and Pten Loss. Cancer Res. 2016; 76:1804–13. https://doi.org/10.1158/0008-5472.CAN-15-2351. [PubMed].
36. Tsujino M, Fujii M, Okabe K, Mori T, Fukushima N, Tsujiuchi T. Differential expressions and DNA methylation patterns of lysophosphatidic acid receptor genes in human colon cancer cells. Virchows Arch. 2010; 457:669–76. https://doi.org/10.1007/s00428-010-0960-2. [PubMed].
37. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009; 4:127–50. https://doi.org/10.1146/annurev.pathol.4.110807.092311. [PubMed].
38. Murugan AK, Munirajan AK, Tsuchida N. Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer Lett. 2013; 338:193–203. https://doi.org/10.1016/j.canlet.2013.04.005. [PubMed].
39. Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins JM, McLendon RE, Fults DW, Velculescu VE, Bigner DD, Yan H. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004; 64:5048–50. https://doi.org/10.1158/0008-5472.CAN-04-1170. [PubMed].
40. Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005; 65:10669–73. https://doi.org/10.1158/0008-5472.CAN-05-2620. [PubMed].
41. Li G, Guo X, Chen M, Tang L, Jiang H, Day JX, Xie Y, Peng L, Xu X, Li J, Wang S, Xiao Z, Dai L, Wang J. Prevalence and spectrum of AKT1, PIK3CA, PTEN and TP53 somatic mutations in Chinese breast cancer patients. PLoS One. 2018; 13:e0203495. https://doi.org/10.1371/journal.pone.0203495. [PubMed].
42. Kinross KM, Montgomery KG, Kleinschmidt M, Waring P, Ivetac I, Tikoo A, Saad M, Hare L, Roh V, Mantamadiotis T, Sheppard KE, Ryland GL, Campbell IG, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012; 122:553–57. https://doi.org/10.1172/JCI59309. [PubMed].
43. Murugan AK, Humudh EA, Qasem E, Al-Hindi H, Almohanna M, Hassan ZK, Alzahrani AS. Absence of somatic mutations of the mTOR gene in differentiated thyroid cancer. Meta Gene. 2015; 6:69–71. https://doi.org/10.1016/j.mgene.2015.08.005. [PubMed].
44. Rose AS, Hildebrand PW. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015; 43:W576–9. https://doi.org/10.1093/nar/gkv402. [PubMed].
45. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–52. https://doi.org/10.1093/nar/gku1003. [PubMed].