INTRODUCTION
Genotyping of oncogenic RAS mutations is routinely undertaken as it is an important biomarker used to predict drug resistance to epidermal growth factor receptor (EGFR)-targeted monoclonal antibodies in patients with metastatic colorectal cancer (mCRC) [1, 2, 3]. In this approach, tumor tissues are used to explore representative genomic profiles of the tumor. However, discrepancies in the genomic profile can occur because of the heterogeneous nature of a tumor (intratumor heterogeneity) [4–7]. Differences in genomic profiles between primary tumors and distant metastases have also been reported in 10% of mCRC [4]. The genomic profile of the tumor, which is representative of the tumor molecular landscape, can be altered during chemotherapy with commonly used cytotoxic agents [8] as well as targeted drugs [9–12]. Because of the possible implications of these factors on the molecular profile, tumor tissue-based genotyping has some limitations in attempts to identify the molecular features of the tumor.
A blood-based technology platform that tracks circulating tumor DNA (ctDNA), known as liquid biopsy, could be an ideal alternative to a biopsy of tumor tissue [13], and may remove the restrictions associated with the use of tissue samples [14]. This technique reflects tumor dynamics [15] and allows multiple testing over time, monitoring real-time changes within the tumor and evaluation of therapeutic responses [9-11, 16-19, 20]. BEAMing technology and digital PCR, one of the platforms of the liquid biopsy using micro-compartmentalization of PCR, can detect rare mutant alleles in blood with a high sensitivity of 0.01 to 0.001% [21, 22]. These blood-based platforms with their high sensitivity enable monitoring of tumor dynamics by tracking ctDNA during treatment in patients with mCRC [15]. Tumor dynamics obtained from KRAS monitoring could provide important information about treatment strategies for patients with mCRC, such as detection of drug resistance to anti-EGFR antibody before radiographic documentation of disease progression [10, 9, 19]. Additionally, they raise the possibility of an alternative molecular explanation for the efficacy of re-challenge therapies based on EGFR blockade [19].
Despite the clinical advantages obtained by tracking KRAS mutated ctDNA (MctDNA), the dynamics of MctDNA during regimens currently in use in clinical practice are not well known in patients with mCRC. Details and the clinical significance are important to help determine the best anti-cancer treatment as a precision medicine. Further exploration is required for clinical application. In this study, we examined the dynamics of MctDNA during various regimens for mCRC and determined the characteristics and clinical significance of the method.
RESULTS
Assessment of KRAS mutations in blood and tissue
A KRAS monitoring image from mCRC patients treated with various drugs during the treatment lines is shown in Figure 1A. KRAS assessment in tumor tissues identified 29 patients with the mutant-type (MT) and 56 patients with the wild-type (WT). Assessment of KRAS status in blood incorporated both the number of MctDNA and the ratio of MctDNA.
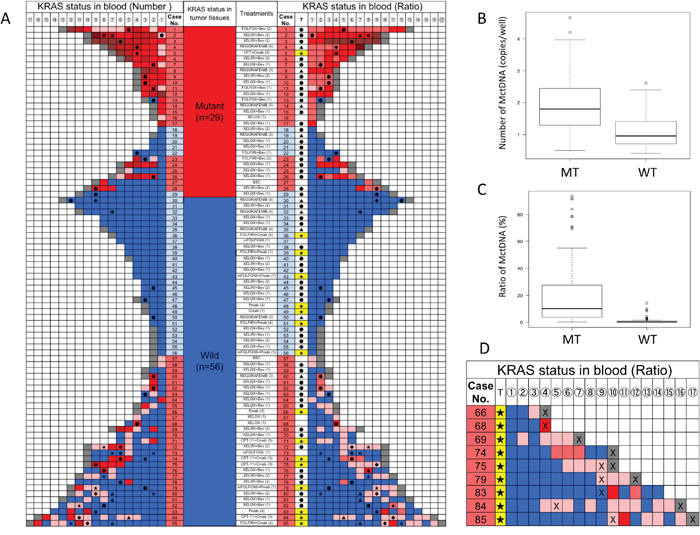
Figure 1: KRAS monitoring of mCRC patients and comparison of MctDNA between MT and WT (A) KRAS monitoring of mCRC patients treated with various drugs across several treatment lines. Initial assessments for circulating tumor DNA with KRAS mutations (MctDNA) varied by treatment line and regimen and are shown under “treatment (lines)”; (XELOX (1) means that XELOX was given as the first-line treatment). KRAS status in tumor tissues is shown. Patients with mutations (red), those without (blue). KRAS assessment in tumor tissues are under “KRAS status in tumor tissues” with red for patients with the mutant-type (MT) and blue for patients with the wild-type (WT). Monitoring MctDNA is shown under “KRAS status in blood”, ordered by timing of blood examination (①→⑯). MctDNA was assessed using two methods for “KRAS status in blood”. Left column under “KRAS status in blood (number)” indicates the number of MctDNA. MctDNA not detected (blue); detection of MctDNA in fewer than 10 copies/well (pink); 10 ≤ MctDNA < 50 copies/well (light red); 50 ≤ MctDNA < 100 copies/well (red); 100 ≤ MctDNA < 100 copies/well (light brown); MctDNA ≥ 1000 copies/well (brown); end of treatment because of disease progression (gray). Right column under “KRAS status in blood (ratio)” shows ratio of MctDNA among total circulating cell-free DNA (MctDNA and circulating cell-free DNA without KRAS mutations). The mutation ratio was calculated by fractional abundance (MctDNA/ total circulating cell-free DNA). MctDNA not detected (blue); detection of MctDNA less than 1% (pink). MctDNA ≤ 1 < 10% (light red); 10 ≤ MctDNA < 30% (red); 30 ≤ MctDNA < 50% (light brown); MctDNA ≤ 100% (brown); end of treatment because of disease progression (gray). ●: anti-VEGF antibody; ▲: regorafenib; ★: anti-EGFR antibody; ◆: TAS-102. (B) Comparison of number of MctDNA between patients with MT and WT in tumor tissues. Vertical axis represents logarithm. (C) Comparison of ratio of MctDNA between patients with MT and WT in tumor tissues. The mutation ratio was calculated by fractional abundance (MctDNA / total circulating cell-free DNA). Vertical axis represents mutation ratio × 102. (D) Initial detection of MctDNA in patients with WT treated with anti-EGFR antibody. T: treatment; ★: anti-EGFR antibody; X: detection of radiological disease progression. Detection of MctDNA less than 1% (pink); MctDNA ≤ 1 < 10% (light red); 10 ≤ MctDNA < 30% (red).
Dynamics of KRAS mutated circulating tumor DNA and its impact on outcome in patients with the mutant-type
In 29 patients with the MT, MctDNA was detected in 23 patients (79.3%) (Figure 1A). Among 18 patients who underwent second-line or subsequent treatment lines, MctDNA was detected in 17 patients (94.4%). Details of the clinical course of these 29 patients is shown in Table 1. The median value of MctDNA was 64.0 copies/well (3.2–45800) for the number and 10.10% (0.26–93.60) for the ratio in these 29 patients (Figure 1B and 1C). Comparing progression-free survival (PFS) of the first-line treatment between patients with MctDNA and without, there was a significant difference in PFS (Figure 2A), with a worse outcome in patients with MctDNA (22.0 vs 3.0 months, p = 0.0007). Most patients showed a continuous change with increasing MctDNA (Figure 3A and 3C). Four patients had a stable MctDNA level, suggesting a long stable disease (Figures 3B and 4A), and two patients showed a quick decrease in MctDNA with shrinkage of tumors in response to treatment (Figure 3D and Figure 4G).
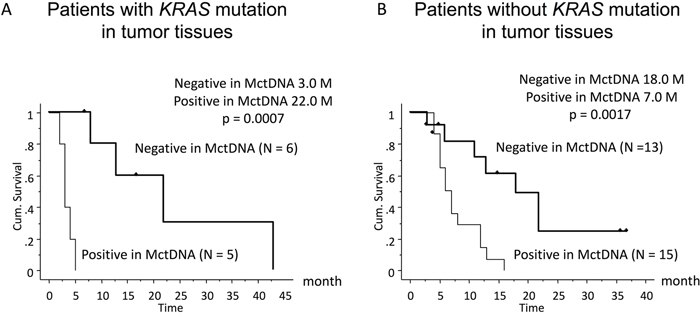
Figure 2: Comparison of progression-free survival (PFS) in patients treated with the first-line therapy according to KRAS status in blood. Patients with KRAS mutations in tumor tissues (left) and those without KRAS mutations (right).
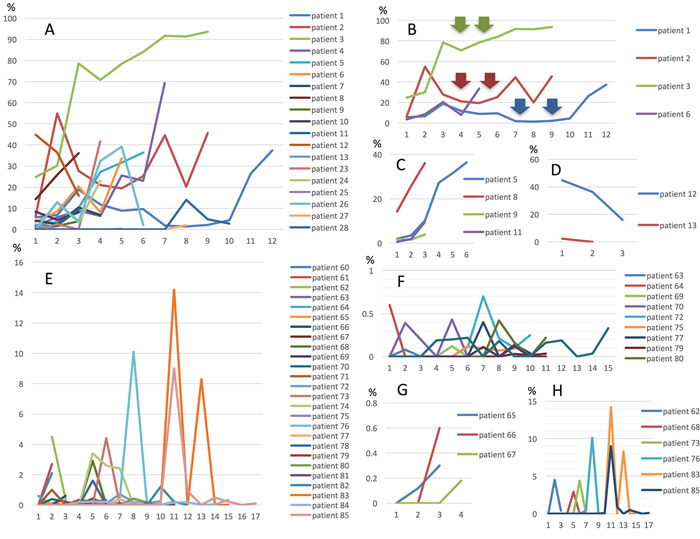
Figure 3: Changes in MctDNA during treatment in 29 patients with mutations in tumor tissues (A), representative graph of four patients with stable levels of MctDNA (B), four patients with increases (C), and two patients with decreases (D). Changes in MctDNA during treatment in 28 patients without mutations in tumor tissues (E), representative graph of nine patients with intermittent changes in MctDNA (F), three patients with increases (G), and seven patients with a spike in elevation (H). Y axis shows ratio of MctDNA (%) and X axis shows timing of blood examination.
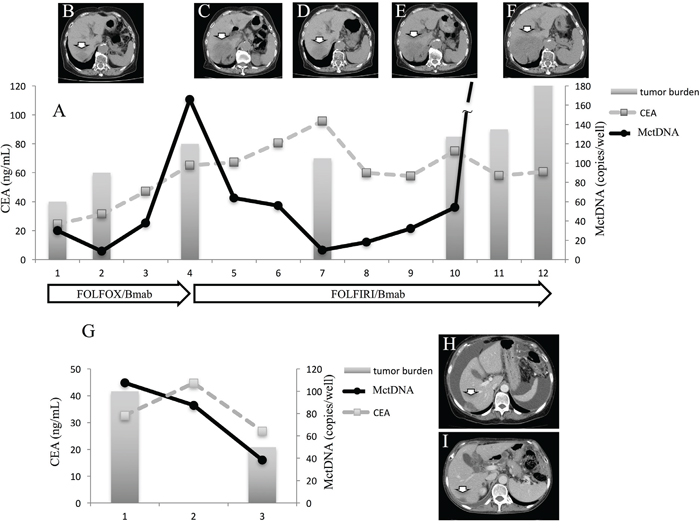
Figure 4: Clinical course of a mCRC patient with multiple liver metastases with a long stable disease (A and G) and computed tomography (CT) (B–F). A 75-year-old woman with multiple liver metastases is denoted as patient 1. She was treated with FOLFOX + bevacizumab for the first-line treatment. (A) and (B) show CT images before and after treatment with FOLFOX + bevacizumab. Increased levels of MctDNA were observed before radiological progression (B). FOLFIRI + bevacizumab was administrated as the second-line treatment. The tumor did not change in size (C, D, and E) and levels of MctDNA were stable for a time with the second-line treatment. Progression-free survival of 6 months was achieved with stable levels of MctDNA (A), followed by progression detected by CT (F). A 66-year-old woman with multiple liver metastases and ascites is denoted as patient 12. She was treated with XELOX + bevacizumab for the first-line treatment. (H) and (I) show CT images before and after treatment with XELOX + bevacizumab. The tumor shrank and levels of MctDNA decreased (I). Arrow shows liver metastasis. CEA: carcinoembryonic antigen.
Table 1: Clinical information for patients with the mutant-type
Case |
Sex |
Age |
Primary site |
Metastatic site |
KRAS primary tissue |
KRAS MctDNA |
1st line |
2nd line |
3rd line |
4th line |
5th line |
|---|---|---|---|---|---|---|---|---|---|---|---|
1 |
f |
78 |
S/C |
Liver, lung |
G12S |
G12S |
mFOLFOX6/Bev |
FOLFIRI/Bev |
BSC |
||
2 |
m |
49 |
Rectum |
Liver |
G12V |
G12V |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
Lonsurf |
BSC |
3 |
f |
73 |
T/C |
Liver |
G12C |
G12C |
XELOX/Bev |
XELIRI/Bev |
Lonsurf |
BSC |
|
4 |
m |
69 |
A/C |
Liver |
G13D |
G13D |
FOLFIRI |
CPT-11/Cmab |
Regorafenib |
Lonsurf |
BSC |
5 |
m |
80 |
Rectum |
Lung, LN |
G13D |
G13D |
XELOX |
XELIRI |
CPT-11/Cmab |
Lonsurf |
BSC |
6 |
f |
67 |
Rectum |
Liver, lung, LN |
G12D |
G12D |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
Lonsurf |
BSC |
7 |
m |
19 |
S/C |
LN |
G12D |
G12D |
XELIRI/Bev |
XELOX/Bev |
|||
8 |
m |
52 |
A/C |
Liver |
G12D |
G12D |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
BSC |
|
9 |
f |
67 |
A/C, S/C |
LN |
G12C |
G12C |
XELOX/Bev |
XELIRI/Bev |
Lonsurf |
BSC |
|
10 |
m |
78 |
A/C |
Liver |
G13D |
G13D |
XELOX/Bev |
XELIRI/Bev |
BSC |
||
11 |
m |
65 |
A/C |
Lung |
G12C |
G12C |
mFOLFOX6 |
FOLFIRI/Bev |
Lonsurf |
BSC |
|
12 |
f |
66 |
A/C |
Peritoneum |
G12S |
G12S |
XELOX/Bev |
||||
13 |
m |
79 |
A/C |
Liver, peritoneum |
G13D |
G13D |
mFOLFOX6/Bev |
FOLFIRI/Bev |
Lonsurf |
Regorafenib |
BSC |
14 |
f |
76 |
Rectum |
Liver, peritoneum |
G12D |
G12D |
XELOX/Bev |
XELIRI |
Regorafenib |
BSC |
|
15 |
m |
76 |
Rectum |
Liver, LN |
G12D |
G12D |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
Lonsurf |
BSC |
16 |
f |
66 |
Rectum |
Lung, LN |
G12S |
G12S |
XELOX |
XELIRI |
BSC |
||
17 |
m |
71 |
T/C |
Liver, lung, peritoneum |
G12D |
G12D |
XELOX/Bev |
Lonsurf/Bev |
|||
18 |
m |
59 |
Rectum |
Lung |
G12V |
N.D. |
XELOX/Bev |
XELIRI/Bev |
|||
19 |
m |
72 |
A/C, D/C |
Liver, peritoneum |
G12C |
N.D. |
FOLFIRI/Bev |
Regorafenib |
BSC |
||
20 |
f |
82 |
Rectum |
Liver, lung |
G12D |
N.D. |
XELOX/Bev |
||||
21 |
f |
73 |
S/C |
Liver, peritoneum |
G12V |
N.D. |
XELOX/Bev |
||||
22 |
m |
76 |
A/C |
Peritoneum, LN |
G12C |
N.D. |
mFOLFOX6 |
FOLFIRI/Bev |
Regorafenib |
||
23 |
f |
71 |
Cecum |
Peritoneum |
G12A |
G12A |
mFOLFOX6 |
FOLFIRI/Bev |
Lonsurf |
||
24 |
f |
68 |
A/C |
Liver, lung |
G12D |
G12D |
XELOX/Bev |
Pmab |
BSC |
||
25 |
m |
62 |
Rectum |
Liver, LN |
G12D |
G12D |
XELOX/Bev |
BSC |
|||
26 |
m |
33 |
Rectum |
Liver, peritoneum |
G12D |
G12D |
XELOX/Bev |
XELIRI/Bev |
BSC |
||
27 |
m |
67 |
Rectum |
Lung |
G12V |
G12V |
BSC |
||||
28 |
f |
61 |
Rectum |
Lung |
G12C |
G12C |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
BSC |
|
29 |
m |
50 |
Rectum |
Liver, LN |
G12V |
N.D. |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
Lonsurf |
BSC |
A/C: ascending colon; T/C: transverse colon; D/C: descending colon; S/C: sigmoid colon; LN: lymph node; mFOLFOX6: oxaliplatin, folinic acid, and fluorouracil; FOLFIRI: irinotecan, folinic acid, and fluorouracil; XELOX: capecitabine and oxaliplatin; XELIRI: capecitabine and irinotecan; Bev: bevacizumab; Pmab: panitumumab; Cmab: cetuximab; CPT-11: irinotecan; BSC: best supportive care.
Dynamics of mutated circulating tumor DNA and outcome in patients with the wild-type
In 56 patients with the WT, MctDNA was detected in 28 patients (50.0%) (Figure 1A). Details of the clinical course of these 56 patients is shown in Table 2. MctDNA was detected in patients treated with anti-EGFR antibody as well as those treated with other drugs such as anti-vascular endothelial growth factor (VEGF) antibody, regorafenib, and TAS-102. Cytotoxic agents without targeted therapies also showed the emergence of MctDNA (Table 3). The median value of MctDNA was 9.1 (2.6–414) for the number and 0.22% (0.002–14.2) for the ratio (Figure 1B and 1C). The median value of MctDNA for the number detected in patients with the WT was 1/7 of that in patients with the MT, suggesting that about 1/7 of tumor cells (14.7%) in patients with the WT might have the KRAS mutation. Comparing first-line treatment between patients with MctDNA and those without, there was a significant difference in PFS (Figure 2B), showing a worse outcome in patients with MctDNA (18.0 vs 7.0 months, p = 0.0017). Most patients showed intermittent changes in elevation; with low levels of MctDNA generally (Figure 3E and 3F), four patients showed a continuous change with an increase (Figure 3G), and five patients showed a transient change with a spike in elevation (quick elevation and disappearance) (Figure 3H), with close to 10 times the amount of MctDNA as the intermittent change in elevation (Figure 3F). Initial detection of MctDNA in WT patients treated with anti-EGFR antibody was likely prior to radiological disease progression (Figure 1D).
Table 2: Clinical information of patients with the wild-type
Case |
Sex |
Age |
Primary site |
Metastatic site |
KRAS Primary tumor |
KRAS MctDNA |
1st line |
2nd line |
3rd line |
4th line |
5th line |
6th line |
7th line |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
30 |
m |
69 |
Rectum |
LN |
WILD |
N.D. |
XELOX/Bev |
XELOX |
Pmab |
Regorafenib |
Lonsurf |
Pmab |
BSC |
31 |
m |
64 |
S/C |
Liver |
WILD |
N.D. |
XELOX |
XELIRI/Bev |
|||||
32 |
m |
70 |
Rectum |
Lung, LN |
WILD |
N.D. |
XELOX |
XELIRI/Bev |
CPT-11/Cmab |
Regorafenib |
Lonsurf |
BSC |
|
33 |
m |
58 |
Rectum |
Liver, peritoneum |
WILD |
N.D. |
XELOX/Bev |
||||||
34 |
m |
74 |
S/C |
Lung, LN |
WILD |
N.D. |
XELOX/Bev |
||||||
35 |
m |
74 |
Cecum |
Liver, lung, LN |
WILD |
N.D. |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Regorafenib |
Lonsurf |
BSC |
|
36 |
m |
68 |
S/C |
Peritoneum |
WILD |
N.D. |
FOLFIRI/Cmab |
BSC |
|||||
37 |
f |
47 |
Rectum |
Liver |
WILD |
N.D. |
mFOLFOX6/Bev |
||||||
38 |
f |
75 |
S/C |
Peritoneum |
WILD |
N.D. |
XELOX/Bev |
||||||
39 |
f |
74 |
S/C |
Peritoneum |
WILD |
N.D. |
FOLFIRI/Pmab |
||||||
40 |
m |
47 |
A/C |
Liver, LN |
WILD |
N.D. |
XELOX/Bev |
BSC |
|||||
41 |
m |
72 |
Rectum |
Liver |
WILD |
N.D. |
XELIRI/Bev |
||||||
42 |
m |
52 |
T/C, Rectum |
Liver |
WILD |
N.D. |
XELOX/Bev |
||||||
43 |
f |
47 |
Rectum |
Liver, LN |
WILD |
N.D. |
mFOLFOX6/Pmab |
||||||
44 |
m |
74 |
Cecum |
Liver |
WILD |
N.D. |
XELOX |
XELIRI/Bev |
|||||
45 |
m |
78 |
Rectum |
LN |
WILD |
N.D. |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Pmab |
||||
46 |
f |
55 |
S/C |
Liver |
WILD |
N.D. |
XELOX/Bev |
XELIRI/Bev |
|||||
47 |
f |
70 |
D/C |
Peritoneum |
WILD |
N.D. |
FOLFIRI/Bev |
mFOLFOX6/Bev |
CPT-11/Cmab |
Regorafenib |
BSC |
||
48 |
m |
58 |
Rectum |
Liver |
WILD |
N.D. |
mFOLFOX6/Bev |
FOLFIRI/Bev |
Pmab |
BSC |
|||
49 |
f |
75 |
A/C |
Liver |
WILD |
N.D. |
Cmab |
BSC |
|||||
50 |
f |
68 |
A/C |
Peritoneum |
WILD |
N.D. |
mFOLFOX6/Bev |
FOLFIRI/Pmab |
Lonsurf |
Regorafenib |
|||
51 |
m |
70 |
S/C |
LN |
WILD |
N.D. |
XELOX/Bev |
||||||
52 |
m |
70 |
S/C |
Bone, LN |
WILD |
N.D. |
XELOX |
BSC |
|||||
53 |
m |
72 |
A/C, Rectum |
Liver, lung |
WILD |
N.D. |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Regorafenib |
Lonsurf |
BSC |
|
54 |
m |
67 |
Cecum |
Peritoneum |
WILD |
N.D. |
XELOX/Bev |
FOLFIRI/Pmab |
BSC |
||||
55 |
f |
57 |
Rectum |
Liver, peritoneum |
WILD |
N.D. |
mFOLFOX6/Pmab |
BSC |
|||||
56 |
f |
66 |
S/C |
Liver, lung |
WILD |
N.D. |
BSC |
||||||
57 |
m |
70 |
A/C |
Liver, peritoneum |
WILD |
N.D. |
XELOX/Bev |
XELIRI/Bev |
BSC |
||||
58 |
f |
62 |
S/C |
Peritoneum |
WILD |
12V |
XELOX/Bev |
XELIRI/Bev |
Regorafenib |
BSC |
|||
59 |
f |
75 |
Rectum |
Liver |
WILD |
12V |
XELOX |
FOLFIRI/Pmab |
Regorafenib |
BSC |
|||
60 |
m |
52 |
S/C |
Liver, lung |
WILD |
12D |
XELIRI/Bev |
FOLFIRI/Cmab |
BSC |
||||
61 |
f |
47 |
Rectum |
Liver |
WILD |
12C, 12R |
XELOX/Bev |
||||||
62 |
f |
69 |
Rectum |
Liver, LN |
WILD |
12S |
XELOX/Bev |
XELIRI/Bev |
|||||
63 |
f |
72 |
T/C |
Liver |
WILD |
12D |
XELOX/Bev |
XELIRI/Bev |
Lonsurf/Bev |
Regorafenib |
|||
64 |
m |
74 |
Rectum |
Lung |
WILD |
12R |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Pmab |
||||
65 |
m |
61 |
S/C |
Liver |
WILD |
12C, 13D |
XELOX/Bev |
BSC |
|||||
66 |
m |
53 |
A/C |
Peritoneum |
WILD |
12D |
XELOX/Bev |
XELIRI/Bev |
Pmab |
BSC |
|||
67 |
f |
67 |
S/C |
Locally advanced |
WILD |
12S |
XELOX |
||||||
68 |
m |
65 |
T/C |
Peritoneum |
WILD |
12S |
FOLFIRI/Bev |
FOLFIRI/Pmab |
|||||
69 |
m |
49 |
S/C |
Liver, peritoneum |
WILD |
12A, 12C, 12S |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Lonsurf |
BSC |
||
70 |
f |
64 |
Rectum |
Liver, LN |
WILD |
13D |
XELIRI/Bev |
Lonsurf/Bev |
|||||
71 |
f |
79 |
Rectum |
LN |
WILD |
12D, 13D |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
||||
72 |
m |
57 |
Rectum |
Liver |
WILD |
12V |
SOX |
XELIRI/Bev |
Lonsurf/Bev |
CPT-11/Pmab |
BSC |
||
73 |
f |
53 |
T/C |
Liver |
WILD |
12V |
FOLFIRI/Cmab |
Regorafenib |
Lonsurf |
Pmab |
BSC |
||
74 |
f |
71 |
Rectum |
Lung |
WILD |
12S |
FOLFIRI |
CPT-11/Pmab |
Lonsurf |
BSC |
|||
75 |
m |
62 |
Rectum |
Liver |
WILD |
12A, 12S |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Lonsurf |
BSC |
||
76 |
m |
60 |
S/C |
Liver |
WILD |
12C, 13D |
XELOX/Bev |
Lonsurf/Bev |
FOLFIRI/Pmab |
||||
77 |
m |
72 |
Rectum |
Liver, lung, LN |
WILD |
12S, 12V |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
BSC |
|||
78 |
f |
65 |
S/C |
Liver, peritoneum |
WILD |
12D, 13D |
XELOX/Bev |
XELIRI/Bev |
Lonsurf/Bev |
||||
79 |
f |
74 |
S/C |
Liver |
WILD |
12D, 12S |
mFOLFOX6/Pmab |
FOLFIRI/Pmab |
BSC |
||||
80 |
m |
72 |
A/C |
Liver, LN |
WILD |
12V |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Regorafenib |
Lonsurf |
BSC |
|
81 |
f |
56 |
Rectum |
Liver |
WILD |
13D |
XELOX/Bev |
XELIRI/Bev |
BSC |
||||
82 |
f |
29 |
S/C |
Peritoneum |
WILD |
12D |
XELOX/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Lonsurf |
|||
83 |
f |
74 |
Rectum |
Liver |
WILD |
12D |
mFOLFOX6 |
IRIS |
Pmab |
Lonsurf |
|||
84 |
f |
50 |
Rectum |
Liver |
WILD |
12A, 12C, 12D, 12V, 13D |
mFOLFOX6/Bev |
XELIRI/Bev |
CPT-11/Cmab |
Regorafenib |
Lonsurf |
Cmab |
BSC |
85 |
m |
65 |
Rectum |
Liver, lung |
WILD |
12R |
XELOX/Bev |
FOLFIRI/Cmab |
Regorafenib |
Lonsurf |
CPT-11/Cmab |
BSC |
A/C: ascending colon; T/C: transverse colon; D/C: descending colon; S/C: sigmoid colon; LN: lymph node; N.D.: not detected; mFOLFOX6: oxaliplatin, folinic acid, and fluorouracil; FOLFIRI: irinotecan, folinic acid, and fluorouracil; XELOX: capecitabine and oxaliplatin; XELIRI: capecitabine and irinotecan; Bev: bevacizumab; Pmab: panitumumab; Cmab: cetuximab; CPT-11: irinotecan; BSC: best supportive care.
Table 3: Emergence during each regimen in patients with the wild-type
Number of patients (n) |
Emergence of MctDNA (n) |
Percentage of patients with emergence of MctDNA (%)* |
|
|---|---|---|---|
Chemotherapy |
4 |
1 |
25.0 |
Anti-VEGF antibody + Chemotherapy |
33 |
13 |
39.4 |
Anti-EGFR antibody |
25 |
9 |
36.0 |
Regorafenib |
10 |
3 |
30.0 |
TAS-102 |
10 |
7 |
70.0 |
* Emergence of MctDNA / Number of Patients *100
Reproducibility and sensitivity of KRAS monitoring
To ensure reproducibility of these dynamic changes in mutated circulating tumor DNA recognized in patients (e.g., patient 85), we conducted an additional experiment. DNA samples from patient 3 with a known KRAS G12C mutation were mixed with DNA samples from patient 41 at varying dilutions. DNA samples with the G12C mutation were used because no patients carried the KRAS G12R mutation in tumor tissues. Patient 41 tumor tissues harbored KRAS wild-type and showed no mutations in the blood during treatments. The number of KRAS G12C mutations declined as the dilution series progressed to one copy in 20000 reference copies (0.005%) and then was not detectable (data not shown). Reproducibility of the emergence of new mutations was confirmed in triplicate.
Clinical course of two patients who showed a spike in elevation in mutated circulating tumor DNA
A spike in elevation was seen in six patients treated with anti-VEGF antibody or TAS-102. Although detection of MctDNA was generally seen in patients with disease progression, a spike in elevation of MctDNA was observed in patients who likely responded to drug treatments, followed by a quick disappearance. A patient who showed a drug response with a spike in elevation is shown in Figure 5. Despite there being no change in tumor size, tumors did show changes in morphology induced by TAS-102 + bevacizumab as the second-line treatment. It is reported that patients with a change in morphology show as good a drug response as those patients with a change in size, similar to a partial response and a complete response, estimated using Response Evaluation Criteria in Solid Tumours (RECIST) in not only colorectal cancer but other types of tumors such as gastrointestinal stromal tumors [23, 24, 25]. Interestingly, this patient also showed a change in tumor morphology with first-line treatment using XELOX + bevacizumab. There were some patients with a decline in MctDNA, suggesting recovery of drug sensitivity, who were then treated with re-introduction of anti-EGFR antibody. Figure 6 shows a representative image of a patient who responded to re-introduction of anti-EGFR antibody. The patient achieved a partial response and 7 months PFS with the sixth-line treatment.
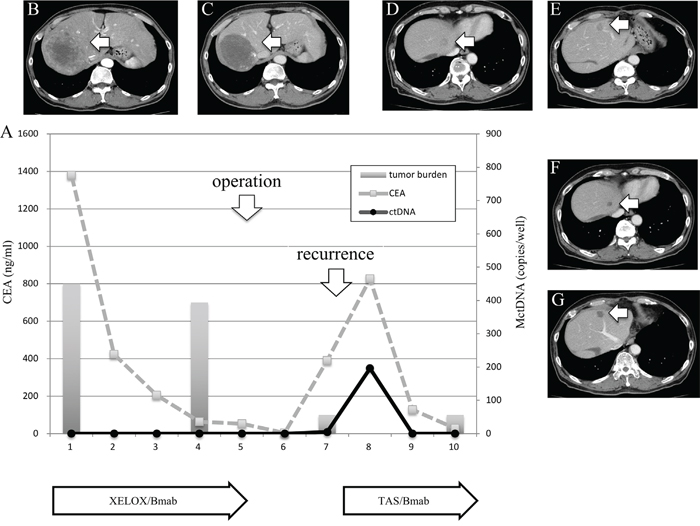
Figure 5: Clinical course of a mCRC patient with multiple liver metastases (A) with morphological changes seen with computed tomography (CT) (B–G) and a spike in elevation in MctDNA. A 60-year-old male with multiple liver metastases is denoted as patient 76. He was treated with XELOX + bevacizumab as the first-line treatment. (B) and (C) show CT images before and after treatment with XELOX + bevacizumab. A change in tumor morphology from heterogeneous to homogeneous low-attenuation was seen in the liver metastases four cycles after treatment with XELOX + bevacizumab despite no change in tumor size. The patient found XELOX + bevacizumab treatment difficult because of severe adverse events and he underwent surgery (right lobectomy for the main tumor and partial resection for other multiple metastases). Soon after surgery, a recurrent liver tumor was found along with increased levels of carcinoembryonic antigen (CEA). Chemotherapy was suggested but the patient refused because of the previous severe adverse events with the first-line treatment. TAS-102 + bevacizumab was then suggested because TAS-102 did not show severe adverse events. Soon after treatment with TAS-102 + bevacizumab, CEA drastically decreased and liver tumors showed morphological changes, which were also seen with the first-line treatment. Additionally, a spike in elevation in MctDNA was observed during this drug response.
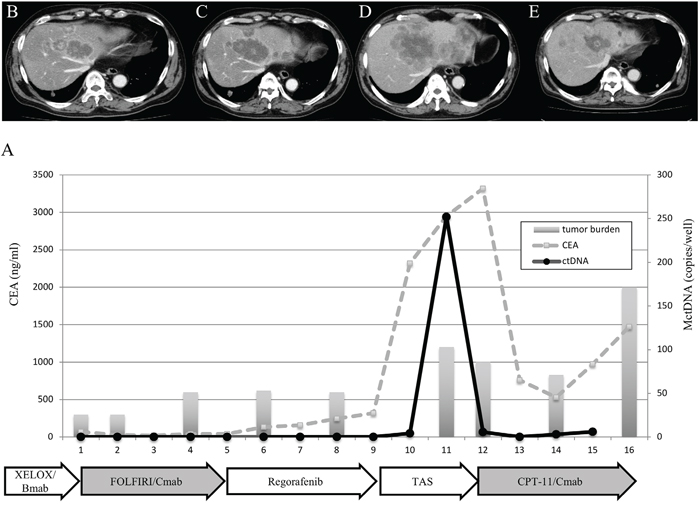
Figure 6: Clinical course of a mCRC patient with multiple liver metastases (A) treated with reintroduction of CPT + cetuximab because of the disappearance of MctDNA. A 65-year-old male with multiple liver metastases is denoted as patient 85. He was treated with XELOX + bevacizumab as the first-line treatment, FOLFIRI + cetuximab as the second-line treatment, regorafenib as the third-line treatment, and TAS-102 as the fourth-line treatment.
Comparison of KRAS mutations between blood and tumor tissues
In tumors of patients with WT KRAS who showed KRAS mutations in blood, the presence or absence of identical mutations were investigated in primary tumors using droplet digital PCR (ddPCR). Table 4 shows a comparison of point mutations in KRAS codon 12/13 between blood and tumor tissues in WT patients. Mutations shared between blood and tumor tissues were seen in 11 patients (73.3%). We also confirmed the accuracy of ddPCR by exploring each mutation using matched normal colorectal tissues from 33 CRC patients as negative controls. No mutations, except G12C and G12D, were detected in matched normal colorectal tissues of all 33 CRC patients. G12C was detected in one patient (3.0%) and G12D was detected in two (6.1%). These results indicated that some cases could be false positive by appearing to harbor G12C or G12D mutations; therefore, results should be interpreted with caution. In cases where G12C or G12D mutations are detected, this should be reevaluated using other samples collected on a different day.
Table 4: Comparison between blood and primary tissues for mutations in 15 patients
No. |
Mutation in blood |
Mutation in primary tissues |
|---|---|---|
58 |
12S |
12S |
59 |
12V |
N.D. |
62 |
12S |
12S |
64 |
12R |
12D, 12S, 13D |
65 |
12C, 13D |
12S, 13D |
68 |
12S |
12S |
69 |
13D |
12S, 13D |
75 |
12A, 12S |
12S |
76 |
12C, 13D |
13D |
77 |
12S, 12V |
N.D. |
78 |
12D, 13D |
12D, 13D |
81 |
13D |
12D, 12S, 13D |
83 |
12D |
12D |
84 |
12A, 12C, 12D, 12V, 13D |
12D |
85 |
12R |
12S, 13D |
N.D.: not detected.
Correlation between mutated circulating tumor DNA and carcinoembryonic antigen
To clarify the characteristics of MctDNA, we examined correlations between MctDNA and carcinoembryonic antigen, a conventional tumor marker used to assess disease progression. The correlation for MT was stronger than that for WT for both number (rs = 0.53, p < 0.01 and rs = 0.33, p < 0.01, respectively) and ratio (rs = 0.50, p < 0.01 and rs = 0.31, p < 0.01, respectively).
DISCUSSION
We investigated dynamic changes in MctDNA during various regimens to provide useful information for the treatment of patients with mCRC.
MctDNA was seen in the blood of patients with not only the MT but also the WT in tumor tissues. MctDNA was observed during treatments with various drugs such as anti-VEGF antibody, regorafenib, TAS-102, and anti-EGFR antibody. Regardless of the KRAS status in tumor tissues, patients with MctDNA in blood showed poor PFS with first-line treatment. KRAS monitoring identified dynamic changes in MctDNA, such as continuous, intermittent, and transient changes, which corresponded with drug response or resistance. It is possible that the disappearance of MctDNA could be involved in recovery of sensitivity to anti-EGFR antibody [19].
MctDNA number and ratio were compared between MT and WT patients. Median values for MT patients were 64.0 copies/well for the number and 10.10% for the ratio, and were 9.1 copies/well for the number and 0.22% for the ratio for WT patients (Figure 1B and 1C). WT patients showed 1/7 the level of MctDNA compared with MT patients (64.0 vs. 9.1), suggesting that WT patients had KRAS mutant cells in 1/7 (14.7%) of tumors. Some smaller pieces of these mutant cells may reach the blood, resulted in the low ratio of KRAS mutant cells (0.22%) in blood of WT patients. The lowest ratio (0.002%) among total cell-free DNA was detected using ddPCR in this analysis. WT patients showed fluctuating changes in the ratio of MctDNA, at around 0.01% of the detection sensitivity of ddPCR, which may have resulted in intermittent detection of MctDNA in WT patients. In contrast, MT patients showed a high ratio of MctDNA (10.10% (0.26–93.60)), allowing for the continuous detection of MctDNA. The lowest ratio (0.26%) was well within the detection range for ddPCR, with a detection sensitivity of 0.01%.
MctDNA was observed in blood of WT patients during treatment with different drugs, such as anti-VEGF antibody, regorafenib, TAS-102, and anti-EGFR antibody. Studies have reported that anti-EGFR antibody is likely involved in the emergence of ctDNA. A recent clinical trial reported ctDNA in 20% of patients treated with anti-VEGF antibody in subgroup analysis [26]. This trial was a randomized phase II study to assess FOLFIRI + bevacizumab beyond progression and FOLFIRI + panitumumab as a second-line treatment for patients with KRAS WT mCRC. No significant differences in PFS were observed in patients without the emergence of ctDNA. Twenty percent of patients with ctDNA showed an extremely poor outcome with the second-line treatment when treated with anti-EGFR antibody. The problem is that these patients are good candidates for anti-EGFR antibody treatment because they have no KRAS mutations in tumor tissues before treatment. The trial indicated that 20% of patients are unlikely to respond to anti-EGFR antibody after prior administration of anti-VEGF antibody. An altered KRAS status is implicated in the sub-sequential treatment outcome; therefore, KRAS monitoring is essential for the treatment of mCRC patients to provide appropriate drug strategies.
There are two possible factors that may contribute to the mechanisms underlying the emergence of KRAS mutations in the blood. First, an acquired KRAS mutation in the tumor may travel to the blood. Anti-VEGF antibody is directed against the tumor vasculature, and should destroy the tumor vasculature, thereby depriving the tumor of oxygen and nutrients [27, 28, 29]. Glucose deprivation in tumors was reported to induce KRAS mutations [30], suggesting insufficient oxygen or nutrients in the tumor induced by anti-VEGF antibody may be involved in this mechanism [27, 28, 29]. Regorafenib is a molecular target drug aimed at inhibiting the VEGF signaling pathway. Anti-EGFR antibody is also reported to induce acquired mutations in vitro, but the mechanism is not well understood [10]. Additionally, tumors in WT patients with latent mutant cells, undetectable by conventional PCR methods with a sensitivity of 1% [31, 32, 33], may expand as a consequence of the treatment, becoming detectable in blood. In the current study, ddPCR with a high sensitivity was able to detect latent mutant cells in tumor tissues. Our data revealed mutations that were shared by both tumor tissues and blood, suggesting that tumor cells with acquired KRAS mutations may travel to the blood. Different types of KRAS mutations were also observed suggesting that latent cells from the tumors, with undetected KRAS mutations may undergo clonal expansion during treatment. The possibility that de novo mutations may arise from within some of the various types of blood cells seems unlikely.
KRAS monitoring identified continuous, intermittent, and transient changes in MctDNA. Continuous detection of MctDNA was frequently seen in MT patients, whereas intermittent detection was more often seen in WT patients. These changes may be associated with the different levels of MctDNA observed between WT and MT patients. In WT patients treated with anti-EGFR antibody, initial detection of MctDNA was likely prior to radiological disease progression (Figure 1D) [10, 9, 19]. Although detection of MctDNA was generally seen in patients with disease progression, transient changes with a spike in elevation were seen in patients in association with the drug response (Figures 3H and 5). One patient showed a transient change with a spike in elevation during treatment with TAS-102, followed by disease progression (Figure 6). MctDNA then disappeared in this patient and anti-EGFR antibody was reintroduced, which achieved a partial response and a long PFS of 7 months with the fifth-line treatment. The results suggest that the later treatment lines had a significant effect on improving the outcome for this patient. The rapid disappearance may have been induced by a delayed drug response to TAS-102 [34]. TAS-102 has a unique mechanism and works by being integrated into the DNA of the tumor cells. Such a process requires some time before an effect on the tumor is observed [35, 36]. The spike in elevation followed by disease progression may indicate a delayed drug response of TAS-102 and contributing effects associated with anti-EGFR antibody.
In conclusion, although our results should be interpreted within the study limitations and further examinations are required to draw a definitive conclusion, KRAS monitoring seems to be a useful tool to help determine treatment strategies. The dynamics of ctDNA during KRAS monitoring provide important information that may aid the treatment of mCRC patients.
MATERIALS AND METHODS
Patients and study design
We prospectively recruited 85 patients (47 males and 38 females) with histologically confirmed mCRC with distant metastases and collected 457 blood samples between June 2014 and March 2017 at the Saitama Medical Center, Jichi Medical University, Japan. Patients were aged >18 years, and their Eastern Cooperative Oncology Group performance status was 0, 1, or 2. Disease extension and response were assessed using computed tomography and the clinical response was evaluated according to RECIST 1.1 criteria. The characteristics of the 85 patients are shown in Table 5.
Table 5: Patient characteristics
KRAS codon 12/13 mutation in primary tumor |
No mutation (wild-type) (n = 56) |
Mutation (n = 29) |
p-value |
|
|---|---|---|---|---|
Gender |
0.663 |
|||
Male |
30 |
17 |
||
Female |
26 |
12 |
||
Mean age (range) |
64.1 (29–79) |
65.7 (19–82) |
0.219 |
|
Location (primary CRC) |
0.278 |
|||
Right |
15 |
13 |
||
Left |
25 |
8 |
||
Rectum |
16 |
8 |
||
Treatment line |
0.825 |
|||
1st line |
29 |
14 |
||
2nd line |
13 |
10 |
||
3rd line |
8 |
3 |
||
4th line |
5 |
1 |
||
BSC |
1 |
1 |
||
Treatment change |
0.479 |
|||
0 |
33 |
13 |
||
1 |
16 |
15 |
||
2 |
4 |
1 |
||
3 |
3 |
0 |
||
CEA at initial assessment |
Median (range) |
10.7 (0–1379) |
24.6 (1.8–3110) |
0.262 |
Blood collection |
Median (range) |
4 (1–16) |
4 (1–12) |
0.196 |
Follow-up months |
Median (range) |
16.5 (3–34) |
12 (3 - 28) |
0.192 |
Differentiation |
0.975 |
|||
Pap+well+mod |
52 |
27 |
||
Muc+por+sig |
4 |
2 |
||
Primary tumor |
0.4 |
|||
Not resected |
12 |
4 |
||
Resected |
44 |
25 |
||
Solitary/synchronous |
0.205 |
|||
Solitary |
31 |
20 |
||
Synchronous |
25 |
9 |
||
Metastatic organ |
0.2 |
|||
1 |
31 |
14 |
||
2 |
22 |
10 |
||
3 |
2 |
5 |
||
0 |
1 |
0 |
BSC: best supportive care; CRC: colorectal cancer; CEA: carcinoembryonic antigen; pap: papillary; mod: moderate; muc: mucinous; sig: signet ring.
The study was approved by the Research Ethics Committee at Jichi Medical University and was conducted in accordance with the principles contained within the Declaration of Helsinki. Written informed consent was obtained from the study participants.
Analysis of KRAS status in primary tumor tissues
KRAS status was evaluated using the Scorpion amplified refractory mutation system method or a RASKET kit using formalin-fixed paraffin-embedded tumor tissues from patients. KRAS analysis was performed by a clinical testing company (Special Reference Laboratories, Tokyo, Japan).
Plasma sample collection and extraction of circulating cell-free DNA
Blood samples were processed for plasma within 5 h of collection. Blood (7 mL) was taken from each patient, and plasma was collected by centrifugation at 3000 × g for 20 min at 4°C, followed by centrifugation at 16000 × g for 10 min at 4°C in a fresh tube. The supernatant was immediately collected and stored at −80°C until DNA extraction. Circulating cell-free DNA was extracted from 2 mL of plasma using a QIAamp circulating nucleic acid kit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions.
Droplet digital PCR analyses
The KRAS status in ctDNA was determined using ddPCR (Bio-Rad, Tokyo, Japan). Seven KRAS mutations (G12D, G12V, G12C, G12R, G12A, G12S, and G13D) were assessed. The ddPCR mixture contained 10 μL of 2 × ddPC Supermix, 250 nM of forward and reverse primers, 62.5 nM MT and WT probe, and 8 μL of sample eluted from plasma. The reaction mixture (20 μL) was loaded into a DG8 cartridge (Bio-Rad) with a gasket and the cartridge was placed into the droplet generator, according to the manufacturer’s instructions. The generated droplets were transferred into a 96-well plate and sealed using a foil lid and a thermal plate sealer. After heat sealing, PCR was performed using a Veriti thermal cycler (Thermo Fisher Scientific, Waltham, MA, USA) under the following conditions: 10 min at 95°C, 39 cycles at 94°C for 30 s, then at 60°C for 60 s. Amplified droplets were analyzed using a QX200 droplet reader (Bio-Rad) for fluorescent measurement of FAM probes for WT and HEX for MT. ddPCR data were analyzed using QuantaSoft software. Amplified DNA products were extracted from droplets following PCR for Sanger sequencing. Samples with two or more positive droplets were determined as positive. To explore the reproducibility and sensitivity of the methods, for instance, a tumor with a known mutation that is also found in the plasma could be spiked into DNA samples from other cases to see if the mutation could be detected at varying dilutions.
Threshold values for droplet digital PCR
To determine the number of positive droplets required for a true positive for MctDNA, we confirmed the sequence of the mutation according to the number of droplets obtained. Different numbers of droplets (1, 2, 4, and 5) were sorted using the On-chip Sort system (On-chip Biotechnologies, Tokyo, Japan) and the sequence of the mutation was confirmed using the HCT-116 cell line, which has a KRAS codon 13 mutation. Clinical samples with the KRAS mutation were used to verify the sequence of the mutation. Only one positive droplet obtained by sorting showed a negative result in one out of three examinations (33.3%); whereas two or more positive droplets did not fail to show the mutation in three examinations. Samples with two or more positive droplets were determined as being positive.
Sorting positive droplets using the On-chip Sort system
To verify the sequence of PCR fragments in droplets, the On-chip Sort system was used to sort positive droplets that were labelled with FAM, according to the manufacturer’s instructions. The instrument was a microfluidic chip-based cell sorter that allows for the use of any liquid as a carrier fluid. Selected FAM-positive droplets were confirmed by fluorescent microscopy and DNA fragments were collected from droplets by extraction with chloroform.
TA cloning and Sanger sequencing
Collected DNA fragments were amplified using the prime PCR for ddPCR KRAS assay (Bio-Rad), and then used for TA cloning after elimination of DNA fragments with the WT. PCR products were used with the TOPO TA cloning kit for Sanger sequencing (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. Plasmid DNA was extracted using a QIAprep spin miniprep kit (Qiagen) and Sanger sequencing was performed using an ABI 3130xl genetic analyzer (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
Fisher’s exact test was used to examine the relationship between two categorical variables. Comparison of continuous variables between two groups was performed, with Student’s t-test being used for those variables with a normal distribution and the non-parametric Mann–Whitney–Wilcoxon test being used for those variables without a normal distribution. The association between ctDNA and carcinoembryonic antigen was determined using Spearman’s correlation test. A p-value of 0.05 was considered statistically significant. All analyses were conducted using StatView ver. 5.0 (SAS Institute, Inc., Cary, NC, USA).
Abbreviations
ctDNA: circulating tumor DNA; ddPCR: droplet digital PCR; EGFR: epidermal growth factor receptor; mCRC: metastatic colorectal cancer; MctDNA: KRAS mutated circulating tumor DNA; MT: mutant-type; PFS: progression-free survival; VEGF: vascular endothelial growth factor; WT: wild-type.
Author contributions
All authors contributed to the design of the study. Y.T and K.S drafted the manuscript, and analyzed the date. Y.T performed experiments. All of the other authors contributed to sample and data collection, interpretation, and critically reviewed the manuscript. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This work was supported in part by a grant-in-aid for post-graduate students from Jichi Medical University; a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan; and the JKA Foundation through its promotion funds from Keirin Race.
REFERENCES
1. Laurent-Puig P, Pekin D, Normand C, Kotsopoulos SK, Nizard P, Perez-Toralla K, Rowell R, Olson J, Srinivasan P, Le Corre D, Hor T, El Harrak Z, Li X, et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin Cancer Res. 2015; 21:1087-97. https://doi.org/10.1158/1078-0432.CCR-14-0983.
2. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009; 360:1408-17. https://doi.org/10.1056/NEJMoa0805019.
3. Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015; 26:13-21. https://doi.org/10.1093/annonc/mdu378.
4. Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010; 16:790-9. https://doi.org/10.1158/1078-0432.CCR-09-2446.
5. Kosmidou V, Oikonomou E, Vlassi M, Avlonitis S, Katseli A, Tsipras I, Mourtzoukou D, Kontogeorgos G, Zografos G, Pintzas A. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum Mutat. 2014; 35:329-40. https://doi.org/10.1002/humu.22496.
6. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883-92. https://doi.org/10.1056/NEJMoa1113205.
7. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, Gold KA, Kalhor N, Little L, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014; 346:256-9. https://doi.org/10.1126/science.1256930.
8. Kawamoto Y, Tsuchihara K, Yoshino T, Ogasawara N, Kojima M, Takahashi M, Ochiai A, Bando H, Fuse N, Tahara M, Doi T, Esumi H, Komatsu Y, et al. KRAS mutations in primary tumours and post-FOLFOX metastatic lesions in cases of colorectal cancer. Br J Cancer. 2012; 107:340-4. https://doi.org/10.1038/bjc.2012.218.
9. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537-40. https://doi.org/10.1038/nature11219.
10. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486:532-6. https://doi.org/10.1038/nature11156.
11. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6:224ra24. https://doi.org/10.1126/scitranslmed.3007094.
12. Morelli MP, Overman MJ, Dasari A, Kazmi SM, Mazard T, Vilar E, Morris VK, Lee MS, Herron D, Eng C, Morris J, Kee BK, Janku F, et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann Oncol. 2015; 26:731-6. https://doi.org/10.1093/annonc/mdv005.
13. Olmedillas Lopez S, Garcia-Olmo DC, Garcia-Arranz M, Guadalajara H, Pastor C, Garcia-Olmo D. KRAS G12V Mutation Detection by Droplet Digital PCR in Circulating Cell-Free DNA of Colorectal Cancer Patients. Int J Mol Sci. 2016; 17:484. https://doi.org/10.3390/ijms17040484.
14. Overman MJ, Modak J, Kopetz S, Murthy R, Yao JC, Hicks ME, Abbruzzese JL, Tam AL. Use of research biopsies in clinical trials: are risks and benefits adequately discussed? J Clin Oncol. 2013; 31:17-22. https://doi.org/10.1200/JCO.2012.43.1718.
15. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985-90. https://doi.org/10.1038/nm.1789.
16. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013; 10:472-84. https://doi.org/10.1038/nrclinonc.2013.110.
17. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015; 61:112-23. https://doi.org/10.1373/clinchem.2014.222679.
18. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32:579-86. https://doi.org/10.1200/jco.2012.45.2011.
19. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, Lamba S, Hobor S, Avallone A, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015; 21:795-801. https://doi.org/10.1038/nm.3870.
20. Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, Mussolin B, Kwak EL, Buscarino M, Lazzari L, Valtorta E, Truini M, Jessop NA, et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016; 6:147-53. https://doi.org/10.1158/2159-8290.CD-15-1283.
21. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011; 83:8604-10. https://doi.org/10.1021/ac202028g.
22. Watanabe M, Kawaguchi T, Isa S, Ando M, Tamiya A, Kubo A, Saka H, Takeo S, Adachi H, Tagawa T, Kakegawa S, Yamashita M, Kataoka K, et al. Ultra-Sensitive Detection of the Pretreatment EGFR T790M Mutation in Non-Small Cell Lung Cancer Patients with an EGFR-Activating Mutation Using Droplet Digital PCR. Clin Cancer Res. 2015; 21:3552-60. https://doi.org/10.1158/1078-0432.CCR-14-2151.
23. Chun YS, Vauthey JN, Boonsirikamchai P, Maru DM, Kopetz S, Palavecino M, Curley SA, Abdalla EK, Kaur H, Charnsangavej C, Loyer EM. Association of computed tomography morphologic criteria with pathologic response and survival in patients treated with bevacizumab for colorectal liver metastases. JAMA. 2009; 302:2338-44. https://doi.org/10.1001/jama.2009.1755.
24. Shindoh J, Loyer EM, Kopetz S, Boonsirikamchai P, Maru DM, Chun YS, Zimmitti G, Curley SA, Charnsangavej C, Aloia TA, Vauthey JN. Optimal morphologic response to preoperative chemotherapy: an alternate outcome end point before resection of hepatic colorectal metastases. J Clin Oncol. 2012; 30:4566-72. https://doi.org/10.1200/JCO.2012.45.2854.
25. Suzuki K, Muto Y, Ichida K, Fukui T, Takayama Y, Kakizawa N, Kato T, Hasegawa F, Watanabe F, Kaneda Y, Kikukawa R, Saito M, Tsujinaka S, et al. Morphological response contributes to patient selection for rescue liver resection in chemotherapy patients with initially un-resectable colorectal liver metastasis. Oncol Lett. 2017; 14:1491-9. https://doi.org/10.3892/ol.2017.6338.
26. Shitara K, Yonesaka K, Denda T, Yamazaki K, Moriwaki T, Tsuda M, Takano T, Okuda H, Nishina T, Sakai K, Nishio K, Tokunaga S, Yamanaka T, et al. Randomized study of FOLFIRI plus either panitumumab or bevacizumab for wild-type KRAS colorectal cancer-WJOG 6210G. Cancer Sci. 2016; 107:1843-50. https://doi.org/10.1111/cas.13098.
27. Utsugi T. New challenges and inspired answers for anticancer drug discovery and development. Jpn J Clin Oncol. 2013; 43:945-53. https://doi.org/10.1093/jjco/hyt131.
28. Lenz HJ, Stintzing S, Loupakis F. TAS-102, a novel antitumor agent: a review of the mechanism of action. Cancer Treat Rev. 2015; 41:777-83. https://doi.org/10.1016/j.ctrv.2015.06.001.
29. Matsuoka K, Iimori M, Niimi S, Tsukihara H, Watanabe S, Kiyonari S, Kiniwa M, Ando K, Tokunaga E, Saeki H, Oki E, Maehara Y, Kitao H. Trifluridine Induces p53-Dependent Sustained G2 Phase Arrest with Its Massive Misincorporation into DNA and Few DNA Strand Breaks. Mol Cancer Ther. 2015; 14:1004-13. https://doi.org/10.1158/1535-7163.MCT-14-0236.
30. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005; 307:58-62. https://doi.org/10.1126/science.1104819.
31. Gerber HP, Ferrara N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res. 2005; 65:671-80.
32. Marques I, Araujo A, de Mello RA. Anti-angiogenic therapies for metastatic colorectal cancer: current and future perspectives. World J Gastroenterol. 2013; 19:7955-71. https://doi.org/10.3748/wjg.v19.i44.7955.
33. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009; 325:1555-9. https://doi.org/10.1126/science.1174229.
34. Yoshino T, Muro K, Yamaguchi K, Nishina T, Denda T, Kudo T, Okamoto W, Taniguchi H, Akagi K, Kajiwara T, Hironaka S, Satoh T. Clinical Validation of a Multiplex Kit for RAS Mutations in Colorectal Cancer: Results of the RASKET (RAS KEy Testing) Prospective, Multicenter Study. EBioMedicine. 2015; 2:317-23. https://doi.org/10.1016/j.ebiom.2015.02.007.
35. Duan H, Lu J, Lu T, Gao J, Zhang J, Xu Y, Wang M, Wu H, Liang Z, Liu T. Comparison of EGFR mutation status between plasma and tumor tissue in non-small cell lung cancer using the Scorpion ARMS method and the possible prognostic significance of plasma EGFR mutation status. Int J Clin Exp Pathol. 2015; 8:13136-45.
36. Sakai K, Yoneshige A, Ito A, Ueda Y, Kondo S, Nobumasa H, Fujita Y, Togashi Y, Terashima M, De Velasco MA, Tomida S, Nishio K. Performance of a novel KRAS mutation assay for formalin-fixed paraffin embedded tissues of colorectal cancer. Springerplus. 2015; 4:7. https://doi.org/10.1186/2193-1801-4-7.