INTRODUCTION
Recombinant immunotoxins (RITs) are antibody-toxin fusion proteins developed for cancer therapy. SS1P is a RIT composed of a Fv that targets mesothelin and a 38-kDa fragment of Pseudomonas exotoxin A (PE38).SS1P was developed to treat a variety of mesothelin expressing tumors; these include mesothelioma, ovarian, pancreatic, lung, stomach and cervical cancer [1-4].In a phase 1 clinical trial in which SS1P was given QODx3 every 21 days, neutralizing antibodies formed after the first cycle in 90% of patients and no major clinical responses were observed [5]. However, when SS1P was used in combination with an immunosuppressive regimen of cytoxan and pentostatin to kill B- and T-cells, additional treatment cycles could be given and major tumor responses were observed in several patients with advanced refractory mesothelioma [6]. This indicates that producing less immunogenic RITs should allow more treatment cycles and more clinical responses.
Formation of anti-drug antibodies is a major problem in the development of protein therapeutics [7] and specifically foreign proteins like a bacterial toxin [8]. The antibodies involved in the immunogenicity response against SS1P mostly react with PE38, the toxin portion of the RIT [8].The formation of high affinity IgG is primarily dependent on activation of three cellular entities: Antigen presenting cells that process the antigen and present it to T-cells, T-helper cells that secrete cytokines that are required for class switching and affinity maturation of B-cells, which then differentiate and secrete antibodies. Activation of both B-and T-cells is dependent on specific antigenic determinants. B-cells produce antibodies that can bind directly to the surface of the protein, whereas helper T-cells recognize peptides that are derived from the protein and are presented by HLA class II molecules.
Mouse models have shown that elimination of murine B-cell epitopes can significantly reduce the formation of anti-drug antibodies (ADA) against therapeutic foreign proteins [9] and specifically against PE38 [10]. To identify the human B-cell epitopes in PE38, Liu et al. screened a phage display library that contained the Fv portions of antibodies isolated from B-cells of patients who had made anti-SS1P antibodies after treatment with SS1P. These Fvs were used to identify the human B-cell epitopes in domain III and mutations identified that suppressed these epitopes [11]. Finally this information was used to make a new mutant RIT (SS1-LO10-R),which has a deletion of domain II and six mutations in domain III (Figure 1). This immunotoxin has high cytotoxic activity and greatly reduced antigenicity, but it has a short serum half-life, because of its small size. To increase half-life and further decrease immunogenicity, the mouse Fv was replaced with a larger humanized anti-mesothelin Fab, resulting in an immunotoxin (RG7787) with a molecular weight of 72-kDa (Figure 1). RG7787 has recently entered clinical trials.
Elimination of T-cell epitopes is also a well-accepted strategy to de-immunize protein therapeutics. Yeung et al. showed that elimination of a T-cell epitopes in the protein IFNβ resulted in elimination of ADA response in BALB/c mice [12]. Similarly, we recently demonstrated that elimination of two murine T-cell epitopes in SS1P resulted in elimination of anti-SS1P antibodies in mice [13].
We previously reported the location of the eight human T-cell epitopes in the PE38 portion of immunotoxins [14] and used this information to construct LMB-T20, a RIT that targets mesothelin and has 80% of its T-cell epitopes diminished by introducing six point mutations in domain III and deleting a large portion of domain II [15].The goal of this study was to make an immunotoxin reacting with mesothelin expressing cancer cells that has high cytotoxic and anti-tumor activity, and is optimized for minimal reactivity with the adaptive immune system by suppressing both B- and T-cell epitopes.
RESULTS
Design of de-immunized RITs targeting mesothelin
To construct the new de-immunized RIT(LMB-T14) we used the dsFv and toxin present in SS1P, deleted most of domain II and made mutations in domain III as shown Figure 1. SS1P (Figure 1A) is composed of an anti-mesothelin dsFv fused to a 38-kDa fragment of Pseudomonas exotoxin A (PE38). PE38 is made up of two domains; domain II (amino acids 253-364) contains a furin cleavage site necessary for toxin processing, and domain III (amino acids 395-613) contains the ADP ribosylation activity. Previous work showed that modifying SS1P by deletion of the majority of domain II and retaining the 11 amino acid furin cleavage site followed by a GGS spacer results in a RIT (SS1-LR-GGS) with high cytotoxic activity on many cell lines and decreased nonspecific toxicity in mice [16].
SS1-LO10R is derived from SS1-LR-GGS; it has six mutations in amino acids that suppress human B-cell epitopes (Figure 1B). Table 1 shows that SS1-LO10R has good cytotoxic activity with an IC50 of 1.7 pM, which is similar to that of the parent RIT that has no point mutations (SS1-LR-GGS) when evaluated on A431/H9 cells. LMB-T20 (Figure 1C) contains the same deletion in domain II as SS1-LO10R and six mutations in domain III that suppress T-cell epitopes [15]. LMB-T20 also has very good cytotoxic activity on A431/H9 cells (Table 1) with an IC50 of 2.2 pM.
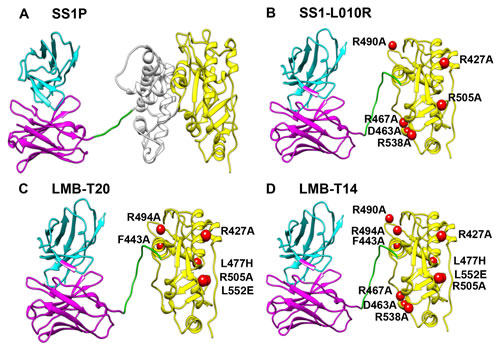
Figure 1: Structural models of RITs.A. SS1P consists of the disulfide-stabilized heavy chain Fv (VH) (magenta) and light chain Fv (VL) (Cyan) of the antibody SS1P. The VH is linked to a 38-kDa fragment of PE38 that is divided into domain II (gray), domain III (yellow), and part of domain Ib from native PE38. B. SS1-LO10R. 24-kDa fragment of PE24 with six point mutations in domain III designed to eliminate binding to B-cell receptor. Point mutations are marked with red balls. C. LMB-T20. PE24 with six point mutations in domain III designed to diminish T-cell epitopes. D. LMB-T14. PE24 with 10 point mutations in domain III designed to diminish B and T cell epitopes. All models are hypothetical arrangements based on the structures of native PE and immunoglobulin G; they do not represent actual structure determinations.
Combination of B- and T-cell mutations
To make a cytotoxic protein with mutations in both B- and T-cell epitopes, we started with SS1-LO10R and introduced amino acid mutations that eliminate T-cell epitopes, usually one at a time as shown in Table 1. We previously observed that introduction of point mutation R494A in CD22 targeting RIT induces a 2-4-fold decrease in relative activity [14]. Here, similarly to the anti CD22 RIT, the mutation R494A (V3) resulted in a 4 fold decrease in activity (Table 1).
The activity of the intermediate construct was improved by the addition of F443A, which by itself induces a positive effect on the activity (V4), and when combined, it moderated the decrease in activity to 2.5-fold.LMB-T14 (Figure 1D) is the most de-immunized RIT. It includes a deletion of domain II and 10 point mutations in amino acids in domain III. Despite all the changes, it maintained very high cytotoxic activity, although a little less than LMB-T20 and LO10R.
Table 1: In vitro cytotoxic activity of various de-immunized RIT constructs in A431/H9 cells
RIT name |
Total mutations |
R427A |
D463A |
R467A |
R490A |
R505A |
F443A |
L477H |
R494A |
R538A |
L552E |
IC50 (pM)* |
Relative activity to LMB-T20 (%) |
Relative activity to LO10R (%) |
B+T |
B |
B |
B |
B+T |
T |
T |
T |
B |
T |
|
|
|
||
LR-GGS |
0 |
|
|
|
|
|
|
|
|
|
|
1.4 |
157 |
121 |
V1 (LO10R) |
6 |
+ |
+ |
+ |
+ |
+ |
|
|
|
+ |
|
1.7 |
129 |
100 |
V2 (LMB-T20) |
6 |
+ |
|
|
|
+ |
+ |
+ |
+ |
|
+ |
2.2 |
100 |
77 |
V3 |
7 |
+ |
+ |
+ |
+ |
+ |
|
|
+ |
+ |
|
7.2 |
31 |
24 |
V4 |
7 |
+ |
+ |
+ |
+ |
+ |
+ |
|
|
+ |
|
1.5 |
146 |
113 |
V5 |
8 |
+ |
+ |
+ |
+ |
+ |
+ |
|
+ |
+ |
|
4.3 |
51 |
40 |
V6 |
9 |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
5.2 |
42 |
33 |
V7 (LMB-T14) |
10 |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
4.2 |
52 |
40 |
V8 (LMB-36) |
8 |
+ |
|
|
+ |
+ |
+ |
+ |
+ |
+ |
+ |
4.5 |
49 |
38 |
* IC50 was evaluated in A431/H9 cells as described in the Experimental Procedures
Cytotoxic activity on cells from mesothelioma patients
Because SS1P has shown anti-tumor activity in patients with mesothelioma [6, 17], we established cell lines from mesothelioma patients and used them to examine the activity of the de-immunized variants. These cells resemble cells growing in patients more closely than established cell lines [18].We found that LMB-T14 and its parent molecules (LMB-T20 and LO10R) were all more cytotoxic than SS1P with IC50s that were less than 100 pM on NCI-Meso16, NCI-Meso19, NCI-Meso21, and NCI-Meso29, (Figure 2A-2D). Figure 2E, which contains averaged data from four assays, shows that LMB-T14 had similar activity to LMB-T20 and LO10R and significantly better cytotoxic activity than SS1P (Figure 2E) (p < 0.05 in one way ANOVA in Dunn’s multiple comparison test).
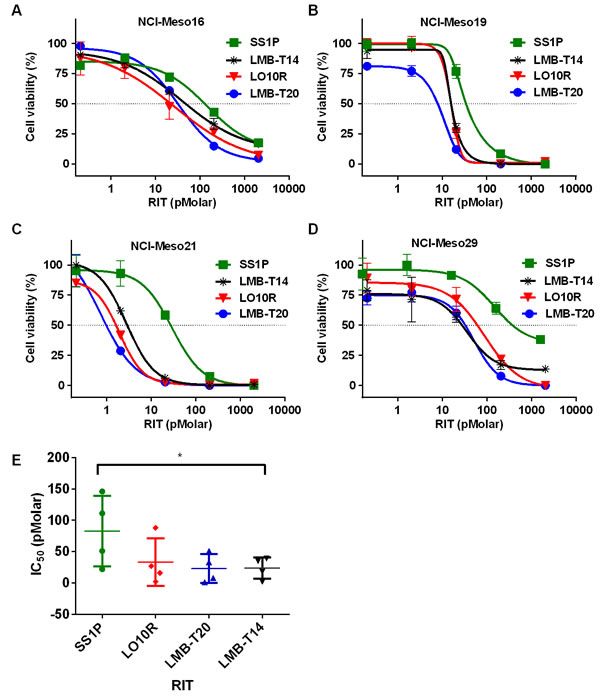
Figure 2: Activity of mesothelin targeting RITs on mesothelioma patients cells. Cells cultured from the pleural fluid or ascites of four mesothelioma patients. NCI-Meso16 A., NCI-Meso19 B., NCI-Meso21 C., and NCI-Meso29 D. were treated with increasing concentrations of RIT. After 72 hr, cells were evaluated for viability using a WST-8 assay and IC50 were calculated. E. Mean of the IC50 value for the four samples. Cells were treated in three replicas; line represents mean; error bar, SEM. Asterisk indicates significant differences of p < 0.05 (*).
Cytotoxic activity on a variety of cells lines
We also compared the activity of LMB-T14 with the three other RITs on several mesothelin expressing cancer cell lines (Figure 3).We found that all cell lines had good responses to LMB-T14; however, a loss in activity between LMB-T14 and its parent molecules (LO10R and LMB-T20) was observed in all cell lines. In stomach cell lines, MKN45 and MKN74, LMB-T14 had a small change in activity compared to LMB-T20 with 1.5-1.8-fold loss in activity. In HAY (mesothelioma cell line), L55 (lung cancer cell line) and KLM1 (pancreatic cell line) LMB-T14 had 3-5-fold lower activity than its parent molecules (LO10R and LMB-T20). Nevertheless, it had better activity than SS1P. Statistically significant differences are shown in Figure 3. In all the cell lines that naturally express mesothelin, SS1P was the least active (p < 0.01 in one way ANOVA with Dunn’s multiple comparison). A431/H9 cells were the only cells that displayed a different pattern in which SS1P had a significantly better or similar activity to all of the variants. This is a transfected cell line that does not normally express mesothelin.
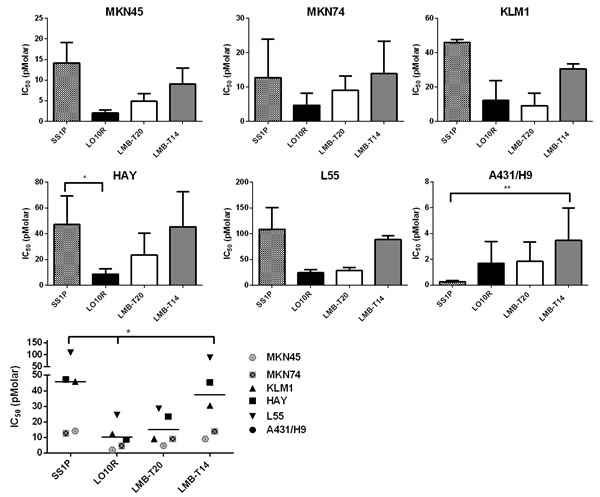
Figure 3: Cytotoxic activity in six mesothelin expressing cell lines. The cytotoxicity of LMB-T14 was compared with SS1P, LMB-T20 and LO10R in a panel of six cells lines: HAY, L55, KLM1, MKN45, MKN74 and A431/H9. For each cell line, the mean IC50 of two or more assays is shown. Summary of all IC50s for all cell lines is shown in the bottom left. Error bars, SEM; p < 0.05 in the Freidman test with Dunn’s multiple comparisons.
Functional stability
To evaluate the stability of LMB-T14, we incubated LMB-T14 and the other RITs at 37oC for 1, 2, 6, and 24 or 72 hr in PBS at 0.5 mg/ml and evaluated their cytotoxic activity on A431/H9 cells. Cells were treated with various concentrations of each immunotoxin and an IC50was calculated for each time point. We found that LMB-T14 was very stable with no loss in activity after 24 hr (Figure 4A). LO10R also had excellent stability, whereas LMB-T20 was less stable and lost 2-fold activity in the time interval between 6 and 24 hr. To simulate the stability of LMB-T14 in the circulation of humans, we diluted it and the other variants in 100% human AB serum to 15µg/ml, incubated at 37oC for various times and measured cytotoxic activity. We found that LMB-T14 and LO10R were stable for 72 hr under these conditions and that LMB-T20 was less stable, losing 2-fold activity after 24hr and more after 72 hr (Figure 4B).
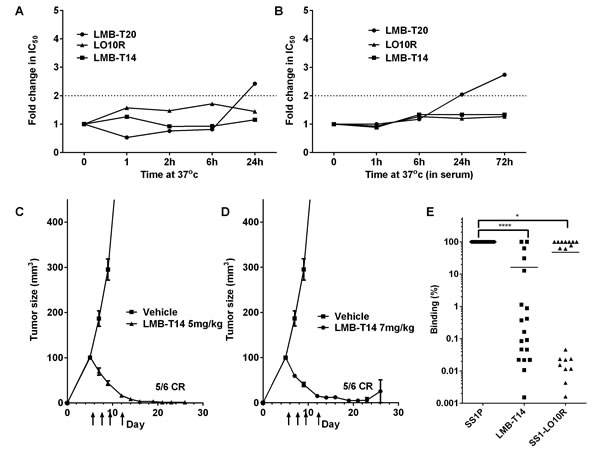
Figure 4: Stability, anti-tumor activity and antigenicity of LMB-T14. A., B. Stability of RITs. RITs were warmed to 37oC for indicated durations and used to treat A431/H9 cells at serial concentrations. Cell viability was assayed, a curve fit was created for each RIT using a 4 parameter curve fit and IC50 was calculated. A. Fold change in IC50 after 0, 1, 2, 6 and 24 hr. B. Fold change in IC50 after incubation at 37oC in 100% human serum for 0, 1, 6, 24, and 72 hr. Cytotoxic activity was evaluated in six replicas for each data point with a standard deviation < 5% for all replicas. C., D.Anti-tumor activity of LMB-T14 in mouse xenograft. A-thymic nude mice were innoculated 106 A431/H9 cells at time 0. Intravenous treatment with LMB-T14 with a dose of 5 mg/kg C. or 7 mg/kg D.or vehicle began on day 5 and continued every other day for a total of four doses. On day 30 the experiment was terminated when 5/6 mice in both dose groups were tumor free. Arrows indicate the days that treatment was administered. E. Human antigenicity of SS1P and variant RITs. The reactivity of SS1P, LMB-T14 and LO10R with preexisting antibodies in human sera from 19 patients with neutralizing antibodies were compared using a binding assay to determine the concentration at which time the RITs reduced the signal of an ELISA to detect serum antibodies by 50% (IC50). The IC50 values of the RITs relative to SS1P are plotted. Line represents mean. Asterisks indicate significant differences of p < 0.0001 (****), p < 0.05 (*) in Friedman’s test and Dunn’s multiple comparisons.
Mouse toxicity
To evaluate the non-specific toxicity of LMB-T14, we treated small groups of Swiss mice with single doses of LMB-T14 and LMB-T20 (Table S1). We found that LMB-T14 was better tolerated than LMB-T20. There was no significant weight loss at a dose of 20 mg/kg, where as a similar dose of LMB-T20 was toxic in 4/4 mice.LMB-T14 was also well tolerated at a dose of 22 mg/kg, which is the highest tolerated dose reported for active immunotoxins in our lab. We found that 28 mg/kg was toxic for 4/4 mice. Interestingly, four QOD doses of 7 mg/kg (which adds up to 28 mg/kg) were well tolerated, with no weight loss. This indicates that 28 mg/kg is not toxic when administered over a period of time and suggests that LMB-T14 would be more efficacious if given in multiple small doses than in a single large dose.
Efficacy of LMB-T14 in a mouse xenograft model
To evaluate the anti-tumor activity of LMB-T14, we implanted A431/H9 tumors into the flank of athymic nude mice. Mice were treated with LMB-T14 on days 5, 7, 9 and 12 after tumor implantation with doses of 5mg/kg or 7 mg/kg (Figure 4C and 4D). While the tumors treated with vehicle grew rapidly, reaching an average of 800 mm3 within 14 days, the treated groups had a significant decrease in tumor size as early as two days after the first dose. The tumors continued to decrease in size and by day 16, 5/6 tumors in both groups were undetectable. The complete tumor regressions persisted until day 30 when the experiment was terminated. In addition, this dose was well tolerated with no weight loss in the treated animals (Table S1).
Antigenicity
Because LMB-T14 has mutations that are designed to diminish binding to B-cell receptors and to antibodies in human serum, we compared the reactivity of LMB-T14 and SS1-LO10R with SS1P using serum from 19 patients, who had developed neutralizing antibodies after treatment with SS1P. Figure 4D shows that LMB-T14 had significantly reduced binding to human anti-sera, and the magnitude of the decrease ranged from very little to more than a 3-log decrease with a mean of 16% (p < 0.001 in Friedman’s test and Dunn’s multiple comparisons).LO10R also had significantly reduced binding compared to SS1P (p < 0.05 in Friedman’s test and Dunn’s multiple comparisons) (Figure 4D). These findings indicate that the B-cell epitopes in SS1P that bind to patients sera are significantly diminished in these proteins. No significant difference between LO10R and LMB-T14 was observed, which indicates that the addition of four T-cell mutations did not significantly affect the structure of the molecule.
T-cell activation
To investigate the magnitude of the decrease in T-cell immunogenicity and to determine whether the four additional mutations (designed to remove B-cell epitopes) induced formation of new T-cell epitopes, we stimulated peripheral blood mononuclear cells (PBMC) from 10 normal donors with SS1P or the variants LMB-T20 and LMB-T14. After 14 days of in vitro expansion, the cells that were stimulated with SS1P were re-stimulated with 111 peptides spanning the sequence of PE38 and the cells that were stimulated with LMB-T14 or LMB-T20 were re-stimulated with 76 peptides spanning the sequence of LMB-T14 or LMB-T20, respectively. T-cell activation was detected using IL-2 ELISpot. As expected, both de-immunized RITs (LMB-T20 and LMB-T14) had a significant reduction in the number of IL-2 specific spots. The decrease was 61% with LMB-T14 and 81% with LMB-T20 (p < 0.01 in Wilcoxon matched-pairs signed rank test) (Figure 5).
Unexpectedly, 4/10 donors had a new and significant response to peptides 63-65 in LMB-T14 (Figure 5). Peptides 63-65 from LMB-T14 differ from WT and LMB-T20 by two separate mutations, which are D463A and R467A (labeled with blue stars). These mutations were introduced to eliminate B-cell epitopes. We further characterized this new epitope in 19 PBMC donors (Table S2) and searched for a correlation for specific HLA alleles. We found that 7/19 donors had a response to this epitope. The donors that responded to this epitope share three HLA DRB1 allele families: 15, 08, and 07.
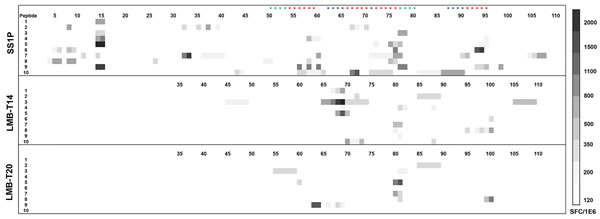
Figure 5: Stimulation of PBMC from 10 donors with LMB-T20, LMB-T14 and SS1P. PBMC from 10 naïve donors were stimulated with either SS1P LMB-T14 or LMB-T20. After 14 days of in vitro expansion cells were re-stimulated with either 111 peptides spanning the sequence of PE38, 76 peptides spanning the sequence of LMB-T14 or LMB-T20, respectively. T-cell activation was detected using IL-2 ELISpot. Response strength is shown in the Spot Forming Cells ladder. Red stars represent mutations of T-cell epitopes, blue stars for B-cell mutations and green stars for both B and T-cell mutations.
DISCUSSION
We describe here the properties of a new RIT, LMB-T14, that has greatly reduced immunogenicity because it contains mutations that suppress both T and B-cell epitopes. The new protein, which contains a large deletion of domain II and 10 amino acid mutations in domain III, is very cytotoxic to mesothelioma cells as well as other cancer cell lines, is well tolerated by mice and produces complete remissions of mesothelin expressing cancers in mice.
We have previously described immunotoxins with mutations in either B- or T-cell epitopes designed to decrease immunogenicity [11, 14]. In principle, elimination of either B- or T-cell epitopes should prevent immunogenicity. Unfortunately, total elimination of all B- or all T-cell epitopes is difficult to accomplish due to the complexity of the humoral immune system. One major obstacle is the polymorphism of HLA class II which makes it difficult to find single point mutations that will completely prevent the binding of the peptide-epitopes to various binding cores on the HLA. In addition, some of the mutations were able to decrease but not completely eliminate T-cell responses (Figure 5). Furthermore, the six mutations that eliminated B-cell epitopes do not decrease the binding of all tested human anti-sera [11]. In an attempt to improve on the properties of immunotoxins with only one arm of the immune system impaired, we have explored the possibility of incorporating mutations that decrease both B- and T-cell epitopes into one molecule. To our knowledge, this is the first time a therapeutic protein has been designed with silencing of both the B- and the T-cell epitopes.
Clinical trials with immune suppressive regimens also indicate a need to address both the B- and T-cells arms of the adoptive immune system. Hassan et al. treated patients with Rituximab to induce depletion of all circulating B-cells and followed with RIT treatment [19]. They found that B-cell depletion with Rituximab was not sufficient to prevent ADA formation against PE38. On the other hand, a combination of pentostatin and cyclophosphamide [6] that depleted both B- and T-cells prior to RIT treatment significantly delayed the ADA response, and allowed additional treatment cycles to be given. This finding indicates that both B- and T-cells are involved in the ADA response against RIT and that de-immunization against both should be beneficial.
In the process of combining six mutations designed to eliminate B-cell epitopes and the six mutations designed to diminish T-cell epitopes, we found that two point mutations (R427A and R505A) diminished both B- and T-cell mediated immunity. R505A and R427A have very large ASAs (150Å and 142Å) indicating that the arginines are located on the surface of the protein. Since B-cell epitopes are known to contain bulky hydrophilic amino acid like arginine [10, 20, 21], it is not surprising that R505A and R427A, which we found to suppress T-cell epitopes, also diminished B-cell epitopes. Others have previously reported that important immunogenic epitopes can be recognized by both B- and T-cells [22-24].
Our finding that the mutations (D463A and R467A) created a new T-cell epitope was unexpected, because alanine substitutions are frequently found to reduce the binding of a peptide to an HLA molecule due to loss of non-polar side chains [25, 26] and not to induce binding. The amino acid sequence that forms the new epitope is ARSQDLAAIWAGFYIAGD (peptides 64-65). A blast search of the mutant sequence (ARSQDLDAIWRGFYIAGD) revealed that the mutant did not resemble peptides found in other proteins except for the wild-type (WT) sequence. Similarly, a search in the immune epitope database (search for known epitopes with similar structure) [27] did not reveal similarity to other known epitopes. To eliminate this new epitope we constructed V8, which reverts residues 463 and 467 back to WT. LMB-36 has similar cytotoxic activity to LMB-T14 (Table 1). However reverting D463A and R467A back to WT also restores the B-cell epitope that those mutations eliminated. At this point it is not possible to determine which epitope (B or T) is more important for de-immunization. We also plan to determine if only one of the mutated amino acids is required for creating the new T-cell epitope.
When incorporating multiple point mutations into a molecule, there is the risk in decreasing its activity. There is a delicate balance between the decrease in activity the molecule will endure and the benefit of the de-immunization. This tradeoff is demonstrated in Table 1 that shows that addition of some of the de-immunizing mutations reduced the cytotoxic activity on several cancer cell lines, but not on cells from patients with mesothelioma (Figure 2). It is possible that LMB-T14 may be more efficacious in patients, due to its good stability, low nonspecific toxicity in animals so higher doses can be given and low immunogenicity will allow it to be given for more cycles.
MATERIALsAND METHODS
Human donor and patient samples
PBMC were isolated from apheresis samples from patients who were previously treated with a PE38-containing RIT and from naïve donors were collected under research protocols approved by the NIH Review Board (08-C-0026) and (99-CC-0168), respectively with informed consent. PBMC were isolated using gradient density separation by Ficoll-Hypaque (GE Healthcare, Piscataway, NJ) according to manufacturer’s instructions. PBMC were frozen in 10% human AB serum (Gemini, Sacramento,CA) RPMI media (Lonza, Walkersville, MD) containing 7.5% DMSO (Cellgro, Manassas, VA) for 12 months in liquid nitrogen. Human sera were obtained under protocols 01-C-0011, 03-C-0243, and 08-C-0026.
Peptide synthesis
Peptides for T-cell assays were synthesized by American Peptides (Sunnyvale, CA). All peptides were purified to 95% homogeneity by HPLC and confirmed by mass spectrometry.
Construction, expression and purification of RIT
All RIT described in this work are composed of a heavy-chain Fv fused to LR-PE24 (VH-PE24) disulfide-linked to the light-chain Fv (VL) of SS1 antibody [28]. The different point mutations described in the constructs were added one by one using PCR overlap extension. The resulting PCR products were cloned back into the parent plasmid, and the mutations were confirmed by DNA sequencing. All RITs were purified by a standard protocol [29].
Antigenicity assay
Binding of RITs to antibodies present in patients sera was assayed as previously described [10]. Briefly, ELISA plates were coated with 100 ng Fc-Mesothelin in 50 µl PBS over night at 4°C. In separate plates, the different RITs were incubated overnight at 4°C with patient’s serum in serial concentrations. After washing of the coated plates, the immune complexes were transferred to the ELISA plates and incubated at room temperature for 1 hr. The human antibodies not bound to the RITs were captured by SS1P and detected. Next HRP-conjugated rabbit anti-human IgG Fc (Jackson Laboratory) was added, followed by TMB substrate (Thermo Scientific). IC50 values were calculated from the binding curves. The IC50 values indicate the concentration of RIT that inhibits 50% of the antibody reactivity with SS1P. The binding ratio was calculated from each IC50 value.
In vitro expansion of PE38-specific cells and ELISpot assay
In vitro expansion using whole RIT and T-cell activation detection using IL-2 ELISpot were performed as previously described [30]. Briefly, PBMC from naïve donors were stimulated with 5µg/ml of SS1P, LMB-T14 or LMB-T20 in separate plates. The cells were supplemented with recombinant human IL-2 every 4 days (Millipore). On day 14, the cells were harvested and washed. They were brought to a concentration of 2x106 cells/ml and 50 µl were plated in pre-coated ELISpot plates (Mabtech). The enriched cells were then restimulated with peptide pools; cells that were expanded with SS1P were restimulated with 22 peptide pools spanning the sequence of WT PE38. Cells that were expanded using LMB-T14 or T20 were restimulated with 15 peptide pools spanning the sequence of the deimmunized RIT. Peptide pools that had a positive responses as defined [31] were fine screened to identify the individual immunogenic peptides by testing individual peptides from the pool.
Cytotoxicity assay
Cytotoxicity of RIT against early passage mesothelioma tumor cells and cell lines
Early passage mesothelioma cells from the ascites or pleural fluid of four patients with mesothelioma seen at the National Cancer Institute on Institutional Review Board-approved protocols (08-C-0026) [18]. Frozen tumor cells were thawed, washed and grown in T75 flasks for 4 days in cell culture media. After reaching confluence (5×103 cells/ well) cells were seeded in a 96 well plate and 24 hr later were treated with various concentrations of the RITs.
Cytotoxic activity in established mesothelin expressing cell lines
Cells were seeded in a 96 well plate at optimal cell concentrations (A431/H9 cells 2.5 ×103/well, KLM1, L55, MKN74, MKN45, and HAY cells at 5 ×103/well) and 24hr later were treated with various concentrations of the RITs. The A431/H9 cell line was transfected in our laboratory and previously described [32]. The KLM1 pancreatic cell line was provided by Dr. U. Rudloff (NCI, Bethesda,MD), the L55 lung adenocarcinoma cell line was provided by Dr. S. Albelda (University of Pennsylvania, PA), MKN74 and MKN45 stomach cell lines were provided by Dr. T. Yamori (Pharmaceuticals and Medical Device Agency, Japan), and the HAY cells was provided by the Stehlin Foundation for Cancer Research (Houston, TX).
Cell viability was determined 72 hr later using WST8 cell counting kit (Dojindo Molecular Technologies Inc,) according to manufacturer’s instructions. Color change was evaluated at O.D. 450nm.
Cell viability was normalized between 0-100 percent. Complete cell death (0%) was obtained by treating the cells with Cyclohexamideor Staurosporine and 100% by no treatment.
Functional stability assays
LMB-T20, LO10R and LMB-T14 were diluted in D-PBS to concentrations of 0.5 mg/ml. RIT variants were distributed in five aliquots and placed in an incubator at 37oC. Vials were taken out of the incubator and placed on dry ice for 15 min and transferred to -80oC at the following time points: 0, 1, 2, 6 and 24 hr. A431/H9 cells were plated in a concentration of 2.5 ×103 cells/well and 24 hr later were treated using serial dilutions of the treated RITs in six replicas. Cell viability was detected 72 hr later as described above.
Serum stability
RIT variants were diluted to a concentration of 15 µg/ml in 100% human AB Serum (Gemini Bio-products). Five aliquots of 60 µl each were made for each protein and placed in 37oC.Vials were taken out of the incubator and placed in -20oC in the following time points: 0, 1, 6, 24 and 72 hr. Functional activity was evaluated as described above.
Mouse xenograft tumor model
All animal experiments were performed in accordance with NIH guidelines and approved by the NCI Animal Care and Use Committee. Female athymic nude mice were injected subcutaneously in the flank with 1.0 × 106 A431/H9 cells in 0.2 mL RPMI with 4 mg/mL Matrigel (BD Biosciences) on day 0. After 7 days, when the tumors reached 100 mm3, mice were injected IV with RITs in the indicated concentrations and the indicated schedules. Body weight and tumor size were observed for 30 days. Mice were euthanized if they experienced rapid weight loss or tumor burden greater than 10% body weight. No animals were excluded from statistical analysis. Tumor-size evaluation was evaluated blindly using a caliper.
Nonspecific toxicity
Nonspecific toxicity was evaluated by IV injections of indicated doses to Swiss mice.
Statistical analysis
Statistical analysis and plots were done using Graph Pad Prism software. For comparisons between two parametric variables we used Student T test. For comparisons between two non- parametric variables we used Wilcoxon matched rank test. For comparisons of multiple parametric variables we used One way ANOVA followed by Holm-Sidak’s multiple comparisons test. For comparisons of multiple non-parametric variables we used Friedman test followed by Dunn’s multiple comparison test. For comparisons between different cell lines and different RIT we used two way ANOVA with multiple comparisons with Dunnet test.
ACKNOWLEDGMENTS
The authors wish to thank Dr. B.K. Sathyanarayana for creating the structural model of RIT for Figure 1, Dr. Prince Awuah and Richard Beers for expression and purification of LMB-T20 and LMB-T14, and Dr. John Weldon for providing SS1-LO10R.
SUPPORT
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Hassan R, Kreitman RJ, Pastan I and Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13:243-247.
2. Tang Z, Qian M and Ho M. The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med Chem. 2013; 13:276-280.
3. Hollevoet K, Mason-Osann E, Liu XF, Imhof-Jung S, Niederfellner G and Pastan I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. 2014;13:2040-2049.
4. Alewine C, Xiang L, Yamori T, Niederfellner G, Bosslet K and Pastan I. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol Cancer Ther. 2014; 13:2653-2661.
5. Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC and Pastan I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007; 13:5144-5149.
6. Hassan R, Miller AC, Sharon E, Thomas A, Reynolds JC, Ling A, Kreitman RJ, Miettinen MM, Steinberg SM, Fowler DH and Pastan I. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. 2013; 5:208ra147.
7. Krishna M and Nadler SG. Immunogenicity to biotherapeutics - the role of anti-drug immune complexes. Front Immunol. 2016; 7:21.
8. Mazor R, Onda M and Pastan I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev. 2016; 270:152-164.
9. Collen D, Bernaerts R, Declerck P, De Cock F, Demarsin E, Jenne S, Laroche Y, Lijnen HR, Silence K and Verstreken M. Recombinant staphylokinase variants with altered immunoreactivity. I: Construction and characterization. Circulation. 1996; 94:197-206.
10. Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ and Pastan I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci U S A. 2011; 108:5742-5747.
11. Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L and Pastan I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci U S A. 2012; 109:11782-11787.
12. Yeung VP, Chang J, Miller J, Barnett C, Stickler M and Harding FA. Elimination of an immunodominant CD4+ T cell epitope in human IFN-beta does not result in an in vivo response directed at the subdominant epitope. J Immunol. 2004; 172:6658-6665.
13. Mazor R, Crown D, Addissie S, Jang Y, Kaplan G and Pastan I. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol Immunol. 2015; doi: 10.1038/cmi.2015.91.
14. Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, Lee EC, Kreitman RJ, Lee B, Baker D, King C, Hassan R, Benhar I and Pastan I. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci U S A. 2014; 111:8571-8576.
15. Mazor R, Zhang J, Xiang L, Addissie S, Awuah P, Beers R, Hassan R and Pastan I. Recombinant immunotoxin with T-cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol Cancer Ther. 2015; 14:2789-2796.
16. Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ and Pastan I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009; 113:3792-3800.
17. Pastan I and Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014; 74:2907-2912.
18. Zhang J, Qiu S, Zhang Y, Merino M, Fetsch P, Avital I, Filie A, Pastan I and Hassan R. Loss of mesothelin expression by mesothelioma cells grown in vitro determines sensitivity to anti-mesothelin immunotoxin SS1P. Anticancer Res. 2012; 32:5151-5158.
19. Hassan R, Williams-Gould J, Watson T, Pai-Scherf L and Pastan I. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB-1. Clin Cancer Res. 2004; 10(1 Pt 1):16-18.
20. Ansari HR and Raghava GP. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res. 2010; 6:6.
21. Bogan AA and Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998; 280:1-9.
22. Kaliyaperumal A, Michaels MA and Datta SK. Naturally processed chromatin peptides reveal a major autoepitope that primes pathogenic T and B cells of lupus. J Immunol. 2002; 168:2530-2537.
23. Ratto-Kim S, de Souza MS, Currier JR, Karasavvas N, Sidney J, Rolland M, Valencia-Micolta A, Madnote S, Sette A, Nitayaphan S, Pitisuttuthum P, Kaewkungwal J, Rerks-Ngarm S, O’Connell R, Michael N, Robb ML, et al. Identification of immunodominant CD4-restricted epitopes co-located with antibody binding sites in individuals vaccinated with ALVAC-HIV and AIDSVAX B/E. PloS One. 2015; 10:e0115582.
24. Barnett BC, Burt DS, Graham CM, Warren AP, Skehel JJ and Thomas DB. I-Ad restricted T cell recognition of influenza hemagglutinin. Synthetic peptides identify multiple epitopes corresponding to antibody-binding regions of the HA1 subunit. J Immunol. 1989; 143:2663-2669.
25. Allen PM, Matsueda GR, Evans RJ, Dunbar JB, Jr., Marshall GR and Unanue ER. Identification of the T-cell and Ia contact residues of a T-cell antigenic epitope. Nature. 1987; 327:713-715.
26. Johansen BH, Vartdal F, Eriksen JA, Thorsby E and Sollid LM. Identification of a putative motif for binding of peptides to HLA-DQ2. Int Immunol. 1996; 8:177-182.
27. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A and Peters B. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015; 43:D405-412.
28. Chowdhury PS, Chang K and Pastan I. Isolation of anti-mesothelin antibodies from a phage display library. MolImmunol. 1997; 34:9-20.
29. Pastan I, Beers R and Bera TK. Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol. 2004; 248:503-518.
30. Oseroff C, Sidney J, Kotturi MF, Kolla R, Alam R, Broide DH, Wasserman SI, Weiskopf D, McKinney DM, Chung JL, Petersen A, Grey H, Peters B and Sette A. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol. 2010; 185:943-955.
31. Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, Tsang KY, Benhar I and Pastan I. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc Natl Acad Sci U S A. 2012; 109:E3597-3603.
32. Ho M, Hassan R, Zhang J, Wang QC, Onda M, Bera T and Pastan I. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 2005; 11:3814-3820.