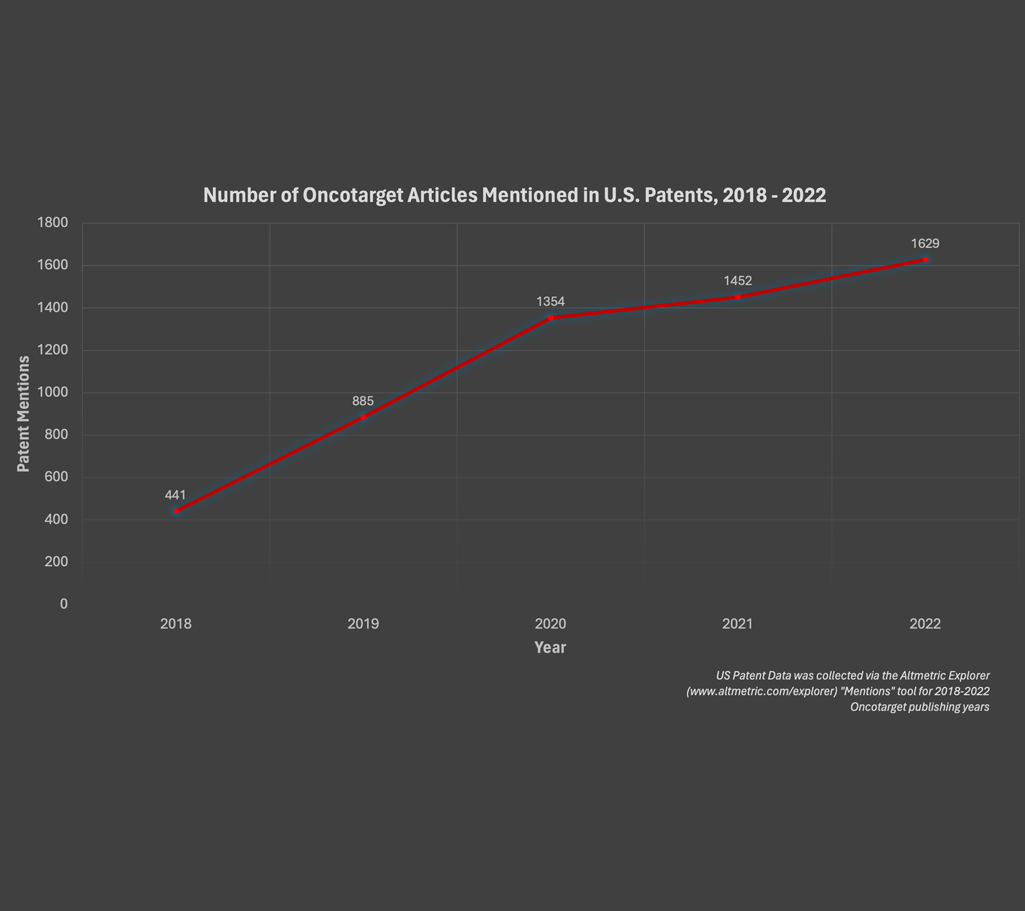
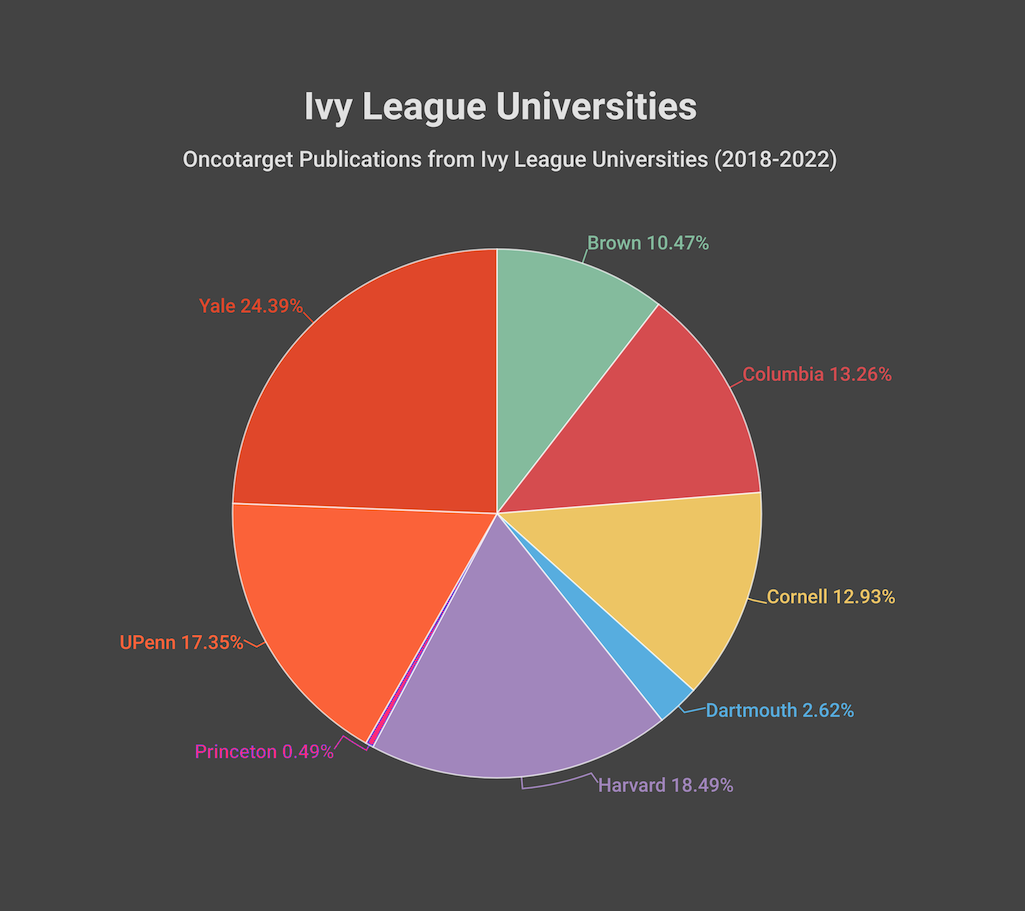
Oncotarget
Oncotarget (a primarily oncology-focused, peer-reviewed, open access journal) aims to maximize research impact through insightful peer-review; eliminate borders between specialties by linking different fields of oncology, cancer research and biomedical sciences; and foster application of basic and clinical science.
Its scope is unique. The term "oncotarget" encompasses all molecules, pathways, cellular functions, cell types, and even tissues that can be viewed as targets relevant to cancer as well as other diseases. The term was introduced in the inaugural Editorial, Introducing Oncotarget.
As of January 1, 2022, Oncotarget has shifted to a continuous publishing model. Papers will now be published continuously within yearly volumes in their final and complete form and then quickly released to Pubmed.
Publication Alerts
Post-Publication Promotion
Learn about our FREE
Rapamycin Press LLC dba Impact Journals is the publisher of Oncotarget: www.impactjournals.com.
Impact Journals is a member of the Wellcome Trust List of Compliant Publishers.
Impact Journals is a member of the Society for Scholarly Publishing.
On December 23, 2022, Oncotarget server experienced a DDoS attack. As a result, Oncotarget site was inaccessible for a few hours. Oncotarget team swiftly dealt with the situation and took it under control. This malicious action will be reported to the FBI.