
INTRODUCTION
Among evolutionarily conserved pathways, mitogen-activated protein kinase (MAPK) pathways link extracellular signals to their intracellular targets and controls fundamental cellular processes such as cell proliferation, cell growth, cell migration, cell differentiation, embryogenesis and cell death [1–3]. The first MAP kinases were identified in the pheromone pathway of the budding yeast (Saccharomyces cerevisiae), Kss1p and Fus3p in between 1989 and 1991 [4]. Till date, 14 MAPKs have been recognized in mammals belonging to seven groups. These seven groups may further be classified into two broad categories, either conventional or atypical MAPKs [5]. Conventional MAPKs includes extracellular signal-regulated kinase (ERK)1/2, ERK5, Jun N-terminal kinase (JNK)1/2/3, p38 isoforms α/β/γ(ERK6)/δ while atypical MAPKs comprises of ERK3/4, ERK7 and Nemo like Kinases (NLK). Since, MAPK pathways are crucial for the every aspect of cell survival and growth, any irregularities may impose cancerous properties to cells, like independence from proliferation signals, infinite replicative potential, capability to invade and metastasize, attract and endure angiogenesis for nutrient supply, evasion of apoptosis, attainment of drug resistance and evasion of oncogene induced senescence. To illustrate the significance of MAPK in cancers, a number of reviews have highlighted their role in various types of cancers [1, 6–8].
Chronic myeloid leukemia (CML) is a rare clonal myeloproliferative malignancy of pluripotent hematopoietic stem cells [9, 10]. It was perhaps the first form of leukemia to be acknowledged and may account for up to 15% of reported cases of leukemia in the developed world [11], though global prevalence is not known. CML is caused due to a reciprocal translocation between chromosomes 9 and 22 t(9;22) (q34;q11) which generate an abnormal fusion gene, BCR-ABL. Gene product of BCR-ABL exerts a constitutive tyrosine kinase activity crucial for the function of several signaling pathways involved in various malignancies, including CML [12]. Accordingly, a modifications in most of the members of the MAPKs have been observed due to BCR/ABL transformation and have been found associated with cell survival or drug resistance [8, 13, 14]. Taking into consideration the importance of MAPK in CML, specific inhibitors for BCR-ABL tyrosine kinase activity have been designed and are being developed for treating CML [15].
microRNAs (miRNAs) consist of a large family of short (~22-nucleotides in length) noncoding RNAs [16, 17] that are not translated into proteins and control target gene expression in metazoan animals, plants, and protozoa, especially through post-transcriptional and translational regulation [18]. miRNAs regulate gene expression by cleaving the target mRNAs directly or inhibiting translation through perfect or nearly perfect complementary base pairing to targeted mRNAs at the 3′ untranslated regions (UTRs) [19–23]. This class of RNA was initially discovered in 1993 by Ambros and colleagues, who described a 22-nucleotides RNA in Caenorhabditis elegans encoded by the lin-4 gene [24]. However, miRNAs that might be regulating various crucial cellular functions and pathways of a given cell are yet be revealed completely. Recent studies on miRNAs associated with human diseases have indicated that these tiny molecules play a crucial role in controlling cellular signal transduction cascades. The expression profile of miRNAs in CML was first studied by Zhu et al., describing that the regulatory mechanism of miRNAs can regulate the expression of several CML targets [25]. From then, several miRNAs have been identified and are known to be associated with the signaling cascade pathways related to CML.
As the role of various miRNAs are being identified in the pathogenesis of CML and MAPK signaling pathways is known to play a crucial role in the development of CML, in this review, we have tried to discuss the regulatory role of miRNAs in MAPK signaling cascade. Particular attention has been paid to explain that how the expression pattern of the small miRNAs might regulate the components of MAPK signaling pathway related to pathogenesis of CML. For this, we have highlighted the miRNA mediated regulation of different proteins involved in MAPK signaling cascade, such as BCR-ABL, CRK, CRKL, KRAS, RAF1, as well as MAPK1. An understanding of the expression pattern and regulative role of miRNAs in controlling the components of MAPK signaling pathway might help to design the approaches required to combat CML.
miRNAs and MAPK signaling pathway in CML
CML involves an extremely complex network of signaling cascade mechanism. Cytogenetically, it is characterized by the presence of the Philadelphia chromosome (Ph), which originates from the reciprocal translocation between chromosome 9 and 22 [26–30]. Approximately 90% of patients with CML have this acquired genetic abnormality [28]. Due to translocation, the BCR gene from chromosome 22 is fused to the ABL gene on chromosome 9 which generate an abnormal BCR-ABL fusion gene. This fusion gene encodes a fusion protein with tyrosine kinase activity and transforming ability which activates downstream signal transduction pathways involved in CML [26]. It is well known that the MAPK pathway is an important downstream signaling cascade in several types of cancer [31] as well as various other cellular mechanisms. The MAPK signaling cascade is a highly conserved component and plays a central role in CML (Figure 1). This pathway is necessary for the transcription of genes involved in cell proliferation and survival [32, 33]. In CML, auto-phosphorylation of tyrosine 177 on BCR-ABL fusion protein provides a docking site for the adapter molecule, growth factor receptor-bound protein 2 (GRB-2) [34]. Subsequently, GRB-2, binds to the SOS protein, which stabilizes a GTPase, RAS, in its dynamic GTP-bound form. Other two adapter molecules, Src homology 2 domain containing (SHC), and CRKL, can also activate RAS. Both acts as substrates for BCR-ABL [35, 36] and bind BCR-ABL through their Src Homology (SH) 2 (SHC) or SH3 (CRKL) domains. Activated RAS prompts the kinase activity of RAF. Activated RAF in turn initiates a signaling cascade through the serine–threonine kinases MEK1/MEK2 and ERK (MAPK), which eventually leads to the activation of gene transcription [37].
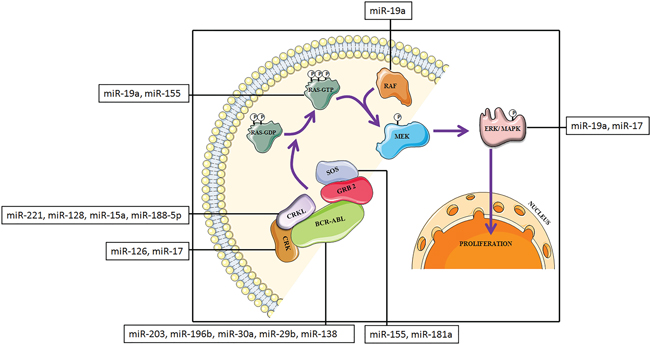
Figure 1: Schematic representation of MAPK protein cascade downstream of BCR-ABL transformed cells with miRNAs that target MAPK signaling pathway components.
In these circumstances, miRNAs may regulate the expression of proteins at different levels by binding with the complementary sequence of 3’ untranslated region of the target mRNAs, thereby controlling the signal transduction processes [38]. Several miRNAs play significant role in the MAPK signaling pathway and among them, the following miR-203, miR-196b, miR-29b, miR-30a, miR-138, miR-155, miR-19a, miR-17, miR-126, miR-128, miR-221, miR-15a, miR-188-5p and miR-181a are well studied (Table 1, 2) [39].
Table 1: Different Human miRNA and Their Chromosomal Location, Gene Location, pre-miRNA Length, Mature Sequence Associated with the MAPK signaling pathway in CML
Name of miRNA |
Chromosomal (Ch) Location |
Gene location (EXON/INTRON/UTR) |
Pre-miRNA length |
Mature Sequence |
|---|---|---|---|---|
miR-203 |
Ch14q32.33 |
intergenic |
110 nt |
65| 5’- GUGAAAUGUUUAGGACCACUAG -3’ |86 |
miR-196b |
Ch7 p15.2 |
3UTR + 3/ intron + 1/ exon + 1/ intron + 1/ exon -1/ exon -2 |
84 nt |
15| 5’- UAGGUAGUUUCCUGUUGUUGG -3’ |35 |
miR-29b-1 |
Ch7 q32.3 |
Intergenic |
81 nt |
51| 5’- UAGCACCAUUUGAAAUCAGUGUU -3’ |73 |
miR-29b-2 |
Ch1 q32.2 |
intergenic |
81 nt |
52| 5’- UAGCACCAUUUGAAAUCAGUGUU -3’ |74 |
miR-30a |
Ch6 q13 |
intron + 3/ intron + 3 |
71 nt |
6| 5’- UGUAAACAUCCUCGACUGGAAG -3’ |27 |
miR-138-1 |
Ch3p21.32 |
intergenic |
99 nt |
23| 5’- AGCUGGUGUUGUGAAUC -3’ |39 |
miR-138-2 |
Ch16q13 |
intergenic |
84 nt |
10| 5’- AGCUGGUGUUGUGAAUC -3’ |26 |
miR-155 |
Ch21 q21.3 |
intergenic |
65 nt |
4| 5’- UUAAUGCUAAUCGUGAUAGGGG -3’ |25 |
miR-19a |
Ch13 q31.3 |
3UTR + 2/ intron + 3/ intron + 2/ intron + 3 |
82 nt |
49| 5’- UGUGCAAAUCUAUGCAAAACUGA -3’ |71 |
miR-17 |
Ch13 q31.3 |
3UTR + 2/ intron + 3/ intron + 2/ intron + 3 |
84 nt |
14| 5’- CAAAGUGCUUACAGUGCAGGUAGU -3’ |37 |
miR-221 |
ChX p11.3 |
intergenic |
110 nt |
65| 5’- AGCUACAUUGUCUGCUGGGUUUC -3’ |87 |
miR-181a-1 |
Ch1 q32.1 |
intergenic |
110 nt |
24| 5’- AACAUUCAACGCUGUCGGUGAGU -3’ |46 |
miR-181a-2 |
Ch9 q33.3 |
intron -2/ intron -2/ intron -2/ intron + 1 |
110 nt |
39| 5’- AACAUUCAACGCUGUCGGUGAGU -3’ |61 |
miR-126 |
Ch9q34.3 |
Intron +7/ Intron +7/ Intron +6/ Intron +6/ Intron +5/ Intron +7/ Intron +7 |
85 nt |
15| 5’- CAUUAUUACUUUUGGUACGCG -3’ |35 |
miR-128a |
Ch2q21.3 |
Intron +15/ Intron +18 |
82 nt |
50| 5’- UCACAGUGAACCGGUCUCUUUU -3’ |71 |
miR-128b |
Ch3p22.3 |
Intron +17/ Intron +18/ Intron +18/ Intron +11/ Intron +17 |
84 nt |
52| 5’- UCACAGUGAACCGGUCUCUUUC -3’ |73 |
miR-15a |
Ch13q14.2 |
intron + 4/ intron + 4/ intron + 5/ intron + 5/ intron + 4/ intron + 3/ intron + 4/ intron + 3 |
83 nt |
14| 5’- UAGCAGCACAUAAUGGUUUGUG -3’ |35 |
miR-188 |
ChXp11.23 |
intron + 3/ intron + 3/ intron + 3/ intron + 3/ intron + 3 |
86 nt |
15| 5’- CAUCCCUUGCAUGGUGGAGGGU -3’ |36 |
Table 2: Various human miRNA and their synonym, miRNA Map accession no. and HGNC ID associated with MAPK signaling pathway in CML
Name of miRNA |
miRNAs synonym |
miRNA Map accession no. |
HGNC |
|---|---|---|---|
miR-203 |
hsa-miR-203 |
MI0000283 |
HGNC:31581 |
miR-196b |
hsa-miR-196b |
MI0001150 |
HGNC:31790 |
miR-29b-1 |
hsa-miR-29b-1 |
MI0000105 |
HGNC:31619 |
miR-29b-2 |
hsa-miR-29b-2 |
MI0000107 |
HGNC:31620 |
miR-30a |
hsa-miR-30a |
MI0000088 |
HGNC:31624 |
miR-138-1 |
hsa-miR-138-1 |
MI0000476 |
HGNC:31524 |
miR-138-2 |
hsa-miR-138-2 |
MI0000455 |
HGNC:31525 |
miR-155 |
hsa-miR-155 |
MI0000681 |
HGNC:31542 |
miR-19a |
hsa-miR-19a |
MI0000073 |
HGNC:31574 |
miR-17 |
hsa-miR-17 |
MI0000071 |
HGNC:31547 |
miR-221 |
hsa-miR-221 |
MI0000298 |
HGNC:31601 |
miR-181a-1 |
hsa-miR-181a-1 |
MI0000289 |
HGNC:31590 |
miR-181a-2 |
hsa-miR-181a-2 |
MI0000269 |
HGNC:31549 |
miR-126 |
hsa-miR-126 |
MI0000471 |
HGNC:31508 |
miR-128a |
hsa-miR-128-1, hsa-miR-128a |
MI0000447 |
HGNC:31510 |
miR-128b |
hsa-miR-128-2, hsa-miR-128b |
MI0000727 |
HGNC:31511 |
miR-15a |
hsa-miR-15a |
MI0000069 |
HGNC:31543 |
miR-188 |
hsa-miR-188 |
MI0000484 |
HGNC:31559 |
miRNAs and cascades of MAPK signaling
BCR-ABL fusion protein
It is well recognized that BCR-ABL tyrosine kinase activity plays a crucial role in CML [40]. Most of the patients with CML possess breakpoints in intron 1 or 2 of the ABL gene and in the main breakpoint cluster region (M-bcr) of the BCR gene, either amongst exons 13 and 14 (b2), or else 14 and 15 (b3) [41]. These breakpoints produce BCR/ABL fusion genes that transcribe either a b2a2 or b3a2 mRNA. The final product of this gene reorganization is a 210 kDa cytoplasmic fusion protein, p210BCR/ABL. This fusion protein is vital and adequate for the malignant transformation of CML, and is liable for the phenotypic anomalies of chronic phase CML [42–44]. Several functional domains have been established in the BCR-ABL protein that may contribute to cellular transformation. In the ABL portion, these domains are; SH1 (tyrosine kinase), SH2, and actin-binding domains. Whereas in BCR portion, there are coiled-coil oligomerization domain (amino acids (aa) 1–63), GRB-2 binding site (tyrosine at location 177) and the phosphoserine/threonine rich SH2 binding domain [41, 45].
A number of miRNAs regulate the expression of BCR-ABL, of which, miR-203 has been extensively studied. It essentially regulates ABL1 and BCR-ABL1 levels and inhibit cell proliferation [39, 46]. miR-203 is located intergenically on human chromosome 14 (Ch14q32.33) [47] and has been identified as a skin-specific miRNA. In normal condition it promotes epidermal differentiation by inducing cell-cycle exit and restricting proliferative potential [48]. It is exclusively expressed in keratinocytes (most common cell type in the epidermis), but not in the hair follicles of the skin. It has been demonstrated that over expression of miR-203 reduces ABL1 and BCR-ABL1 fusion protein levels in an ABL1-dependent manner [46]. A recent study of combined treatment effects of miR-203 and imatinib (small molecule kinase inhibitor) on imatinib-resistant cell lines demonstrated that miR-203 can serve as a novel target for CML treatment [49]. Another study also revealed that imatinib provoke the demethylation of the miR-203 promoter region, resulting in low expression of targeted BCR-ABL1 genes, and loss of proliferation of leukemic cells [50]. After noticing that both, the murine and the human, 3’ UTR of ABL1 genes contain miR-203 target sequences and this target site is well conserved in other vertebrates, Bueno et al. suggested that miR-203 may control ABL1 levels in a variety of organisms [46]. It was further suggested that miR-203 functions as a tumor-suppressor miRNA, targeting BCR-ABL and ABL kinases, which is epigenetically silenced in human Ph-positive leukemia cell lines [51, 52].
miRNA-196b is a vertebrate-specific miRNA, which appears to be expressed from intergenic regions in Homobox (HOX) gene clusters in many vertebrate species [53]. It belongs to the miR-196 family. Three miR-196 genes have been found so far i.e., miR-196a-1, miR-196a-2 and miR-196b [54]. In humans, the gene for miR-196b is located in a highly evolutionarily conserved region between HOXA9 and HOXA10 genes, on chromosome 7 (Ch7 p15.2)[55]. It has been reported that the expression of miR-196b is lower in CML patients than in healthy individuals. Recently, a study demonstrated that low level of expression of the tumor-suppressor, miR-196b can cause up-regulation of BCR-ABL1 expression which leads to the development of CML [55]. The dual luciferase reporter assay system also showed that BCR-ABL1 is the target genes of miR-196b. Furthermore, the study reported that a decline in the expression of miRNA-196b, in the cells overexpressing it, can restore BCR-ABL1 protein levels, enhance cell multiplication, and impeded the synthesis (S) phase of the cell cycle. In addition, down-regulation of BCR-ABL1 gene by small interfering (si) RNAs reduced the BCR-ABL1 protein levels and obstructed proliferation, similar to what was observed in cells displaying over-expression of miRNA-196b and a retarded G1 stage. It was also suggested that as regulation of miRNA-196b by DNA methylation is known to be involved in the progress of many other cancers it may hold true even for CML [55].
The miR-30a, generated from an intronic transcriptional unit, is located on human chromosome 6 (Ch6 q13) and belongs to miR-30 family [56, 57]. The miRNAs of miR-30 family has been found to be highly expressed in cardiac cells [58]. In an investigation, bone marrow samples of 16 CML patients and 10 normal patients, collected for the diagnosis of CML because of clinical features (hematological characters and presence of Philadelphia Chromosome), demonstrated that the expression of miR-30a is lower in bone marrow from CML patients than in normal control subjects [59]. It was also revealed that overexpression of the miR-30a in K562 cells (human immortalized myelogenous leukemia line) decreases the BCR-ABL1 protein levels, reduces cell proliferation and arrests the cells between G1 and S phase of the cell cycle. In contrast, inhibiting the expression of miR-30a in these cells notably increased the BCR-ABL1 protein levels, cell proliferation and restores the cell cycle. Furthermore, functional genomics studies in K562 cells verified that miR-30a played a tumor suppression role in CML by regulating the expression of BCR-ABL1 [59]. In another study, it was suggested that imatinib considerably inhibits expression of miR-30a in human CML cells. In contrast, reduction of miR-30a by antagomiR-30a surges the expression of Beclin 1 and Autophagy protein 5 (ATG5), and inhibits imatinib-induced cytotoxicity [60].
The miR-29 is a family of small RNA molecule in the shape of a stem-loop or hairpin. The miR-29 family in human includes hsa-miR-29a, hsa-miR-29b-1, hsa-miR-29b-2, and hsa-miR-29c. miR-29b-1 and miR-29b-2 have identical mature sequences, which are together called miR-29b [61, 62]. Mature miR-29s are highly conserved in rat, mouse and human, and share identical seed sequences at 2 to 7 nucleotide positions [63]. The genes coding for the precursors of miR-29b-1 and miR-29b-2 are located on chromosome 7 (Ch7 q32.3) [33] and chromosome 1 (Ch1 q32.2), respectively [64]. A recent study utilizing luciferase reporter assay demonstrated that miR-29b considerably reduces the activity of a luciferase reporter containing ABL1-3’UTR [65]. Another investigation showed that forced expression of miR-29b in K562 cells inhibits cell growth, colony formation ability and induces apoptosis through cleavage of procaspase 3 and Poly ADP ribose polymerase (PARP). Which suggests that miR-29b induced reduction of BCR-ABL1 protein in K562 cells is sufficient to trigger apoptotic response [66]. Thus, a prominent reduction of miR-29b in CML might implicate miR-29b as a potential tumor suppressor in CML by targeting ABL1 and BCR/ABL1 [67]. It had also been documented that miR-29b could impact CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein and Ribonuclease latent (RNase-L) [68]. The microarray studies of miRNAs downregulated in CML blast crisis revealed that miR-29b expression was significantly lower in CML patient samples as compared with normal volunteers [39]. In CML, abnormal expression of miR-29 family has been described [69] and a recently performed qPCR analysis of miR-29b expression further suggested that miR-29b was significantly downregulated in CML patient samples, suggesting that miR-29b negatively regulates ABL1 and BCR/ABL1, post transcriptionally [70, 71].
The miR-138 family was first detected in humans (Homo sapiens) [72]. miR-138 is usually considered as an example of the post-transcriptional control of miRNAs. Precursor form of miR-138 (pre-miR-138-2) is ubiquitously expressed in all tissues but, the mature miR-138 is spatially restricted to only certain tissue and cell types. It was observed that pre-miR-138-2 is cleaved to its mature form by Dicer in nucleus and is exported to cytoplasm only in distinct cells [73]. In the human genome, there are two miR-138 associated genes which are not located in any cluster. In particular, the miR-138-1 and miR-138-2 gene is located on chromosome 3 (Ch3p21.32) and chromosome 16 (Ch16q13), respectively [74]. In a current study, it was revealed that miR-138 binds to the coding region of ABL protein instead of 3’UTR of ABL mRNA. However, this binding downregulated the expression levels of ABL and BCR-ABL proteins which then causes inhibition of cell proliferation. Moreover, study demonstrated that the expression of miR-138 is triggered by treatment of imatinib which enhances the activity of GATA-binding factor 1(GATA1) and promotes its binding to miR-138 promoter. Generally, this expression of miR-138 is repressed by BCR-ABL. Therefore, miR-138, by the advantage of a BCR-ABL/GATA1/miR-138 integrated circuitry, acts as a tumor suppressor miRNA involved in the pathogenesis of CML and its clinical response to imatinib. The tumor suppressor activity of miR-138 was demonstrated in K562 and Ku812 cells, over expressing miR-138, by the initiation of G0/G1 cell cycle arrest, inhibition of cell proliferation and enhanced imatinib-induced apoptosis. Though de-regulated expression of this miRNA has been documented in a diverse array of tumors, it was revealed that miR-138 expression is down regulated in K562 cells and primary CML samples, which can be restored after imatinib treatment. Moreover, overexpression of miR-138 leads to the down regulation of BCR-ABL suggesting that there is negative regulatory loop between miR-138 and BCR-ABL. [75].
CRK family proteins
The first member of CRK family (v-CRK) of adaptor proteins was detected in late 1980s as the oncogene fusion product of the avian sarcoma virus, CT10 [76]. The CRK family is known to comprise of five members namely, v-CRK, CRKI, CRKII, CRKIII and CRK-like protein (CRKL) [77]. The cellular homolog of v-CRK were found to have an SH2 domain and either one (for CRKI) or two (for CRKII) SH3 domains [78]. These domains (SH2 and SH3) bind to phosphorylated tyrosine and proline-rich motifs, respectively [79]. CRKIII is predicted to encode a protein which have truncated C-terminal SH3 domain [80]. CRKL shares overall 60% of homology with CRKII, with one SH2 and two SH3 domains [81]. In adult murine tissues, CRKL expression is highest in adult hematopoietic tissues and lower in many epithelial tissues, whereas CRK displays elevated expression in the brain, lung, and kidney while exhibits low expression in bone marrow [79]. CRK proteins are prevalent phosphorylation substrates for the BCR-ABL fusion oncogene and are found in more than 95% of CML and 20% to 30% of acute lymphoblastic leukemia cases. CRKL is a key tyrosine-phosphorylated protein present in neutrophils of CML patient [82]. The amino terminal end (N-) of CRKL (SH3N domain) binds precisely to a proline-rich region in the carboxyl (C-) terminus of BCR-ABL protein. CRKL was also determined as a constitutively phosphorylated 39 kD tyrosine phosphoprotein in CML cells [83]. Guanine nucleotide exchange factor (GEF) or SOS1 protein is a major CRK SH3N- domain binding protein, which causes the activation of RAS and allow CRK to couple diversified upstream signals [80, 84].
miRNAs play an essential role in regulating CRK and CRKL. miR-126 is located within the 7th intron of the EGFL7 gene, residing on human chromosome 9 (9q34.3) [85] while, miR-17 is positioned on chromosome 13 (13q31.3) and belongs to the miR-17-92 cluster [86]. The putative target gene of these two miRNAs is CRK protein, which is involved in the MAPK signaling pathway. It was also suggested that miR-126 in humans is expressed only in endothelial cells, throughout capillaries as well as larger blood vessels [87], and acts upon various transcripts to control angiogenesis [85]. Recently, a study demonstrated that both, miR-126 and miR-17 were up-regulated in blast crisis (BC) samples of CML patients [88]. Study also suggested that miR-17 may be able to regulate MAPK expression level in leukocytes.
miR-221 is a tiny RNA molecule whose gene is located on the X chromosome (Xp11.3) [89]. This human miRNA was detected by a computational approach using conservation with mouse and Fugu rubripes sequences [90]. Expression of the excised miRNA were first validated in zebrafish, and later on in human promyelocytic leukemia cell 60 (HL-60) [91]. Recent study showed that the miR-221 level was up-regulated in BC samples of CML patients [88]. Research have demonstrated that miR-221 have an important role in the regulation of apoptosis by directly affecting the pro-apoptotic molecule, p53 upregulated modulator of apoptosis (PUMA), in vitro and xenograft mice model [92]. In a separate study, miR-128, miR-15a and miR-188-5p were suggested to suppress CML via CRKL encoding v-CRK avian sarcoma virus CT10 oncogene homolog-like [93]. The sequence of miR-188 was predicted based on homology to a documented miRNA from mouse [94]. This miRNA is located on the X chromosome (Xp11.23) [95]. Similarly, miR-128a sequence was also predicted based on homology to a verified miRNA from mouse [96]. Later on, the expression of miR-221 was even reported in HL-60 cells [91]. The miR-128a is located on chromosome 2 (2q21.3). The sequence of miR-15 was retitled as miR-15a, which can form a cluster with miR-16 within 0.5 kb at chromosome position 13 (13q14.2) and belongs to the miR-15 family [97]. It is found to be overwhelmed in chronic lymphocytic leukemia [98]. Information about the significance of miRNAs in the pathogenesis of CML is limited to the description of the abnormal expression of miR-15a in the CML cell line K562 [99].
SOS proteins
SOS specify to a set of genes that were first identified in Drosophila melanogaster as a functional gene product downstream of sevenless protein-tyrosine kinase in the RAS/MAP kinase pathway [100, 101]. It encodes GEF that acts upon the RAS subfamily of small GTPases. Mammalian cells possess two types of SOS homologs, SOS1 (~170 kDa) and SOS2 (~150 kDa), derived from divergent genetic loci. The N-terminal end of SOS1 encodes a Dbl homology (Dbl) and Pleckstrin homology (PH) duo that exchanges GTP for GDP on RAS. While, tandem C-terminal proline-rich motifs interacts with several adaptor proteins, including Grb2 and E3b1 (a Rac1 GEF) [102]. Both SOS1 and SOS2 carry repetitive proline motifs that conform to consensus CRK SH3 binding motifs and display a stable association between CRK and SOS.
Recently, it was predicted by TargetScan analysis that miR-155 is a putative target of SOS1 gene. This miRNA is processed from an exon of a noncoding RNA transcribed from the B-cell Integration Cluster (BIC), located intergenically on chromosome 21 (21q21.3), expressed in activated B cells, T cells, monocytes and macrophages [103]. BIC shows robust sequence homology amongst human, mouse and chicken and is highly, although not exclusively, expressed in lymphoid organs suggesting an evolutionary conserved function [96, 104, 105]. It was revealed from miRNA-based microarray and miR–quantitative Polymerase Chain Reaction (qPCR) analysis that the miR-155 is abnormally downregulated in K562 cells, in CML cell lines, and in patients with CML as compared to non-CML cell lines and blood samples from healthy patients [103].
miRNA-181a (miR-181a), one of the copious miRNAs conserved among vertebrates, is differentially expressed in a variety of leukemia. The miR-181 family contains four immensely conserved mature members, i.e., miR-181a, miR-181b, miR-181c and miR-181d, which are derived separately from 6 precursors located on 3 different chromosomes [106]. The miR-181a-1 and miR-181b-1 are clustered together and located on chromosome 1, miR-181a-2 and miR-181b-2 are clustered together and located on chromosome 9 [107], and miR-181c and miR-181d are clustered together and located on chromosome 19 [108, 109]. It had been found that the expression of miR-181a is very low, so low that it cannot be detected by qPCR in K562 cells, indicating that downregulation of miR-181a might play a major role in leukemogenesis. The combination of TargetScan and miRNAmap software prediction proposed that SOS1 is the putative target genes of miR-181a. Moreover, it also revealed that the expression pattern of miR-181a was deregulated in CML patients [110].
K-RAS proteins
RAS proteins are small GTPases. These proteins are known to be preserved across species and play key roles in numerous basic cellular functions, including control of proliferation, cell growth differentiation, and apoptosis [111]. RAS proteins acts as molecular switches that cycle between two conformational states: one when they are bound to GTP (the active form) and another one when bound to GDP (the inactive form) [112–115]. GEFs promote the formation of GTP-bound RAS [116] whereas, GTPase-activating proteins, or GAPs, stimulate the hydrolysis of GTP on RAS, returning them to their inactive state [117]. Transgenic and cell-biological studies [118–121] complemented by clinical observations [122] actively pointed out that RAS has different iso-forms i.e., H-RAS, N-RAS and K-RAS. These isoforms can provoke distinct signal outputs, although interacting with a prevalent set of activators and effectors. This biological disparity is probably considered due to the C-terminal amino acids (25 in numbers) of the Hypervariable domain (Hvr), which is the only region that differs significantly [111]. Physiological and oncogenic activation of RAS trigger a broad range of downstream signaling pathways, of which, the RAF-MEK-ERK pathway was the first to be identified as RAS effector pathway [123–126].
Bioinformatics analysis utilizing TargetScan revealed that KRAS is the probable target of miR-155 and miR-19a. As discussed earlier, miR-155 is encoded from the gene located on miRNA chromosome 21 (21q21.3) [127, 128]. In humans, this miRNA is transcribed from the MIR155 parent gene or MIR155HG [129, 130] and the MIR155HG RNA transcript does not have a long open reading frame (ORF), however, it does consists of an imperfectly base-paired stem loop that is conserved across species. Subsequent studies established that the MIR155HG was composed of three exons that span a 13 kb region [131]. In a study Rokah et al. utilized miRNA microarray to identify miRNA expression in CML cell lines and patient samples. Among several dysregulated miRNAs identified in CML cell lines, expression levels of miR-155 was too found downregulated [103]. A decrease in miR-155 may establish an additional mechanism leading to deregulated expression of RAS. From PCR analysis, K-RAS has been shown to cooperate with ABL to induce full in-vitro and in-vivo transformation of cells leading to tumorigenesis. Moreover, it was found that the expression level of K-RAS was significantly higher in K562 cells compared to normal blood samples [103]. Downregulated expression of miR-155 and higher levels of K-RAS in CML patients points toward the possible regulation of K-RAS by miR-155. The potential of miR-155 in regulating K-RAS may be a unique pathway of regulation that can be targeted for therapeutic purpose and hence, requires further researches for determining the possibilities.
miR-19 is the key oncogenic component of the miR-17-92 cluster, and, therefore, miR-19-specific targets are likely to mediate the oncogenic effects of the miR-17-92 cluster (also known as oncomiR-1) [132]. The miR-17-92 miRNA cluster produces a single polycistronic primary transcript that yields six mature miRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a. This distinctive structural feature of miR-17-92, common in vast number of miRNA genes in mammalian genomes, may be responsible for the molecular basis for its pleiotropic functions in a cell type-dependent and context-dependent manner [133]. miR-19a is located on human chromosome 13 (13q31.3). Targetscan analysis identified that, K-RAS, which is involved in MAPK signaling, is a predicted target of miR-19a. Abnormal expression of onco-miR, miR-19a was described in CML CD34+ cells. Study further revealed that the level of miR-19a is up-regulated in the CML cell line and it may act as oncomiRs [134].
RAF1 protein
The cytoplasmic protein, RAF1 has been verified as a key molecule in MAPK signaling pathway. RAS subfamily of membrane associated GTPases are known to activate downstream RAF1 proteins [135]. In humans it is encoded by the RAF1 gene [136]. Therefore, RAF1 is an interesting target for molecular therapies and target-based therapies which are widely considered to be the future of cancer treatment. The RAF1 gene is positioned on the short (p) arm of human chromosome 3 at position 25 (3p25) [137]. As discussed in above section, miR-19a is located on human chromosome 13 and belongs to the miR-19 family of the miR-17-92 cluster. It was recently demonstrated that the expression pattern of miR-19a was found up-regulated in CML patients [133]. The confirmed increase of miR-19a was also identified in samples of BC pool [134]. In addition, TargetScan analysis identified RAF1 as a probable target of the oncogenic miR-19a. However, future studies are needed to authenticate the role miR-19 in CML via regulation of RAF-1.
MAPK/ERK protein
MAPKs are eukaryotic protein Ser/Thr kinases, which is encoded by MAPK1 gene and activated by an upstream activator, RAF, in the MAPK signaling pathway [5]. The conventional MAP kinases can be assembled into three major families. These are ERKs (extracellular-signal-regulated kinases), JNKs (Jun amino-terminal kinases), and p38/SAPKs (stress-activated protein kinases) [138]. All MAPKs comprise a Serine/Threonine kinase domain flanked by N- and C-terminal regions of diverse lengths. Different additional domains also exists in some MAPKs, comprising a transactivation domain (TAD), a region conserved in ERK3 and ERK4 (C34), a nuclear localization sequence (NLS), and a domain rich in alanine, histidine, and glutamine (AHQr) [5]. It is well known that regulation of both RAS and RAF is crucial for the proper maintenance of cell proliferation, as activating mutations in these genes lead to oncogenesis [1, 139]. Activated RAF binds to and phosphorylates the dual specificity kinases MEK1/2, which in turn, phosphorylate ERK1/2 within a conserved Thr-Glu-Tyr (TEY) motif in their activation loop [5, 32, 136].
Several miRNAs play regulatory role in MAPK1 expression, of which, miR-17 and miR-19a can directly regulate the MAPK1. miR-17 and miR-19a belongs to miR-17 and miR-19 miRNA family, respectively and are members of highly conserved miR-17-92 cluster in vertebrates [140]. Both of these miRNAs are located on human chromosome 13 (13q31.3). The 17-92 miRNA clusters is recognized as an important CML-associated oncogene. An investigation reported that overexpression of this cluster stimulates cell proliferation in K562 cell line [141]. Moreover, this cluster is important for cell cycle, apoptosis and other pivotal processes. Often miR-17-92 cluster is found dysregylated in hematopoietic and solid tumors. A study also reported that miR-17 is expressed from the 3’ arm of the hairpin precursor in human epithelial carcinoma cell line (HeLa) cells [97]. Interestingly, it was described that RAS/MAPK signaling may contribute to the survival of BCR-ABL+ cells under imatinib selection pressure [142]. PCR analysis has demonstrated that the levels of miR-17 and miR-19a are up-regulated in CML patients as compared to non-CML patients [88]. Another study dealing with miRNA expression in CML demonstrated abnormal expression of the miR-17 and miR-19a in CML CD34+ cells [134].
Future direction
CML has appeared as one of the significant challenges for the scientific community who are dealing with cancer and new drug discovery. Over the last few years, several interesting studies have been performed on the regulatory mechanism of CML and its pathogenesis. Recently, studies are paying attention on the identification of small miRNAs and there contribution toward regulating key events of the cells. Current review will help to understand the contribution of these recently identified miRNAs in the regulatory complexities of CML. Our comprehensive study will also help the researchers to target and verify the novel miRNA-based diagnostics for this leukemia. Furthermore, these miRNAs may be utilized as therapeutics to upregulate or downregulate the differentially expressed proteins related to MAPK pathways in CML in upcoming time.
CONCLUSIONS
Growing evidences indicates the significance of miRNAs in modulating signal transduction pathways in CML (Figure 1). With the aid of computational tools, we have summarized a number of miRNAs that are being identified to regulate proteins cascades during CML (Figure 2). Our study has highlighted the role of miRNAs in CML and at the same time has explored new possibilities in the field of leukemia. In summary, we have depicted an extensive connection between miRNA expression and human CML which shows the dual functions of miRNAs as oncogene and tumor suppressor. We can conclude that the ability of miRNAs to control different cellular processes in various tissues at multiple levels makes them one of the most competent therapeutic agents in modern medicine. However, more fundamental understanding of miRNA regulated MAPK signal transduction pathways is required to address deeper insight into the mechanism of CML which can help to develop novel miRNA/anti-miRNA-based therapeutics in the near future.
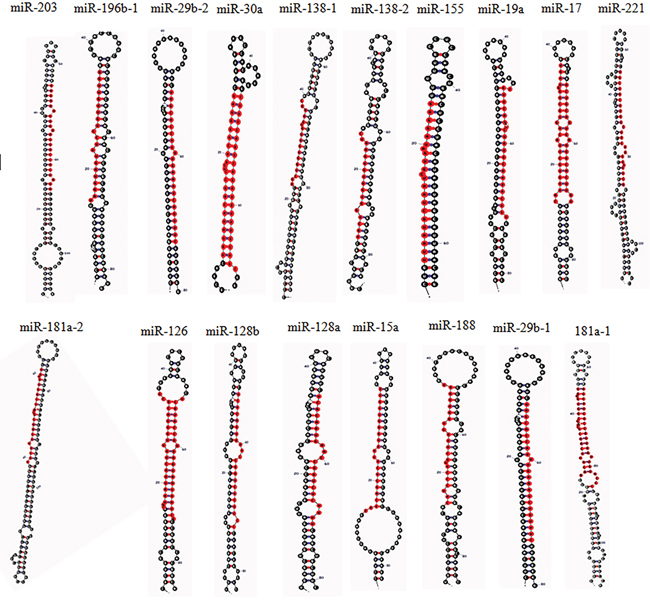
Figure 2: The Stem-loop structure of human miRNAs related to MAPK signaling pathway in CML, (determined by miRNAMAP; http://mirnamap.mbc.nctu.edu.tw/) [143].
Abbreviations
BCR-ABL: breakpoint cluster region- Abelson murine leukemia viral oncogene homolog, non-receptor tyrosine kinase; CRK: chicken tumour virus no. 10 [CT10] regulator of kinase; CRKL: v-crk avian sarcoma virus CT10 oncogene homolog-like; KRAS: v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; NRAS: neuroblastoma RAS viral (v-ras) oncogene homolog; HRAS: Harvey rat sarcoma viral oncogene homolog; RAF1: Raf-1 proto-oncogene, serine/threonine kinase; SOS1,2: son of sevenless homolog 1 (Drosophila), 2; Mek1,2: mitogen-activated protein/extracellular signal-regulated kinase kinase 1, 2; ERK: extracellular signal-regulated kinase; v-Crk: V-Crk Avian Sarcoma Virus CT10 Oncogene Homolog; EGFL7: Multiple Epidermal Growth Factor-Like Domains Protein 7; GTPase: guanosine triphosphatase.
ACKNOWLEDGMENTS
This research was supported by Hallym University Research Fund, by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2014R1A1A4A03009388) and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health &Welfare, Republic of Korea (HI12C1265).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007; 26:3279-3290.
2. Meister M, Tomasovic A, Banning A, Tikkanen R. Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount. Int J Mol Sci. 2013; 14:4854-4884.
3. Pearson G, Robinson F, Beers Gibson T, Xu B-e, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions 1. Endocrine reviews. 2001; 22:153-183.
4. Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998; 62:1264-1300.
5. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011; 75:50-83.
6. Cossa G, Gatti L, Cassinelli G, Lanzi C, Zaffaroni N, Perego P. Modulation of sensitivity to antitumor agents by targeting the MAPK survival pathway. Curr Pharm Des. 2013; 19:883-894.
7. De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012; 16:S17-27.
8. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013; 49:1297-1304.
9. Fialkow PJ, Jacobson RJ, Papayannopoulou T. Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am J Med. 1977; 63:125-130.
10. Comert M, Baran Y, Saydam G. Changes in molecular biology of chronic myeloid leukemia in tyrosine kinase inhibitor era. Am J Blood Res. 2013; 3:191-200.
11. Leitner AA, Hochhaus A, Muller MC. Current treatment concepts of CML. Curr Cancer Drug Targets. 2011; 11:31-43.
12. Garcia-Manero G, Faderl S, O’Brien S, Cortes J, Talpaz M, Kantarjian HM. Chronic myelogenous leukemia: a review and update of therapeutic strategies. Cancer. 2003; 98:437-457.
13. Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Basecke J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM, McCubrey JA. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008; 22:686-707.
14. Sanchez-Arevalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Tipping AJ, Viniegra JG, Hernandez Losa J, Parada Cobo C, Galan Moya EM, Gayoso Cruz J, Melo JV, Ramon y Cajal S, Sanchez-Prieto R. Modulation of the p38 MAPK (mitogen-activated protein kinase) pathway through Bcr/Abl: implications in the cellular response to Ara-C. Biochem J. 2005; 387:231-238.
15. Aceves-Luquero CI, Agarwal A, Callejas-Valera JL, Arias-Gonzalez L, Esparis-Ogando A, del Peso Ovalle L, Bellon-Echeverria I, de la Cruz-Morcillo MA, Galan Moya EM, Moreno Gimeno I, Gomez JC, Deininger MW, Pandiella A, Sanchez Prieto R. ERK2, but not ERK1, mediates acquired and ‘de novo’ resistance to imatinib mesylate: implication for CML therapy. PLoS One. 2009; 4:e6124.
16. Ambros V. The functions of animal microRNAs. Nature. 2004; 431:350-355.
17. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-297.
18. Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010; 79:351-379.
19. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007; 302:1-12.
20. de Moor CH, Meijer H, Lissenden S. Mechanisms of translational control by the 3’ UTR in development and differentiation. Semin Cell Dev Biol. 2005; 16:49-58.
21. Lai EC. Micro RNAs are complementary to 3’ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002; 30:363-364.
22. Robins H, Press WH. Human microRNAs target a functionally distinct population of genes with AT-rich 3’ UTRs. Proc Natl Acad Sci U S A. 2005; 102:15557-15562.
23. Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3’UTR evolution. Cell. 2005; 123:1133-1146.
24. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75:843-854.
25. Zhu X, Lin Z, Du J, Zhou X, Yang L, Liu G. Studies on microRNAs that are correlated with the cancer stem cells in chronic myeloid leukemia. Mol Cell Biochem. 2014; 390:75-84.
26. Melo JV, Gordon DE, Cross NC, Goldman JM. The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood. 1993; 81:158-165.
27. Shet AS, Jahagirdar BN, Verfaillie CM. Chronic myelogenous leukemia: mechanisms underlying disease progression. Leukemia. 2002; 16:1402-1411.
28. Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Landmarks in Medical Genetics: Classic Papers with Commentaries. 2004; 132:103.
29. Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973; 243:290-293.
30. Lynch RG. The Philadelphia Chromosome. Science. 1960; 132:1497-1501.
31. Koul HK, Pal M, Koul S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer. 2013; 4:342-359. doi: 10.1177/1947601913507951.
32. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010; 1802:396-405.
33. Torii S, Yamamoto T, Tsuchiya Y, Nishida E. ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci. 2006; 97:697-702.
34. Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, Gishizky ML. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993; 75:175-185.
35. Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J Biol Chem. 1994; 269:22925-22928.
36. Pelicci G, Lanfrancone L, Salcini AE, Romano A, Mele S, Grazia Borrello M, Segatto O, Di Fiore PP, Pelicci PG. Constitutive phosphorylation of Shc proteins in human tumors. Oncogene. 1995; 11:899-907.
37. Cahill MA, Janknecht R, Nordheim A. Signalling pathways: jack of all cascades. Curr Biol. 1996; 6:16-19.
38. Kumar A, Asaf V, Srivastava K, Rahim A, Chaudhary J, Panigrahi M. MicroRNA: biogenesis and computational target identification: a review. Veterinary World. 2013; 6:761-765.
39. Polakova KM, Lopotová T, Klamová H, Burda P, Trněný M, Stopka T, Moravcová J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Molecular cancer. 2011; 10:41-53.
40. Goldman JM, Melo JV. Chronic myeloid leukemia—advances in biology and new approaches to treatment. New England Journal of Medicine. 2003; 349:1451-1464.
41. Salesse S, Verfaillie CM. BCR/ABL: from molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene. 2002; 21:8547-8559.
42. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990; 247:824-830.
43. Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proceedings of the National Academy of Sciences. 1988; 85:9312-9316.
44. Gishizky ML, Johnson-White J, Witte ON. Efficient transplantation of BCR-ABL-induced chronic myelogenous leukemia-like syndrome in mice. Proceedings of the National Academy of Sciences. 1993; 90:3755-3759.
45. Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000; 96:3343-3356.
46. Bueno MJ, de Castro IP, de Cedrón MG, Santos J, Calin GA, Cigudosa JC, Croce CM, Fernández-Piqueras J, Malumbres M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer cell. 2008; 13:496-506.
47. Xu M, Gu M, Zhang K, Zhou J, Wang Z, Da J. miR-203 inhibition of renal cancer cell proliferation, migration and invasion by targeting of FGF2. Diagnostic pathology. 2015; 10:24.
48. Yi R, Poy MN, Stoffel M, Fuchs E. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature. 2008; 452:225-229.
49. Li Y, Yuan Y, Tao K, Wang X, Xiao Q, Huang Z, Zhong L, Cao W, Wen J, Feng W. Inhibition of BCR/ABL protein expression by miR-203 sensitizes for imatinib mesylate. PLoS One. 2013; 8:e61858.
50. Shibuta T, Honda E, Shiotsu H, Tanaka Y, Vellasamy S, Shiratsuchi M, Umemura T. Imatinib induces demethylation of miR-203 gene: An epigenetic mechanism of anti-tumor effect of imatinib. Leukemia research. 2013; 37:1278-1286.
51. Chim CS, Wong KY, Leung CY, Chung LP, Hui PK, Chan SY, Yu L. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. Journal of cellular and molecular medicine. 2011; 15:2760-2767.
52. Wang L-S, Li L, Li L, Chu S, Shiang K-D, Li M, Sun H-Y, Xu J, Xiao F-J, Sun G. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood. 2015; 125:1302-1313.
53. Yekta S, Shih I-h, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004; 304:594-596.
54. Chen C, Zhang Y, Zhang L, Weakley SM, Yao Q. MicroRNA-196: critical roles and clinical applications in development and cancer. Journal of cellular and molecular medicine. 2011; 15:14-23.
55. Liu Y, Zheng W, Song Y, Ma W, Yin H. Low expression of miR-196b enhances the expression of BCR-ABL1 and HOXA9 oncogenes in chronic myeloid leukemogenesis. PloS one. 2013; 8:e68442.
56. Patnaik SK, Kannisto E, Yendamuri S. Overexpression of microRNA miR-30a or miR-191 in A549 lung cancer or BEAS-2B normal lung cell lines does not alter phenotype. PloS one. 2010; 5:e9219.
57. Vosa U, Vooder T, Kolde R, Vilo J, Metspalu A, Annilo T. Meta-analysis of microRNA expression in lung cancer. International Journal of Cancer. 2013; 132:2884-2893.
58. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P. miR-133 and miR-30 regulate connective tissue growth factor Implications for a role of microRNAs in myocardial matrix remodeling. Circulation research. 2009; 104:170-178.
59. Liu Y, Song Y, Ma W, Zheng W, Yin H. Decreased microRNA-30a levels are associated with enhanced ABL1 and BCR-ABL1 expression in chronic myeloid leukemia. Leukemia research. 2013; 37:349-356.
60. Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, Tang D, Cao L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia. 2012; 26:1752-1760.
61. Jiang H, Zhang G, Wu J-H, Jiang C-P. Diverse roles of miR-29 in cancer (Review). Oncology reports. 2014; 31:1509-1516.
62. O’Rourke JR, Olson EN. Modulating the MicroRNArchitecture of an aging aorta. Circulation research. 2011; 109:1098-1099.
63. Kriegel AJ, Liu Y, Fang Y, Ding X, Liang M. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiological genomics. 2012; 44:237-244.
64. Eyholzer M, Schmid S, Wilkens L, Mueller B, Pabst T. The tumour-suppressive miR-29a/b1 cluster is regulated by CEBPA and blocked in human AML. British journal of cancer. 2010; 103:275-284.
65. Li Y, Wang H, Tao K, Xiao Q, Huang Z, Zhong L, Cao W, Wen J, Feng W. miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp Cell Res. 2013; 319:1094-1101.
66. Rangatia J, Bonnet D. Transient or long-term silencing of BCR-ABL alone induces cell cycle and proliferation arrest, apoptosis and differentiation. Leukemia. 2006; 20:68-76.
67. Long L, Yu P, Liu Y, Wang S, Li R, Shi J, Zhang X, Li Y, Sun X, Zhou B, Cui L, Li Z. Upregulated microRNA-155 expression in peripheral blood mononuclear cells and fibroblast-like synoviocytes in rheumatoid arthritis. Clin Dev Immunol. 2013; 2013:296139.
68. Lee TY, Ezelle HJ, Venkataraman T, Lapidus RG, Scheibner KA, Hassel BA. Regulation of human RNase-L by the miR-29 family reveals a novel oncogenic role in chronic myelogenous leukemia. Journal of Interferon & Cytokine Research. 2013; 33:34-42.
69. San José-Enériz E, Román-Gómez J, Jiménez-Velasco A, Garate L, Martin V, Cordeu L, Vilas-Zornoza A, Rodríguez-Otero P, Calasanz MJ, Prósper F. MicroRNA expression profiling in Imatinib-resistant Chronic Myeloid Leukemia patients without clinically significant ABL1-mutations. Mol Cancer. 2009; 8:69-72.
70. Li Y, Wang H, Tao K, Xiao Q, Huang Z, Zhong L, Cao W, Wen J, Feng W. miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Experimental cell research. 2013; 319:1094-1101.
71. Kollinerova S, Vassanelli S, Modriansky M. The role of miR-29 family members in malignant hematopoiesis. Biomedical Papers. 2014; 158:489-501.
72. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Current Biology. 2002; 12:735-739.
73. Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. RNA. 2006; 12:1161-1167.
74. Liu X, Jiang L, Wang A, Yu J, Shi F, Zhou X. MicroRNA-138 suppresses invasion and promotes apoptosis in head and neck squamous cell carcinoma cell lines. Cancer letters. 2009; 286:217-222.
75. Xu C, Fu H, Gao L, Wang L, Wang W, Li J, Li Y, Dou L, Gao X, Luo X. BCR-ABL/GATA1/miR-138 mini circuitry contributes to the leukemogenesis of chronic myeloid leukemia. Oncogene. 2014; 33:44-54.
76. Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988; 332:272-5.
77. Prosser S, Sorokina E, Pratt P, Sorokin A. CrkIII: a novel and biologically distinct member of the Crk family of adaptor proteins. Oncogene. 2003; 22:4799-4806.
78. Matsuda M, Tanaka S, Nagata S, Kojima A, Kurata T, Shibuya M. Two species of human CRK cDNA encode proteins with distinct biological activities. Molecular and Cellular Biology. 1992; 12:3482-3489.
79. de Jong R, Haataja L, Voncken JW, Heisterkamp N, Groffen J. Tyrosine phosphorylation of murine Crkl. Oncogene. 1995; 11:1469-1474.
80. Bell ES, Park M. Models of crk adaptor proteins in cancer. Genes Cancer. 2012; 3:341-52. doi: 10.1177/1947601912459951.
81. Ten Hoeve J, Morris C, Heisterkamp N, Groffen J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene. 1993; 8:2469-2474.
82. Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. Journal of Biological Chemistry. 1994; 269:22925-22928.
83. Nichols GL, Raines M, Vera JC, Lacomis L, Tempst Pa, Golde D. Identification of CRKL as the constitutively phosphorylated 39-kD tyrosine phosphoprotein in chronic myelogenous leukemia cells. Blood. 1994; 84:2912-2918.
84. Matsuda M, Hashimoto Y, Muroya K, Hasegawa H, Kurata T, Tanaka S, Nakamura S, Hattori S. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Molecular and Cellular Biology. 1994; 14:5495-5500.
85. Meister J, Schmidt MH. miR-126 and miR-126*: new players in cancer. ScientificWorldJournal. 2010; 10:2090-2100.
86. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008; 132:875-886.
87. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008; 15:261-271.
88. Machova Polakova K, Lopotova T, Klamova H, Burda P, Trneny M, Stopka T, Moravcova J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011; 10:41.
89. Zhang J, Han L, Ge Y, Zhou X, Zhang A, Zhang C, Zhong Y, You Y, Pu P, Kang C. miR-221/222 promote malignant progression of glioma through activation of the Akt pathway. Int J Oncol. 2010; 36:913-920.
90. Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003; 299:1540.
91. Kasashima K, Nakamura Y, Kozu T. Altered expression profiles of microRNAs during TPA-induced differentiation of HL-60 cells. Biochem Biophys Res Commun. 2004; 322:403-410.
92. Zhang CZ, Zhang JX, Zhang AL, Shi ZD, Han L, Jia ZF, Yang WD, Wang GX, Jiang T, You YP, Pu PY, Cheng JQ, Kang CS. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol Cancer. 2010; 9:229.
93. Xiong Q, Yang Y, Wang H, Li J, Wang S, Li Y, Yang Y, Cai K, Ruan X, Yan J, Hu S, Fang X. Characterization of miRNomes in acute and chronic myeloid leukemia cell lines. Genomics Proteomics Bioinformatics. 2014; 12:79-91.
94. Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003; 9:175-179.
95. Chi J, Ballabio E, Chen XH, Kusec R, Taylor S, Hay D, Tramonti D, Saunders NJ, Littlewood T, Pezzella F, Boultwood J, Wainscoat JS, Hatton CS, Lawrie CH. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol Direct. 2011; 6:23.
96. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002; 12:735-739.
97. Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001; 294:853-858.
98. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002; 99:15524-15529.
99. Agirre X, Jimenez-Velasco A, San Jose-Eneriz E, Garate L, Bandres E, Cordeu L, Aparicio O, Saez B, Navarro G, Vilas-Zornoza A, Perez-Roger I, Garcia-Foncillas J, Torres A, Heiniger A, Calasanz MJ, Fortes P, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res. 2008; 6:1830-1840.
100. Simon MA, Dodson GS, Rubin GM. An SH3-SH2-SH3 protein is required for p21Ras1 activation and binds to sevenless and Sos proteins in vitro. Cell. 1993; 73:169-177.
101. Rogge RD, Karlovich CA, Banerjee U. Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell. 1991; 64:39-48.
102. Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009; 7:13.
103. Rokah OH, Granot G, Ovcharenko A, Modai S, Pasmanik-Chor M, Toren A, Shomron N, Shpilberg O. Downregulation of miR-31, miR-155, and miR-564 in chronic myeloid leukemia cells. PLoS One. 2012; 7:e35501.
104. Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009; 28:264-284.
105. Higgs G, Slack F. The multiple roles of microRNA-155 in oncogenesis. J Clin Bioinforma. 2013; 3:17.
106. Fei J, Li Y, Zhu X, Luo X. miR-181a post-transcriptionally downregulates oncogenic RalA and contributes to growth inhibition and apoptosis in chronic myelogenous leukemia (CML). PLoS One. 2012; 7:e32834.
107. Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007; 67:6031-6043.
108. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007; 129:1401-1414.
109. Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005; 37:766-770.
110. Yendamuri S, Calin GA. The role of microRNA in human leukemia: a review. Leukemia. 2009; 23:1257-1263.
111. Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003; 4:373-384.
112. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990; 348:125-132.
113. Field J, Broek D, Kataoka T, Wigler M. Guanine nucleotide activation of, and competition between, RAS proteins from Saccharomyces cerevisiae. Mol Cell Biol. 1987; 7:2128-2133.
114. Satoh T, Nakamura S, Kaziro Y. Induction of neurite formation in PC12 cells by microinjection of proto-oncogenic Ha-ras protein preincubated with guanosine-5’-O-(3-thiotriphosphate). Mol Cell Biol. 1987; 7:4553-4556.
115. Wittinghofer A, Pai EF. The structure of Ras protein: a model for a universal molecular switch. Trends Biochem Sci. 1991; 16:382-387.
116. Wolfman A, Macara IG. A cytosolic protein catalyzes the release of GDP from p21ras. Science. 1990; 248:67-69.
117. Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987; 238:542-545.
118. Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998; 273:24052-24056.
119. Walsh AB, Bar-Sagi D. Differential activation of the Rac pathway by Ha-Ras and K-Ras. J Biol Chem. 2001; 276:15609-15615.
120. Wolfman JC, Wolfman A. Endogenous c-N-Ras provides a steady-state anti-apoptotic signal. J Biol Chem. 2000; 275:19315-19323.
121. Hamilton M, Wolfman A. Ha-ras and N-ras regulate MAPK activity by distinct mechanisms in vivo. Oncogene. 1998; 16:1417-1428.
122. Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989; 49:4682-4689.
123. Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993; 364:352-355.
124. Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993; 74:205-214.
125. Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, Bruder JT, Rapp UR, Avruch J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 1993; 364:308-313.
126. Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993; 260:1658-1661.
127. Zhang CM, Zhao J, Deng HY. MiR-155 promotes proliferation of human breast cancer MCF-7 cells through targeting tumor protein 53-induced nuclear protein 1. J Biomed Sci. 2013; 20:79.
128. Kotlabova K, Doucha J, Chudoba D, Calda P, Dlouha K, Hromadnikova I. Extracellular chromosome 21-derived microRNAs in euploid & aneuploid pregnancies. Indian J Med Res. 2013; 138:935-943.
129. Vargova K, Curik N, Burda P, Basova P, Kulvait V, Pospisil V, Savvulidi F, Kokavec J, Necas E, Berkova A, Obrtlikova P, Karban J, Mraz M, Pospisilova S, Mayer J, Trneny M, et al. MYB transcriptionally regulates the miR-155 host gene in chronic lymphocytic leukemia. Blood. 2011; 117:3816-3825.
130. Chang S, Wang RH, Akagi K, Kim KA, Martin BK, Cavallone L, Kathleen Cuningham Foundation Consortium for Research into Familial Breast C, Haines DC, Basik M, Mai P, Poggi E, Isaacs C, Looi LM, Mun KS, Greene MH, Byers SW, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011; 17:1275-1282.
131. Tam W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene. 2001; 274:157-167.
132. Song DW, Ryu JY, Kim JO, Kwon EJ, Kim do H. The miR-19a/b family positively regulates cardiomyocyte hypertrophy by targeting atrogin-1 and MuRF-1. Biochem J. 2014; 457:151-162.
133. Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L. miR-19 is a key oncogenic component of miR-17-92. Genes Dev. 2009; 23:2839-2849.
134. Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, Muckenthaler MU, Ganser A, Eder M, Scherr M. Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood. 2007; 109:4399-4405.
135. Bonner T, O’Brien SJ, Nash WG, Rapp UR, Morton CC, Leder P. The human homologs of the raf (mil) oncogene are located on human chromosomes 3 and 4. Science. 1984; 223:71-74.
136. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007; 26:3291-3310.
137. Huebner K, ar-Rushdi A, Griffin CA, Isobe M, Kozak C, Emanuel BS, Nagarajan L, Cleveland JL, Bonner TI, Goldsborough MD, Croce CM, Rapp U. Actively transcribed genes in the raf oncogene group, located on the X chromosome in mouse and human. Proc Natl Acad Sci U S A. 1986; 83:3934-3938.
138. Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012; 4.
139. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008; 9:517-531.
140. Mogilyansky E, Rigoutsos I. The miR-17-92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013; 20:1603-1614.
141. Wang XS, Zhang JW. The microRNAs involved in human myeloid differentiation and myelogenous/myeloblastic leukemia. J Cell Mol Med. 2008; 12:1445-1455.
142. Chu S, Holtz M, Gupta M, Bhatia R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004; 103:3167-3174.
143. Hsu SD, Chu CH, Tsou AP, Chen SJ, Chen HC, Hsu PW, Wong YH, Chen YH, Chen GH, Huang HD. miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res. 2008; 36:D165-169.