
INTRODUCTION
Oral squamous cell carcinoma (OSCC) is one of the most common cancers of the head and neck region [1]. Despite advances in surgery, radiotherapy, and chemotherapy, the five-year survival rate is still 50–60% [2, 3]. Many strategies for the development of new OSCC therapeutics have considered the role of the tumor microenvironment in cancer development and progression [4]. In solid tumors such as OSCC, hypoxia and chronic inflammation are two of the most prominent features of tumor progression [5].
Hypoxia is generated by an insufficient blood supply during tumor growth [6]. The oxygen pressure within solid tumors is heterogeneous and can range from approximately 5% O2 in well-vascularized regions to 1% O2 (hypoxic conditions) near necrotic regions [7]. We previously demonstrated that hypoxia was associated with tumor progression and clinical prognosis in OSCC [8]. Hypoxia-inducible factor-1 (HIF-1), a key mediator of the cellular response to hypoxia, is comprised of an oxygen-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit [9]. The HIF-1 dimer regulates the expression of more than 100 downstream genes that protect cells under hypoxic conditions [10].
Inflammation is another aspect of the tumor microenvironment that has a significant role in tumor progression. Under normal conditions, the inflammatory response can promote tissue repair processes such as wound healing. However, an abnormal inflammatory response can ultimately lead to tumorigenesis [11]. In solid tumors, cell growth can exceed the oxygen and nutrient supply resulting in hypoxia. This promotes the release of pro-inflammatory mediators that recruit additional inflammatory cells [12]. Nuclear factor-κB (NF-κB) is a key transcription factor involved in the inflammatory response and is thought to be a critical link between inflammation and cancer [13]. NF-κB is expressed in almost all cell types and is involved in cellular responses to stimuli such as stress, cytokines, apoptosis, and immune reactions [14]. NF-κB signaling is mediated by Toll-like receptors (TLRs), interleukin-1 receptor (IL-1R), and the tumor necrosis factor receptor (TNFR). Inflammation and NF-κB in particular play dual roles in cancer progression. NF-κB activation is part of the immune response and is particularly involved in acute inflammatory processes. NF-κB is constitutively activated in many cancers and can exert a variety of pro-tumorigenic effects [15]. Activation of NF-κB typically results in upregulation of anti-apoptotic genes and activation of cell survival mechanisms [16]. Cancer-related inflammation is a potential target for innovative therapeutic strategies. Therefore, NF-κB suppression could have therapeutic potential. Both in vitro and in vivo studies have shown that targeted NF-κB inhibition sensitized tumor cells to chemotherapy and radiation. However, targeting NF-κB in solid tumors did not achieve optimal treatment outcomes [17]. This may be explained by the tumor microenvironment. Despite inhibition of inflammatory signaling pathways, other pathways could still stimulate NF-κB expression. Recently, hypoxia in a solid tumor microenvironment was shown to activate NF-κB signaling [18]. However, the mechanisms by which hypoxia regulates NF-κB activity are unclear.
TLRs are a family of transmembrane receptors that recognize conserved microbial structures/patterns. It was initially though that TLRs were only expressed in immune cells and that they played a key role in the host defense against infection by recognizing a range of chemicals produced by bacteria, viruses, fungi, and protozoa [19]. However, recent data has indicated that various cancer cells also express TLRs. To date, 13 mammalian TLRs have been described, 11 of which are expressed in humans. TLR3, TLR4, and TLR9 are found in breast, prostate, and colon cancer [20, 21, 22]. TLR7 expression has also been reported in esophageal squamous cell carcinoma [23]. Finally, TLR3 and TLR4 expression was observed in head and neck squamous cell carcinoma (HNSCC) [24, 25]. TLRs are functionally active in various tumors and might induce cancer cell resistance to apoptosis [26].
In the present study, we demonstrated that TLR signaling induced HIF-1 expression via NF-κB. Moreover, HIF-1, as part of a transcriptional response to hypoxia, directly activated the TLR/NF-κB signaling pathway in OSCC. Our results demonstrate that HIF-1 and TLR/NF-κB form a positive feedback loop in OSCC cells that connects hypoxia to inflammation, which contributes to OSCC initiation and progression.
RESULTS
TLRs are highly expressed in OSCC
We investigated the expression of TLR2, TLR3, TLR4, TLR7, and TLR9 in two OSCC cell lines (HSC3 and SCC4). Quantitative reverse transcription-PCR (qRT-PCR) revealed that these two cell lines expressed higher levels of TLR3 and TLR4 mRNA than TLR2, TLR7, and TLR9 (Figure 1A). TLR3 and TLR4 expression in these cells was confirmed by western blotting (Figure 1B). High levels of TLR3 and TLR4 were also observed in OSCC patient samples as shown by immunohistochemistry (Figure 1C).
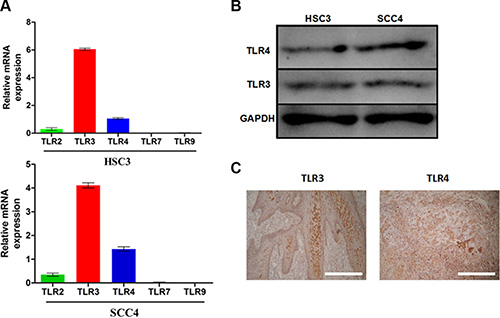
Figure 1: TLR3 and TLR4 are expressed in OSCC. (A) Relative mRNA expression of TLR2, TLR3, TLR4, TLR7, and TLR9 in the SCC4 and HSC3 OSCC cell lines. (B) Expression of TLR3 and TLR4 in HSC3 and SCC4 cells. (C) Expression of TLR3 and TLR4 in tumor tissue from OSCC patients (200×).
TLRs induce HIF-1 expression in OSCC cells
We next examined whether TLR4 pathway activation could alter the expression of HIF-1 in OSCC cells. Our results indicated that lipopolysaccharide (LPS), a TLR activator, induced the expression of HIF-1α and its target gene vascular endothelial growth factor (VEGF) in both HSC3 and SCC4 cells in a dose- and time-dependent manner. Maximum induction was observed after treatment with 10 μg/mL LPS for 24 h (Figure 2A–2B). Polyinosinic-polycytidylic acid [poly (I:C)] (10 μg/mL), a TLR3 ligand, induced expression of HIF-1α and VEGF in HSC3 and SCC4 cells in a time-dependent manner (Figure 2C–2D), suggesting that TLR pathway activation resulted in upregulation of HIF-1α and VEGF expression.
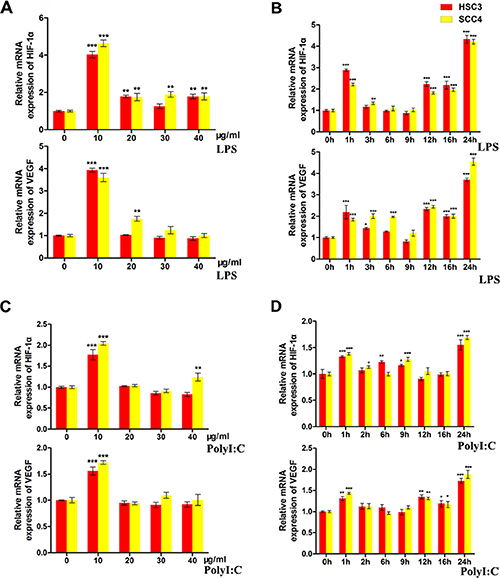
Figure 2: LPS and poly (I:C) induce HIF-1α and VEGF expression in HSC3 and SCC4 cells. (A) Relative mRNA expression of HIF1A and its target gene VEGF in HSC3 and SCC4 cells treated with 0–40 μg/mL LPS for 24 h. (B) Relative mRNA expression of HIF1A and its target gene VEGF in HSC3 and SCC4 cells treated with 10 μg/mL LPS for 0–24 h. (C) Relative mRNA expression of HIF1A and its target gene VEGF in HSC3 and SCC4 cells treated with 0–40 μg/mL poly (I:C) for 24 h. (D) Relative mRNA expression of HIF1A and its target gene VEGF in HSC3 and SCC4 cells treated with 10 μg/mL poly (I:C) for 0–24 h. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
To further investigate whether TLRs regulated HIF-1α and VEGF expression, we knocked down TLR3 and TLR4 using three independent small interfering RNAs (siRNAs). Treatment of the cells with an anti-TLR4 siRNA (siTLR4 1332) decreased TLR4 mRNA levels by > 70% in both HSC3 and SCC4 cell lines (Figure 3A). Concomitantly, this siRNA inhibited LPS-induced HIF-1α and VEGF expression (Figure 3A). Similarly, an siRNA that decreased TLR3 expression by 70% (siTLR3 2658) also resulted in a significant reduction in poly (I:C)- induced HIF-1α and VEGF expression in HSC3 and SCC4 cells (Figure 3B). Our results also demonstrated that hypoxia enhanced LPS- and poly (I:C)-induced HIF-1α and VEGF expression, which was decreased by treatment with siRNAs targeting TLR3 and TLR4 (Figure 3C–3D). These results strongly suggested that TLR3 and TLR4 signaling was involved in regulation of HIF-1α and VEGF expression in OSCC cell lines.
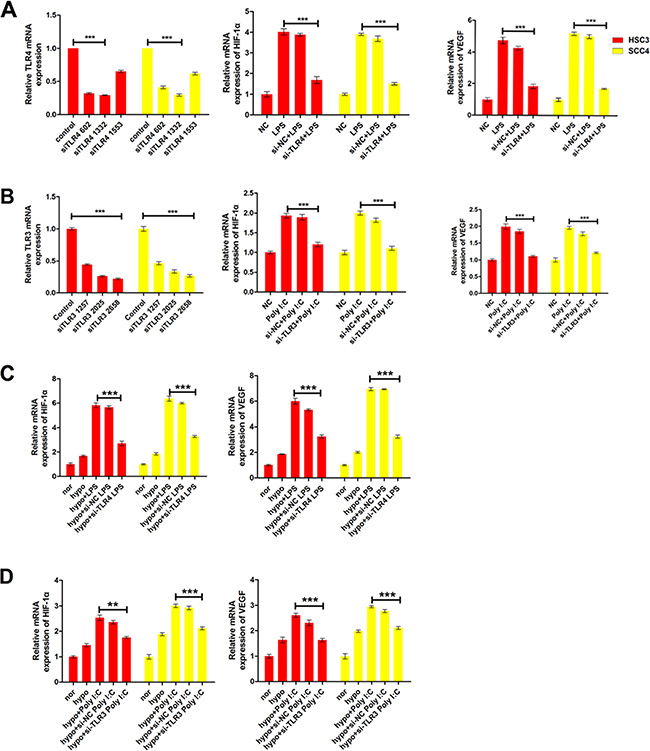
Figure 3: LPS and poly (I:C) induce HIF-1α and VEGF expression via TLR3 or TLR4. (A) Relative mRNA expression of HIF1A and its target gene VEGF in HSC3 and SCC4 cells transfected with 20 nM siTLR4 1332 prior to treatment with 10 μg/mL LPS. (B) Relative mRNA expression of HIF1A and its target gene VEGF in HSC3 and SCC4 cells transfected with 40 nM siTLR3 2658 prior to treatment with 10 μg/mL poly (I:C). (C) Relative mRNA expression of HIF1A and its target gene VEGF under hypoxic conditions (1% O2). Cells transfected with 20 nM siTLR4 1332 prior to treatment with 10 μg/mL LPS. (D) Relative mRNA expression of HIF1A and its target gene VEGF under hypoxic conditions (1% O2). Cells transfected with 40 nM siTLR3 2658 prior to treatment with 10 μg/mL poly(I:C) treatment. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001). NC, negative control.
NF-κB is involved in TLR3- and TLR4-mediated regulation of HIF-1
Given that NF-κB plays a key role in the TLR pathway, we investigated the possible role of NF-κB in TLR-dependent upregulation of HIF-1 using qRT-PCR. These results confirmed increased expression of the p65 subunit of NF-κB after treatment with LPS or poly (I:C) (Figure 4A). Using immunofluorescence, we demonstrated that treatment of HSC-3 cells with LPS and poly (I:C) led to the nuclear localization of p65 and activation of NF-κB signaling, which was not observed in cells transfected with TLR4 siRNA (Figure 4B–4C).
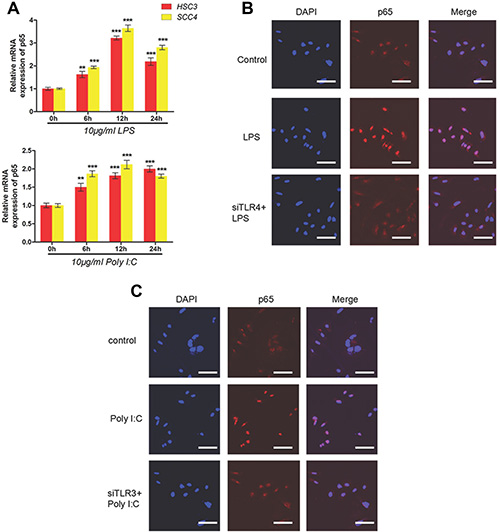
Figure 4: LPS and poly (I:C) induce HIF-1α and VEGF expression via the TLR-NF-κB pathway in OSCC. (A) Relative mRNA expression of p65 in HSC3 and SCC4 cells treated with 10 μg/mL LPS or poly (I:C) for 0–24 h. (B) Localization of p65 in cells treated with 10 μg/mL LPS for 2 h with or without transfection of siTLR4. Blue, nuclei; Red, p65. (C) Localization of p65 in cells treated with 10 μg/mL poly(I:C) for 2 h with or without transfection of siTLR3. Blue, nuclei; Red, p65. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
It was previously reported that NF-κB could regulate HIF-1 expression in endometrial carcinoma and malignant lymphoma [27, 28]. Given that our results indicated both HIF-1 and NF-κB were upregulated through the TLR3 and TLR4 pathways in OSCC cell lines (Figures 3–4), we investigated whether NF-κB could also regulate HIF-1 expression in OSCC cells. After evaluating the knockdown efficiency of three siRNAs against p65 (data not shown), we demonstrated that a highly efficient siRNA (si-p65 665) could inhibit LPS- and poly (I:C)-induced HIF-1α and VEGF expression in both HSC3 and HSC4 cells (Figure 5A).
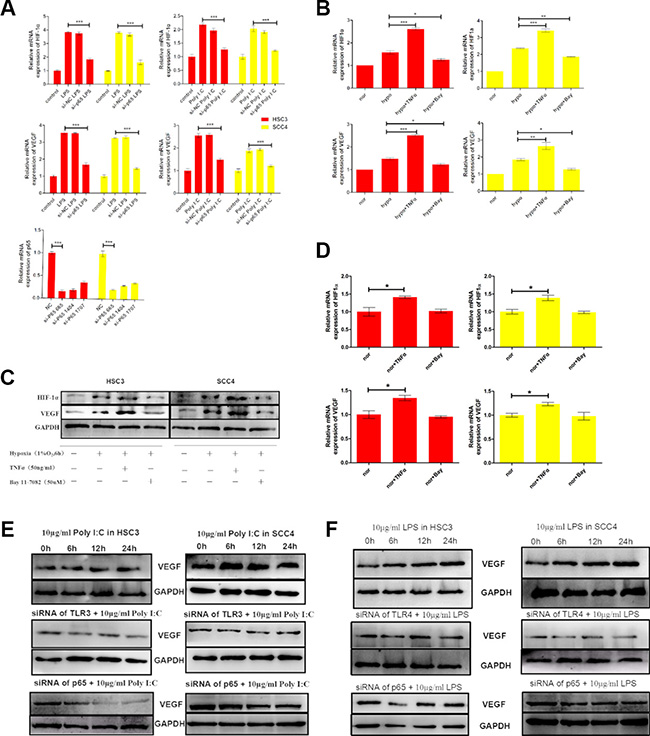
Figure 5: The TLR-NF-κB pathway regulates HIF-1α and VEGF expression in HSC3 and SCC4 cells. (A) (Bottom) Relative mRNA expression of p65 in HSC3 and SCC4 cells transfected with different sip65 siRNAs. We selected siRNA 665 to target p65. Cells were transfected with siRNA 665 prior to stimulation with LPS or poly (I:C). (Top) Relative mRNA expression of HIF1A and VEGF in HSC3 and SCC4 cells following sip65 665 transfection. (B) Relative mRNA expression of HIF1A and VEGF in HSC3 and SCC4 cells cultured in 1% O2 for 6 h with 50 ng/mL TNF-α or 50 μM BAY 11–7082. (C) Western blot analysis of changes in HIF-1α and VEGF protein levels. (D) Relative mRNA expression of HIF1A and VEGF in HSC3 and SCC4 cells cultured in 20% O2 for 6 h with 50 ng/mL TNF-α or 50 μM BAY 11–7082. (E) VEGF protein levels in HSC3 and SCC4 cells that were treated with 10 μg/mL poly (I:C), transfected with siTLR3 or sip65, and then treated once more with 10 μg/mL poly (I:C). (F) VEGF protein levels in HSC3 and SCC4 cells that were treated with 10 μg/mL LPS, transfected with siTLR4 or sip65, and then treated once more with 10 μg/mL LPS. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001). NC, negative control; nor, normoxic conditions; hypo, hypoxic conditions.
We next incubated OSCC cells under hypoxic conditions (1% O2 for 6 h in the presence or absence of either TNF-α, an NF-κB stimulator, or BAY 11–7082, an NF-κB inhibitor). The results indicated that TNF-α enhanced, but BAY 11–7082 inhibited, hypoxia-induced HIF-1α and VEGF expression at both the mRNA and protein levels (Figure 5B–5C). Similarly, when cells were cultured under normoxic conditions (20% O2) for 6 h, TNF-α induced expression of HIF-1α and VEGF (both mRNA and protein), whereas expression was inhibited by BAY 11–7082 (Figure 5D, Supplementary Figure 1A). These results showed that even under normoxic conditions, TNF-α could induce expression of HIF-1α and VEGF, which was inhibited by BAY 11–7082. These data strongly suggested that NF-κB was involved in regulation of TNF-mediated HIF-1α and VEGF expression in OSCC cells.
We also examined the impact of poly (I:C) and LPS on the expression of VEGF and HIF-1α. Consistent with the mRNA levels (Figure 2), both poly (I:C) and LPS increased VEGF expression under normoxic conditions, whereas expression was inhibited by TLR siRNAs (TLR3 and TLR4 siRNA) and p65 siRNA (Figure 5E, 5F). Similarly, expression of both HIF-1α and VEGF was induced by hypoxia, but this induction was inhibited by siRNAs against TLR3 and TLR4 (Supplementary Figure 1B–1C). Finally, to further explore the roles of TLRs in HIF-1 regulation, we inserted a sequence containing three copies of the HIF-1-binding hypoxia response element (HRE) into a pGL6 plasmid (pGL6-3xHRE) (Figure 6A). When HSC3 and SCC4 cells were transfected with pGL6-3xHRE and then cultured under hypoxic conditions, luciferase activity was greatly increased (Figure 6B). Consistent with these results, when cells transfected with pGL6-3xHRE were treated with LPS or poly (I:C) under normoxic conditions, luciferase activity was also significantly increased (Figure 6C), which provided additional evidence that TLR signaling could regulate HIF-1α activity in OSCC cells. Collectively, these data demonstrated that TLR3 and TLR4 were expressed in OSCC and that the TLR activators LPS and poly (I:C) could regulate HIF-1α and VEGF expression through NF-κB.
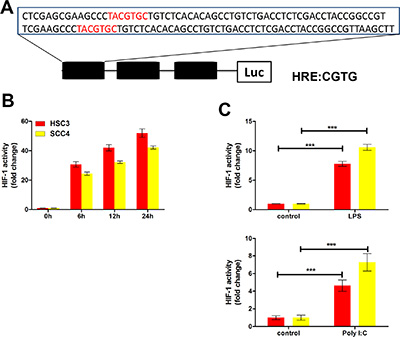
Figure 6: The TLR3 and TLR4 pathways regulate HIF-1 activity in HSC3 and SCC4 cells. (A) Three HRE sequences (3xHRE), each containing two HRE binding sites, were inserted into a pGL6-luciferase reporter plasmid. (B) Luciferase reporter assay in HSC3 and SCC4 cells transfected with the 3xHRE reporter plasmid and cultured under hypoxic conditions for 6, 12, and 24 h. (C) Luciferase reporter assay of HSC3 and SCC4 cells transfected with the 3xHRE reporter plasmid and cultured under normoxic conditions with 10 μg/mL LPS or poly (I:C) for 24 h. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
Hypoxia induces TLR3 and TLR4 expression through HIF-1
To determine whether hypoxia regulated TLR3 and TLR4 expression, we exposed two OSCC cell lines to hypoxic conditions for varying times. Both TLR3/TLR4 mRNA and protein levels were upregulated under hypoxic conditions (Figure 7A–7B). To evaluate the possible role of HIF-1 in the hypoxia-induced upregulation of TLR3 and TLR4, we established HSC3 and SCC4 stable cell lines with low HIF-1α expression using a HIF-1α short hairpin RNA (shRNA) lentiviral vector (data not shown). Hypoxia-induced upregulation of HIF-1 mRNA and protein was inhibited in these cells (Figure 7C–7D), and the upregulation of TLR3/TLR4 mRNA and protein by hypoxia suppressed, indicating that HIF-1 was a key mediator of the hypoxia-induced upregulation of TLR3 and TLR4 mRNA and protein.
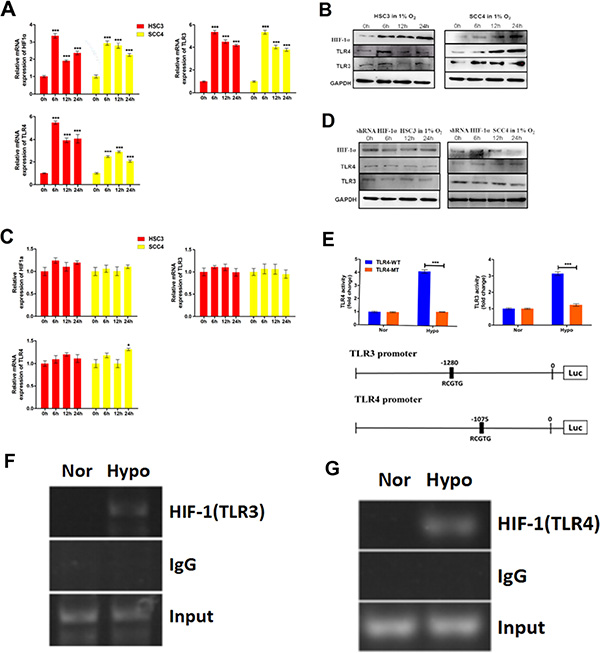
Figure 7: Hypoxia induces TLR3 and TLR4 expression via HIF-1. (A) Relative mRNA expression of HIF1A, TLR3, and TLR4 in HSC3 and SCC4 cells cultured in 1% O2 for 6, 12, and 24 h. (B) Expression of HIF-1α, TLR3, and TLR4 in HSC3 and SCC4 cells cultured in 1% O2 for 6, 12, and 24 h. (C–D) Expression of HIF-1α, TLR3, and TLR4 mRNA (C) and protein (D) in HSC3 and SCC4 cells transfected with or without HIF-1α shRNA and cultured in 1% O2 for 6, 12, and 24 h. (E) Luciferase reporter assay in HSC3 and SCC4 cells transfected with wild-type (WT) or mutant (MT) TLR3 and TLR4 promoter luciferase reporters and cultured under 20% O2 (normoxic, nor) or 1% O2 (hypoxic, hypo) conditions for 24 h. (F) ChIP assay of the TLR3 promoter (−1280 bp). HSC3 cells were cultured in 1% O2 or 20% O2 for 24 h. (G) ChIP assay of the TLR4 promoter (−1075 bp). HSC3 cells were cultured in 1% O2 or 20% O2 for 24 h. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
To investigate whether HIF-1 could directly regulate TLR3 and TLR4 expression, we analyzed the promoter sequences of each gene and identified possible functional HIF-1-binding HRE sequences (5′-RCGTG-3′) located upstream of the TLR3 and TLR4 promoters, respectively. This raised the possibility that these HREs were directly involved in HIF-1-mediated regulation of TLR3 and TLR4. To test this hypothesis, we generated luciferase reporters driven by either wild-type or HRE-mutated (e.g., from 5′-RCGTG-3′ to 5′-RAAAG-3′) TLR3 and TLR4 promoters, respectively (Figure 7E). Consistent with this hypothesis, hypoxia induced luciferase reporter activity in the presence of the wild-type TLR3 or TLR4 promoters, whereas HRE mutations in the TLR3 and TLR4 promoters impaired or abolished luciferase activity under hypoxic conditions (Figure 7E). These results strongly suggested that HIF-1 directly regulated TLR3 and TLR4 promoter activity. Finally, we used a chromatin immunoprecipitation (ChIP) assay to examine the actual binding sites of HIF-1 within the promoter regions of TLR3 and TLR4. Our results revealed that sequences at -1280 bp of the TLR3 promoter and at -1075 bp of the TLR4 promoter were functional HIF-1-binding sites (Figure 7F–7G), providing further support for a direct role of HIF-1 in the regulation of TLR expression.
Hypoxia induces the expression of NF-κB and pro-inflammatory cytokines via HIF-1
NF-κB is a transcription factor that regulates the expression of inflammatory cytokines. It can be activated by many factors including cytokines, ultraviolet irradiation, and bacterial or viral infections. Interestingly, hypoxia induced p65 expression in HSC3 and SCC4 cells (Figure 8A). Moreover, our results showed that this induction was diminuished by HIF-1α shRNA (Figure 8A), suggesting that hypoxia-induced p65 expression was dependent on HIF-1 in OSCC cells. Finally, consistent with the role of NF-κB in inflammation, increased expression of the inflammatory cytokines IL-1β, IL-6, IL-8, and IL-12p70 was also observed in cells cultured under hypoxic conditions (Figure 8B).
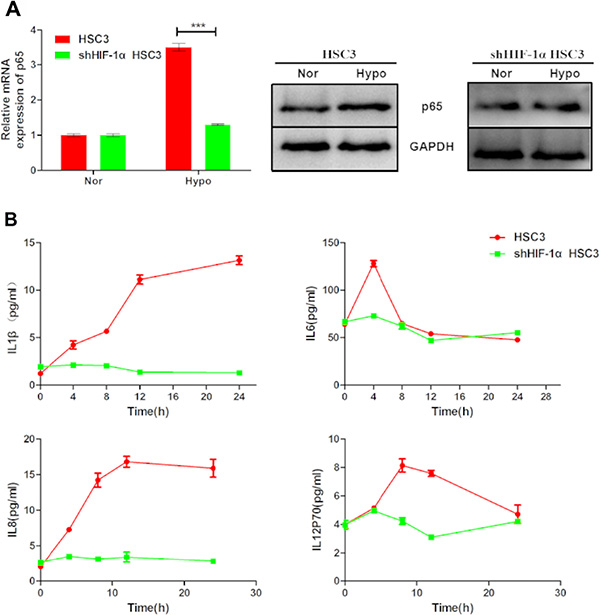
Figure 8: Hypoxia upregulates p65 and pro-inflammatory cytokine expression via HIF-1α. (A) Expression of p65 mRNA and p65 protein in HSC3 cells transfected with or without HIF-1α shRNA under normoxic (nor) or hypoxic (hypo) conditions for 24 h. (B) IL-1β, IL-6, IL-8, and IL-12p70 released by non-transfected and HIF-1α shRNA-transfected HSC3 cells cultured under normoxic (nor) or hypoxic (hypo) conditions for 4, 8, 12, and 24 h. Error bars indicate SE (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
In vivo analysis of human OSCC using a transplantation model in nude mice
To explore the relationship between HIF-1 and TLRs, we established an OSCC transplantation model in nude mice using normal and HIF-1α shRNA-treated HSC3 cells. Inhibition of HIF-1α expression by shRNA significantly reduced tumor growth in mice (Figure 9A). Importantly, HIF-1α expression in tumors was positively correlated with TLR3 and TLR4 expression (Figure 9B). These data confirmed the results of the in vitro studies and suggested that TLR and HIF-1 mutually regulated each other in OSCC cells.
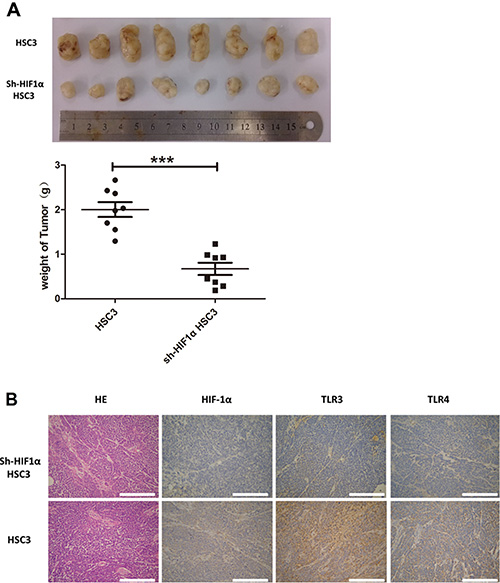
Figure 9: In vivo study of human OSCC using a transplantation model in nude mice. (A) HSC3 and sh-HIF1α HSC3 were injected into nude mice. After 30 days, tumors were collected and weighed. (B) IHC analyses of HIF1α, TLR3, and TLR4 expression in tumor tissue (200×) (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
DISCUSSION
We demonstrated that upregulation of HIF-1α was directly correlated with activation of TLR3 and TLR4 through NF-κB signaling. Importantly, our results suggested that HIF-1 could directly regulate TLR/NF-κB signaling in OSCC cells, suggesting the existence of a positive feedback loop between HIF-1 and TLR/NF-κB signaling in OSCC cells (Figure 10). Cross-talk between HIF-1 and TLR/NF-κB signaling may contribute to OSCC initiation and progression.
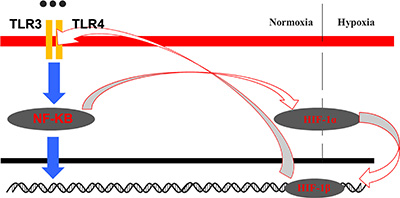
Figure 10: Model for the crosstalk between the HIF-1 and TLR-NF-κB pathways in OSCC.
TLRs are crucial components of the innate immune response to bacterial and viral pathogens [29]. Recent studies have revealed that they can be “double-edged swords” because they might also be expressed in tumor cells [30]. TLR3 was previously detected in papillary thyroid, lung, and breast cancer [31]. TLR4 has been detected in both gastric and colorectal cancer [32]. We demonstrated that TLR3 and TLR4 are highly expressed in OSCC (Figure 1A) which is consistent with previous studies [24, 25, 33]. The expression of TLR4 has been correlated with resistance of human OSCC to chemotherapy [34], and TLR2 expression was shown to be highly correlated with tumor progression [35]. Here, we screened for expression of TLR2, -3, -4, -7, and -9. TLR2 was detected but at a much lower level than TLR3 and TRL4 (data not shown). Thus, TLR3 and TLR4 might be more important for OSCC progression than the other TLRs. HIF-1α may be directly regulated by TLR/NF-κB signaling since the HIF1A promoter contains an active NF-κB binding site in position 197/188, upstream of the transcription start site [36]. Although it has been shown that the TLR-myeloid differentiation primary response 88 (MyD88)-NF-κB signaling pathway could activate NF-κB in immune cells, there have been few reports regarding the functions of TLRs in OSCC. Similarly, although it was reported that HIF-1 could up-regulate TLR4 expression in pancreatic cancer cells under hypoxic conditions [39], to our knowledge this is the first report that HIF-1 can upregulate TLR3 and TLR4 in OSCC cell lines under hypoxia.
Given that HIF-1 regulates more than 100 downstream genes such as VEGF, Bcl-2, and survivin, activation of HIF-1 in oral tumor cells by hypoxia and inflammatory signals will induce target gene expression, thereby promoting cell survival and metastasis. Moreover, our data suggest that bacteria, viruses, fungi, and protozoa in the microenvironment could activate the HIF-1 pathway. Hypoxia could result in increased release of IL-1β, IL-6, IL-8, and IL-12P70 in HSC3 and SCC4 cells and upregulation of the mRNA levels of these cytokines through HIF-1 (Figure 8). In our study, the baseline levels of IL-1β, IL-8, and IL-12P70 in HSC3 and SCC4 cells were much lower than those of IL-6. The release of inflammatory cytokines from OSCC cells could promote cell transformation, survival, proliferation, and metastasis. These cytokines are upregulated by NF-κB, which is further upregulated under hypoxic conditions by HIF-1.
Hypoxia and inflammation are two typical features of the microenvironment in solid tumors [40]. Our studies not only demonstrated that TLR3 and TLR4 are highly expressed in OSCC, but also that TLR3/TLR4–NF-κB signaling is active in oral tumor cells. This signaling pathway forms a TLR3/TLR4–NF-κB–HIF-1 loop that is closely associated with hypoxia and inflammation in the tumor and leads to tumor cell survival, proliferation, angiogenesis, and metastasis. Our findings will help with the development of novel and more effective therapeutic strategies for OSCC based on HIF-1 and TLR/NF-κB inhibition.
MATERIALS AND METHODS
Ethics statement
All experiments involving human samples were performed in accordance with the ethical standards and according to the Declaration of Helsinki. All animal work was conducted according to national and international guidelines. Institutional review board approval was gained for this study from Nanjing Stomatological Hospital Ethics Committee, approval number: 2015NL-009(KS).
Cell culture and reagents
Human OSCC cell lines (HSC3 and SCC4) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were grown in monolayer cultures in Dulbecco’s Modified Eagle’s Medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA), 100 μg/mL streptomycin, and 100 U/mL penicillin in a humidified incubator (5% CO2) at 37°C. Confluent cells were trypsinized with 0.05% trypsin/0.02% EDTA.
Hypoxic treatment
Cells were placed in a humidified hypoxia workstation (Memmert, Germany) equilibrated to 1% O2 and 5% CO2 at 37°C. The experimental chemical compounds were added to the cells immediately before placement in the hypoxia chamber. Cell lysates were collected immediately after cells had been removed from the hypoxia chamber.
Reagents and antibodies
LPS derived from Escherichia coli strain 055: B5, and the NF-κB inhibitor BAY 11–7028 were purchased from Sigma-Aldrich. Recombinant human TNF-α was obtained from PeproTech. Poly (I:C) was obtained from InvivoGen. The following antibodies were used for western blotting: an anti-TLR4 rabbit polyclonal antibody (Abcam); anti-TLR3 rabbit polyclonal antibody (Cell Signaling Technology); anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rabbit polyclonal antibody (Bioworld Technology); anti-HIF-1α rabbit polyclonal antibody (Cell Signaling Technology), anti-VEGF rabbit polyclonal antibody (Abcam). Protein standards were obtained from SunShineBio. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) was purchased from Immunology Consultants Laboratory (Newberg, OR, USA). An anti-p65 rabbit polyclonal antibody (Cell Signaling Technology) and goat anti-rabbit IgG (Bioworld Technology) were used for immunofluorescence.
RNA extraction and qRT-PCR
Total RNA from OSCC cells was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. After RNA quantification, 2 μg of total RNA was reverse-transcribed using the cDNA Reverse Transcription kit (Takara) according to the manufacturer’s instructions. QRT-PCR was performed using a StepOne Real-Time PCR System (Applied Biosystems). The mRNA expression levels of the target genes were assessed by qRT-PCR using iTaq SYBR Green Supermix with ROX (Bio-Rad) and the following cycling conditions: 95°C for 2 min, and 40 cycles of 95°C for 15s and 60°C for 45 s. The comparative threshold cycle (Ct) method was used to evaluate gene expression. The primers used in the experiments are listed in Table 1.
Table 1: Primers of Q-PCR
GENE |
Forward |
Reverse |
|---|---|---|
TLR2 |
ATCCTCCAATCAGGCTTCTCT |
GGACAGGTCAAGGCTTTTTACA |
TLR3 |
TTGCCTTGTATCTACTTTTGGGG |
TCAACACTGTTATGTTTGTGGGT |
TLR4 |
AGACCTGTCCCTGAACCCTAT |
CGATGGACTTCTAAACCAGCCA |
TLR7 |
CACATACCAGACATCTCCCCA |
CCCAGTGGAATAGGTACACAGTT |
TLR9 |
CTGCCACATGACCATCGAG |
GGACAGGGATATGAGGGATTTGG |
HIF-1α |
TTTGCTGAAGACACAGAAGCAAAGA |
TTGAGGACTTGCGCTTTCAGG |
VEGF |
AGGGCAGAATCATCACGAAGT |
AGGGTCTCGATTGGATGGCA |
β-actin |
CTGGGACGACATGGAGAAAA |
AAGGAAGGCTGGAAGAGTGC |
p65 |
ATGTGGAGATCATTGAGCAGC |
CCTGGTCCTGTGTAGCCATT |
Western blotting
Cells cultured under hypoxic or normoxic conditions were harvested and lysed for 20 min using modified RIPA buffer (5 mM EDTA, 2 mM Na3VO4, 5 mM NaF, 1 mM phenylmethylsulfonyl fluoride) supplemented with protease inhibitor cocktail (Sigma-Aldrich). The supernatant (total cellular protein extract) was harvested after centrifugation for analysis. Protein concentrations were determined using a BCA assay (Micro BCA; Pierce, Rockford, IL, USA). Samples containing approximately 40 μg protein were subjected to 10% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis in Tris-Tricine-SDS buffer, and then electrophoretically transferred to polyvinylidene difluoride membranes (Bio-Rad), which were then blocked for 2 h at room temperature in 5% non-fat milk. The membranes were incubated for 1 h with gentle agitation at 4°C with Abs in 2.5% non-fat milk, washed three times with Tris-buffered saline-Tween 20, and incubated with the appropriate HRP-conjugated secondary antibody diluted in secondary antibody dilution buffer (EnoGene) for 1 h at room temperature.
Immunofluorescence microscopy analysis
For immunofluorescence staining, cells were seeded onto 12-mm diameter round glass coverslips in 24-well culture dishes. The cells were fixed with 4% paraformaldehyde for 20 min and then washed and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 20 min. The cells were stained with a primary anti-p65 antibody at 4°C overnight (1:1000 in 1% bovine serum albumin in PBS). Finally, the cells were incubated with the secondary antibody (fluorescein isothiocyanate-conjugated anti-rabbit) for 2 h at room temperature and the nuclei stained with diaminophenylindole (DAPI, 1:100 in PBS) for 15 min. Images were captured using an Olympus FV10i microscope (Olympus).
Knock down of TLR3, TLR4, P65, and HIF-1α with siRNA
The siRNAs used to knock down TLR3, TLR4, and P65 expression in OSCC cells were obtained from GenePharma. We designed three sequences for each target. Cells transfected with different concentrations of each siRNA were then cultured for 24 h. The relative mRNA expression of target gene was measured by qRT-PCR. The siRNAs that achieved the most effective knock-down of the targets were selected for use in experiments. The sequences of each siRNA used are listed in Table 2. The siRNAs and non-silencing controls were transfected into cells using Lipofectamine 2000 (Invitrogen). Transfected cells in fresh media were then incubated under the appropriate experimental conditions and harvested for sample preparation.
Table 2: Sequences of siRNA
GENE |
Target site |
Sense (5′–3′) |
Anti-sense (5′–3′) |
|---|---|---|---|
TLR4 |
602 |
CCACCUCUCUACCUUAAUATT |
UAUUAAGGUAGAGAGGUGGTT |
1332 |
CCCACAUUGAAACUCAAAUTT |
AUUUGAGUUUCAAUGUGGGTT |
|
1553 |
GGGCUUAGAACAACUAGAATT |
UUCUAGUUGUUCUAAGCCCTT |
|
TLR3 |
1757 |
GUCCCAUUUAUUUCCUAAATT |
UUUAGGAAAUAAAUGGGACTT |
2025 |
GCGCUUUAAUCCCUUUGAUTT |
AUCAAAGGGAUUAAAGCGCTT |
|
2658 |
GGAGAUUCCAGAUUAUAAATT |
UUUAUAAUCUGGAAUCUCCTT |
|
P65 |
755 |
GGGAUGAGAUCUUCCUACUTT |
AGUAGGAAGAUCUCAUCCCTT |
1455 |
GCUGCAGUUUGAUGAUGAATT |
UUCAUCAUCAAACUGCAGCTT |
|
1707 |
CCUCCUUUCAGGAGAUGAATT |
UUCAUCUCCUGAAAGGAGGTT |
|
Negative control |
UUCUCCGAACGUGUCACGUdTdT |
ACGUGACACGUUCGGAGAAdTdT |
|
Plasmid construction and luciferase assays
The firefly luciferase reporter plasmid was purchased from Promega (Madison, WI, USA). We designed forward and reverse oligonucleotides containing three copies of the HRE sequence (5′-GTACGTACT-3′) on each end using the NheI and HindIII restriction sites (New England Biolabs, Beverly, MA, USA). The oligonucleotides were annealed and ligated using the NheI and HindIII sites to create Phre-Luc.
Wild type or mutant TLR3 and TLR4 promoters were inserted into each plasmid. Following ligation, transformation, and screening, all plasmids were verified by sequencing.
Cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Luciferase expression was measured using a One-Glo Luciferase Assay kit (Promega) and the luminescence function of a GloMax Multi microplate reader (Promega). The data are presented as the mean and standard deviation (SD) of three experimental replicates.
Enzyme-linked immunosorbent assays
Cytokine levels were quantified using human VEGF, IL-1βa, IL-6, and IL-8 enzyme-linked immunosorbent assays (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. All standards and samples were tested in triplicate.
ShRNA transfection and viral transduction
Recombinant lentivirus pLvx-Luciferase-puro-HIF1a-1+2+3+4 was generated by transfecting HEK293T cells. After 48 h, media containing the viral particles was harvested and passed through a 0.45-μm filter (Millipore, New Bedford, MA, USA). For transduction, 10 μL of the supernatant containing lentivirus was added to HSC-3 cells, which were maintained in 2 mL complete culture media. In addition, 8 μg/mL of polybrene (Sigma-Aldrich) was also added to aid the transduction. After 24 h, the media were replaced by fresh media supplemented with 10% FBS containing 0.5 μg/mL puromycin (Sigma-Aldrich). Cells were maintained in puromycin-containing medium for selection of stable transfectants. The shRNA sequences are listed in Table 3.
Table 3: shRNA sequences of HIF-1α
shRNA1 |
GGGATTAACTCAGTTTGAACT |
shRNA2 |
GCCGAGGAAGAACTATGAACA |
shRNA3 |
GCATTGTATGTGTGAATTACG |
shRNA4 |
GCTGGAGACACAATCATATCT |
Human OSCC transplantation model in nude mice
All mice were maintained under specific pathogen free conditions. The experimental protocols were approved by the Ethics Committee of Nanjing Stomatological Hospital. Immunoincompetent nude mice were injected subcutaneously in the flank with either 100 μL HSC3 or sh-HIF1α HSC3 (1 × 107/mL; 8 mice/group). After 30 days, tumors were collected and weighed to compare tumor growth.
Immunohistochemistry
Xenograft tumors were fixed with 10% formalin, embedded in paraffin, sectioned (5-μm thickness), dewaxed with xylene, and then hydrated in a graded series of ethanol. DAKO was used with the HIF-1α antibody (1:200, Abcam), TLR3 antibody (1:100, Abcam), and TLR4 antibody (1:300, Abcam). Sections were counterstained with Mayer’s hematoxylin, dehydrated through graded ethanol into xylene, and then mounted.
Chromatin immunoprecipitation assays
HSC3 cells were cultured in either 20% or 1% O2 for 24 h. The cells were then cross-liked in 1% formaldehyde for 10 min, quenched in 0.125 M glycine, and lysed with SDS lysis buffer. Chromatin was sheared by sonication (Diagenode). We used a ChIP kit (One Step, Abcam) and a ChIP grade primary antibody against HIF-1α (Abcam). We used the following primers for the TLR3 promoter: Forward 5′-CAAGCCTGGGTAATCGTGTTTG-3′; Reverse 5′-GCAACCCTCTGAGGTAGACTG-3′. The primers for the TLR4 promoter were the following: Forward 5′-CTCACTTCTGAGTACGTATCC-3′; Reverse 5′-GTATAGATCAAAGCCTCCTTCC-3′.
Statistical analysis
All data are presented as the mean ± SD or standard error (SE) of the mean, and n corresponds to the number of experiments. Statistical differences between groups were evaluated using paired Student’s t-tests or one-way analysis of variance followed by a Student’s t-test with Bonferroni correction as indicated. Statistical analyses were performed using the GraphPad Prism 5.0 software (version 5.01 for Windows, GraphPad Software, Inc., San Diego CA, USA). A p < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This study was financially supported by the National Natural Science Foundation of China (81302351), Jiangsu Provincial Natural Science Foundation (BK 20131080), and the Distinguished Young Investigator Project of Nanjing (JQX14010). In addition, we are grateful to Professor Zhou Ge from MD Anderson cancer center, Houston, TX for his help in reviewing the whole article to avoid any improper descriptions.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009; 45:309–316.
2. Warnakulasuriya S. Living with oral cancer: epidemiology with particular reference to prevalence and life-style changes that influence survival. Oral Oncol. 2010; 46:407–410.
3. Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008; 371:1695–1709.
4. Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer cell. 2012; 21:488–503.
5. Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010; 29:285–293.
6. Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology. 2014; 143:512–519.
7. Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002; 2:38–47.
8. Zheng Y, Ni Y, Huang X, Wang Z, Han W. Overexpression of HIF-1alpha indicates a poor prognosis in tongue carcinoma and may be associated with tumour metastasis. Oncol Lett. 2013; 5:1285–1289.
9. Semenza GL. Targeting HIF-1 for cancer therapy. Nature reviews Cancer. 2003; 3:721–732.
10. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010; 29:625–634.
11. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883–899.
12. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436–444.
13. Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008; 18:334–342.
14. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013; 12:86.
15. Disis ML. Immune regulation of cancer. J Clin Oncol. 2010; 28:4531–4538.
16. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011; 12:715–723.
17. Zhu Z, Zhong S, Shen Z. Targeting the inflammatory pathways to enhance chemotherapy of cancer. Cancer Biol Ther. 2011; 12:95–105.
18. Gorlach A, Bonello S. The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. Biochem J. 2008; 412:e17–19.
19. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010; 32:305–315.
20. Gonzalez-Reyes S, Fernandez JM, Gonzalez LO, Aguirre A, Suarez A, Gonzalez JM, Escaff S, Vizoso FJ. Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol Immunother. 2011; 60:217–226.
21. Gonzalez-Reyes S, Marin L, Gonzalez L, Gonzalez LO, del Casar JM, Lamelas ML, Gonzalez-Quintana JM, Vizoso FJ. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer. 2010; 10:665.
22. Eiro N, Gonzalez L, Gonzalez LO, Fernandez-Garcia B, Andicoechea A, Barbon E, Garcia-Muniz JL, Vizoso FJ. Toll-like receptor-4 expression by stromal fibroblasts is associated with poor prognosis in colorectal cancer. J Immunother. 2013; 36:342–349.
23. Sheyhidin I, Nabi G, Hasim A, Zhang RP, Ainiwaer J, Ma H, Wang H. Overexpression of TLR3, TLR4, TLR7 and TLR9 in esophageal squamous cell carcinoma. World J Gastroenterol. 2011; 17:3745–3751.
24. Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski J, Whiteside TL. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009; 69:3105–3113.
25. He Z, Huang X, Ni Y, Shi P, Wang Z, Han W, Hu Q. Functional toll-like receptor 3 expressed by oral squamous cell carcinoma induced cell apoptosis and decreased migration. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014; 118:92–100.
26. Huang B, Zhao J, Shen S, Li H, He KL, Shen GX, Mayer L, Unkeless J, Li D, Yuan Y, Zhang GM, Xiong H, Feng ZH. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007; 67:4346–4352.
27. Qiao Q, Nozaki Y, Sakoe K, Komatsu N, Kirito K. NF-kappaB mediates aberrant activation of HIF-1 in malignant lymphoma. Exp Hematol. 2010; 38:1199–1208.
28. Yoshida T, Hashimura M, Mastumoto T, Tazo Y, Inoue H, Kuwata T, Saegusa M. Transcriptional upregulation of HIF-1alpha by NF-kappaB/p65 and its associations with beta-catenin/p300 complexes in endometrial carcinoma cells. Lab Invest. 2013; 93:1184–1193.
29. Hedayat M, Netea MG, Rezaei N. Targeting of Toll-like receptors: a decade of progress in combating infectious diseases. Lancet Infect Dis. 2011; 11:702–712.
30. Basith S, Manavalan B, Yoo TH, Kim SG, Choi S. Roles of toll-like receptors in cancer: a double-edged sword for defense and offense. Arch Pharm Res. 2012; 35:1297–1316.
31. Li D, Gu R, Yang X, Hu C, Li Y, Gao M, Yu Y. TLR3 correlated with cervical lymph node metastasis in patients with papillary thyroid cancer. Int J Clin Exp Med. 2014; 7:5111–5117.
32. Li TT, Ogino S, Qian ZR. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World J Gastroenterol. 2014; 20:17699–17708.
33. Nair S, Kotrashetti VS, Nayak R, Bhat K, Somannavar P, Hosmani J. HSP70 induces TLR4 signaling in oral squamous cell carcinoma: an immunohistochemical study. J Cancer Res Ther. 2013; 9:624–629.
34. Sun Z, Luo Q, Ye D, Chen W, Chen F. Role of toll-like receptor 4 on the immune escape of human oral squamous cell carcinoma and resistance of cisplatin-induced apoptosis. Mol Cancer. 2012; 11:33.
35. Ng LK, Rich AM, Hussaini HM, Thomson WM, Fisher AL, Horne LS, Seymour GJ. Toll-like receptor 2 is present in the microenvironment of oral squamous cell carcinoma. Br J Cancer. 2011; 104:460–463.
36. Taylor CT, Cummins EP. The role of NF-kappaB in hypoxia-induced gene expression. Ann N Y Acad Sci. 2009; 1177:178–184.
37. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012; 33:207–214.
38. Zhu G, Tang Y, Geng N, Zheng M, Jiang J, Li L, Li K, Lei Z, Chen W, Fan Y, Ma X, Li L, Wang X, et al. HIF-alpha/MIF, NF-kappaB/IL-6 axes contribute to the recruitment of CD11b+Gr-1+ myeloid cells in hypoxic microenvironment of HNSCC. Neoplasia. 2014; 16:168–179.
39. Fan P, Zhang JJ, Wang B, Wu HQ, Zhou SX, Wang CY, Zhang JH, Tian Y, Wu HS. Hypoxia-inducible factor-1 up-regulates the expression of Toll-like receptor 4 in pancreatic cancer cells under hypoxic conditions. Pancreatology. 2012; 12:170–178.
40. Semenza GL. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim Biophys Acta. 2016; 1863:382–391.