
INTRODUCTION
Isochromosome 17q [i(17q)] is a non-random cytogenetic abnormality that involves deletion of the short “p” arm and duplication of the long “q” arm of chromosome 17. It is frequently observed in the setting of a complex karyotype in blast phase of chronic myelogenous leukemia (CML) and acute myeloid leukemia (AML). As an isolated cytogenetic abnormality, isochromosome (17q) in myeloid malignancies is rare with less than 150 reported cases. The presence of isolated i(17q) in myeloid neoplasms is associated with certain distinctive clinicopathologic findings as shown in several recent studies [1–3]. These neoplasms are often classified as myelodysplastic/myeloproliferative neoplasms (MDS/MPN) or high-grade myelodysplastic syndrome (MDS)/AML, and show characteristic morphologic findings, in particular pseudo Pelger-Huet neutrophils and small hypolobated megakaryocytes. Irrespective of the diagnosis or blast count at presentation, myeloid neoplasms with isolated i(17q) have an aggressive clinical course. Most patients undergo rapid progression to AML, often without acquisition of additional clonal cytogenetic abnormalities. Accordingly, i(17q) is classified as “intermediate” cytogenetic prognostic subgroup per the revised International Prognostic Scoring System (IPSS) for MDS [4, 5]; and as an “adverse” cytogenetic prognostic subgroup per revised Medical Research Council classification for AML [6].
Understanding the molecular alterations in myeloid neoplasms associated i(17q) will likely prove to be valuable in the development of targeted therapy for affected patients. The “low-copy repeats” rich breakpoint region, located at 17p11.2 is highly unstable and prone to be affected by large-scale genomic alterations including uniallelic loss of TP53 [7, 8]. Until now, studies have shown that the presence of i(17q) abnormality is associated with wild-type TP53 [1] and mutations in SETBP1 and SRSF2 [3, 9]. However, the molecular consequences of i(17q) are largely unknown. Further, it is unclear if the i(17q) abnormality precedes these gene mutations or represents a secondary event.
In this study, we performed a systematic molecular analysis of myeloid neoplasms with isolated i(17q) and discovered unique molecular alterations that provide insights into underlying pathogenesis and potential therapeutic targets. Using sequential mutation analysis in 5 cases that showed evolution of i(17q) abnormality from a diploid karyotype, we show that SRSF2 and ASXL1 mutations precede the detection of i(17q), whereas SETBP1 mutations are associated with i(17q).
RESULTS
We selected 32 cases of myeloid neoplasm with i(17q) as the primary abnormality that had sufficient DNA for molecular analysis. This group included 13 cases of MDS/MPN, 17 cases of AML (5 of which had a history of myeloid neoplasm), and 1 case each of MDS and MPN (clinical data shown in Table 1). Twenty-nine cases had i(17q) as a sole abnormality (2 acquired +13 subsequently during the course of the disease), 2 had 1 additional abnormality and 1 had multiple additional abnormalities. All cases were negative for BCR/ABL1 rearrangement.
Table 1: Summary of clinicopathologic and cytogenetic findings of 32 myeloid neoplasms with isolated i(17q) cytogenetic abnormality
Variable |
MDS/MPN |
AML |
MDS |
MPN |
|---|---|---|---|---|
Median age (range) |
64 years (51–83) |
66 years (24–78) |
55 years |
60 years |
Gender (female/male) |
9/4 |
8/9 |
0/1 |
0/1 |
Hemoglobin* (g/dL) |
9.4 (6.8–12.1) |
10.1 (6.4–12.8) |
8.6 |
12.9 |
MCV |
88 (78–102) |
88 (70–110) |
91.5 |
104 |
Absolute neutrophil count*, × 109/L |
3.6 (0.5–122.2) |
3.0 (0–24.2) |
1.9 |
5.7 |
Platelet count*, × 109/L |
54 (10–143) |
57 (15–271) |
92 |
141 |
Peripheral blood blasts* (%) |
1 (0–13) |
26 (0–97) |
0 |
1 |
Bone marrow blasts* (%) |
8 (2–19) |
37 (7–93) |
12 |
0 |
Cytogenetic evolution, n (%) |
30.8% |
23.5% |
|
|
Without clonal evolution |
9 |
13 |
0 |
1 |
With clonal evolution |
4 |
4 |
1 |
0 |
Survival |
12 died, 1 lost to follow-up |
14 died, 1 alive, 2 lost to follow-up |
Died |
Died |
2008 WHO sub-classification |
CMML (n = 6) MDS/MPN-U (n = 5) aCML (n = 2) |
De novo/AML MRC (n = 9); sAML: h/o myeloid neoplasm (n = 5) and therapy-related (n = 2); AML, relapsed post-allogeneic SCT, unclassifiable (n = 1) |
RAEB-2 |
Post-ET MF |
aCML, atypical chronic myeloid leukemia; AML, acute myeloid leukemia; AML MRC, AML with myelodysplasia-related changes; CMML, chronic myelomonocytic leukemia; MCV, mean corpuscular volume; MDS, myelodysplastic syndrome; MDS/MPN-U, myelodysplastic/myeloproliferative neoplasm unclassifiable; Post-Et MF, Post-essential thrombocythemia myelofibrosis; RAEB, refractory anemia with excess blasts; sAML, secondary AML; SCT, stem cell transplant.
We analyzed three cases with isolated i(17q) using whole-exome sequencing and found mutations in genes that included SETBP1, SRSF2, and ASXL1 (Supplementary Table 1). We performed mutation analysis of 33 genes using a combination of next-generation sequencing based analysis on all 32 cases targeting the coding regions of 28 genes; and Sanger sequencing for splicing factors, CEBPA, MDM4 and SETBP1. The mutation results are shown in Figure 1. The details are provided in Supplementary Table 2. The median number of mutations per sample was 3. Genes with the highest frequency of mutations included: SETBP1 (17/29; 59%), ASXL1 (17/31; 55%), SRSF2 (16/29; 55%), and NRAS (10/32; 31%). Mutations in genes that directly affect the RAS pathway was noted in 18 (56%) cases and included: NRAS (n = 10, 31%), PTPN11 (n = 4; 13%), KRAS (n = 3; 9%), and HRAS (n = 1; 3%). These four mutations were mutually exclusive with each other, and in all but 1 case, they were exclusive with other genes affecting RAS/MAPK signaling pathway including EGFR, FLT3, KIT, and BRAF, but not SETBP1. There was a significant association between mutations in SETBP1 and RAS (p = 0.003). There was a trend towards association between mutations in SETBP1 and SRSF2 (p = 0.07); and SETBP1 and ASXL1 (p = 0.07). Eight of 28 patients (29%) showed concurrent mutations in ASXL1, SRSF2, SETBP1 and RAS. Ten patients (35%) showed concurrent mutations in SRSF2, SETBP1 and RAS. Notably, mutations in TET2, TP53 and U2AF1 were rare. SF3B1 mutations were absent. SETBP1 and TP53 mutations were mutually exclusive as reported by others [3, 9, 10] except in 1 case where TP53 mutation was observed at 5% variant allelic frequency (VAF) in a post allogeneic stem cell transplant setting. In addition to the findings shown in Figure 1, none of the tested cases had mutations in CEBPA (n = 15) or MDM4 (n = 12).
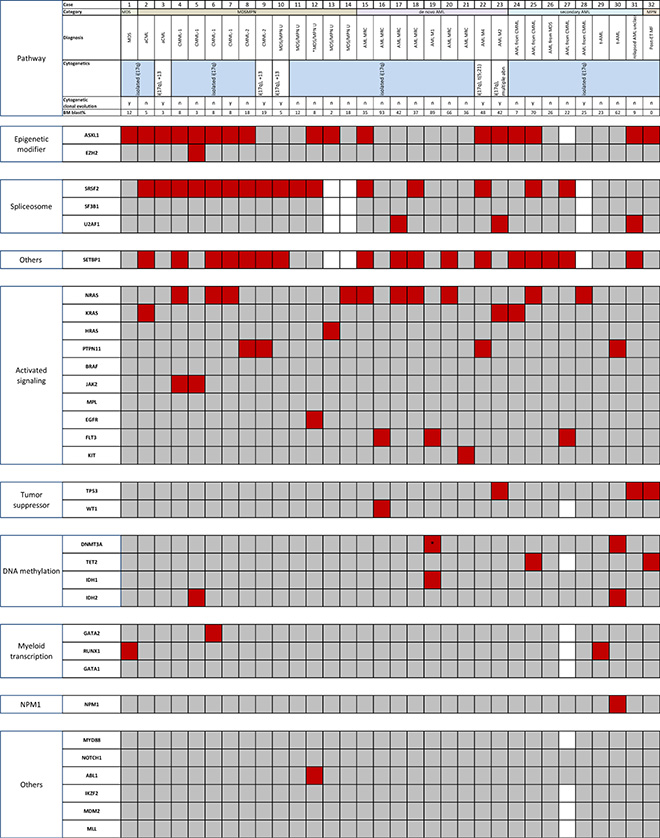
Figure 1: Mutational analysis of 32 cases of myeloid neoplasm with isolated i(17q) (red, mutation; gray, wild-type; white, not tested). Genes are segregated based on the biologic functional categories on the left. The upper panels denote the case number, diagnostic categories per 2008 WHO classification and clonal cytogenetic abnormalities. Case #31 could not be sub-classified. Isochromosome (17q) was detected on a post-allogeneic stem cell transplant sample that showed relapsed AML. The pre-transplant cytogenetic studies were diploid, and molecular studies were unavailable. The patient also had a history of breast carcinoma treated with chemotherapy. Cases 7, 9 and 31 (highlighted in lavender) also underwent whole-exome sequencing. *represents a non-R882 DNMT3A mutation. aCML, atypical chronic myeloid leukemia; AML, acute myeloid leukemia; AML MRC, AML with myelodysplasia-related changes; CMML, chronic myelomonocytic leukemia; ET, Essential thrombocythemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; MDS/MPN, myelodysplastic/myeloproliferative neoplasm; MDS/MPN-U, myelodysplastic/myeloproliferative neoplasm unclassifiable; MF, myelofibrosis; RAEB, refractory anemia with excess blasts; sAML, secondary AML; SCT, stem cell transplant.
Segregation of genes based on the functional classification per Cancer Genome Atlas Research Network showed that mutations occurred in highest frequencies in genes involved in activating signaling pathways (mostly RAS), followed by splicing factor-encoding and epigenetic modifier genes (Supplementary Figure 1) [11]. Of the 4 most frequently mutated genes, VAF could be estimated for the genes of RAS pathway and ASXL1 as these were analyzed by NGS. The median VAF of NRAS and ASXL1 mutations in our cohort were 41.7 (range, 5–49.2) and 32.8 (range, 6–72) respectively. Eleven cases showed mutations in both RAS and ASXL1. In 8 of these cases, the VAF of RAS mutation was either similar to or higher than of ASXL1. The VAFs of SETBP1 and SRSF2 could only be assessed in 3 cases that underwent whole exome sequencing. The results showed that all mutated genes had a similar VAF in all cases.
The median overall survival (OS) from the onset of disease was 22.1 months, and median OS from the onset of i(17q) was 9.4 months (Kaplan-Meier curves shown in Supplementary Figure 3). There were no apparent differences in the mutation profile between the cases of AML and MDS/MPN, although the numbers are too small for significance.
Within this study cohort, 5 cases had an initial diploid karyotype and subsequently acquired i(17q). We performed mutation analysis on these patients at various time points during the evolution from diploid to karyotype showing i(17q) (as shown in Figure 2). Within the interval between the initial analysis to the development of i(17q) abnormality, all patients had undergone treatment with drugs that included decitabine, Ruxolitinib, hydroxyurea, dasatinib and lenalidomide. In one case, the i(17q) abnormality was acquired post-allogeneic stem cell transplantation. Mutations in ASXL1, SRSF2 and RAS appear stable over time. ASXL1 mutations were present in all 5 cases at both diploid and i(17q) stages of karyotypic evolution. SRSF2 mutations were also present at both diploid and i(17q) stages in 3 of 5 patients. Mutations in NRAS (n = 2) and PTPN11 (n = 1) at high allelic frequencies (> 35%) were also stable, being present at the diploid and i(17q) stages. In 3 patients, additional mutations were observed at the time of i(17q) abnormality. These mutations occurred in SETBP1 (n = 2), KRAS (n = 1, subclonal, 7%) and TP53 (n = 1, subclonal, 5%; post allogeneic stem cell transplant) genes. The only case that showed SETBP1 mutation at the time of diploid karytoype developed i(17q) within 5 months.
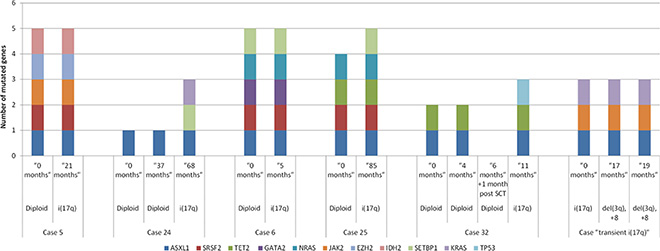
Figure 2: Sequential mutation analysis of myeloid neoplasms during the evolution from a diploid karyotype to the development of isolated i(17q) abnormality. In case #5, i(17q) was observed after allogeneic stem cell transplant. In case #6, i(17q) abnormality was transient (noted for only a duration of 6 months).
DISCUSSION
Myeloid neoplasms associated with isolated i(17q) show characteristic clinicopathologic features and aggressive behavior. In order to study molecular aberrations associated with the presence of i(17q), and to exclude the confounding effects of additional karyotypic aberrations, we included cases showing i(17q) as a sole karyotypic abnormality and performed a comprehensive mutational analysis. The genes with the highest mutation frequency included ASXL1, SRSF2, SETBP1 and RAS signaling pathway genes, noted in more than 50% of the samples. In most of our cases, RAS mutations were detected at high allelic frequencies with a median of 42.1%. We performed pyrosequencing on majority of cases to confirm RAS mutations. These results have clinical implications because patients with myeloid neoplasms associated with i(17q) have a poor outcome and RAS and splicing factor mutations provide potential targets for treatment.
We also observed a high frequency of ASXL1, SETBP1 and SRSF2 mutations in myeloid neoplasms associated with i(17q). Similar observations were presented by Meggendorfer et al. at the 2015 ASH meeting (abstract# 1656; session: 636. Myelodysplastic Syndromes – Basic and Translational Studies: Poster I) [12]. However, the frequency of RAS mutations reported by the authors was much lower. This discordance may be related to the differences in the patient cohort, as their study included 63 patients, of which only 27 had isolated i(17q). The remainder of patients in their study had additional abnormalities including some patients with a complex karyotype.
Mutations in ASXL1 (3/9, 33%), SRSF2 (3/9, 33%), and SETBP1 (5/9, 56%) were also observed in de novo AML cases (n = 9). Lindsley et al. have shown that mutations in ASXL1 and splicing factors, as observed in our cohort, are highly specific for secondary AML [13]. Gene mutations in NPM1, DNMT3A, CEBPA, IDH1 and FLT3, that are reportedly common in de novo AML, were infrequent. Thus, the presence of i(17q) may indicate an underlying MDS or MDS/MPN, at least in a subset of AML cases, even in de novo setting. The only case with an NPM1 mutation was a patient with relapsed AML status post allogeneic stem cell transplant. The patient had a history of breast carcinoma treated with chemotherapy and AML with a diploid karyotype. Cytogenetic studies showed i(17q) in 7 of 20 metaphases. In the presence of NPM1 mutation, i(17q) abnormality may have been transiently acquired due to the damaging effects of chemotherapy.
Another notable finding in our study was the rarity of mutations in TET2 and other genes involved in DNA methylation pathway. Our cohort included 13 MDS/MPN cases and 4 AML cases with a prior history of MDS/MPN. TET2 mutations are highly prevalent in cases of aCML, CMML and MDS/MPN unclassifiable (41%, 62% and 26% respectively) as shown by several authors, although these studies have not looked into cases with isolated i(17q) abnormality in particular [14–25].
A limitation of our study is the low detection sensitivity of Sanger sequencing used for assessment of SRSF2 and SETBP1. The Sanger sequencing technique has a detection sensitivity of 10–20%, compared to NGS-based assay (5%). Hence, the mutational frequencies of SRSF2 and SETBP1 may be underestimated in our results. A strength of this study is that it is the first study to perform sequential mutational analysis of patients over the course of karyotypic progression, from diploid to i(17q). The findings in these 5 patients suggest that ASXL1, SRSF2 and RAS pathway gene mutations occur prior to i(17q). In a subset of cases, SETBP1 mutation may be associated with the development of i(17q) abnormality in these neoplasms. Interestingly, we found 1 patient with a history of primary myelofibrosis who transiently acquired i(17q) abnormality. This abnormality was present in 4/20 and 1/20 metaphases (confirmed by uniallelic TP53 by FISH), at two different time points, 6 months apart. Bone marrow morphology showed myelofibrosis associated with trilineage dysplasia and monocytosis. Subsequently, the i(17q) abnormality disappeared (also confirmed by array based comparative hybridization and FISH studies), and the patient acquired del(3)(q12q24) and trisomy 8. The mutation profile remained the same. NGS sequencing based mutation analysis at 3 different time points showed mutations in JAK2, ASXL1 and KRAS; SRSF2 and SETBP1 were wild-type (Figure 2). These results support that i(17q) abnormality is associated with mutated SRSF2 and SETBP1.
Our results provide insights into the molecular consequences of i(17q), which leads to the obligatory loss of a single TP53 allele located at 17p13.1 [7]. We have confirmed that TP53 mutations, even at low allelic frequencies, are exceedingly rare in myeloid neoplasms with isolated i(17q) using next-generation sequencing based mutation analysis. This finding suggests involvement by mechanisms other than TP53 mutation-induced genomic instability [1, 3, 9, 10]. Alternatively, TP53 dysfunction may be the result of copy number changes or alterations in RNA and protein expression of other molecules of TP53 pathway that have not been explored. At the same time, a potential role for several hematopoiesis-related candidate genes on 17q has not been explored. These genes include NF1, RARA, G-CSF, MPO, ERBB2, and BRCA1 genes and miRNAs. Of note, SRSF2 located at 17q25.1 was mutated in 55% cases in this study. SRSF2 P95 mutation is a gain-of-function mutation that causes alterations in the binding affinity of SRSF2 to a number of target pre-mRNA transcripts thereby affecting alternative splicing and gene expression [26]. Sequential mutational analysis suggests that SRSF2 mutation, along with ASXL1, occurs early during the disease, and is present at the time the karyotype is diploid. In these cases, the formation of i(17q) leads to a higher dosage of mutant SRSF2 and alterations of gene expression on a larger scale. The combination of SRSF2 and ASXL1 mutations, together with mutations in SETBP1 may contribute to the dismal outcome.
In summary, we have performed a comprehensive gene mutational analysis in the largest series of myeloid neoplasms with isolated i(17q) abnormality to date. Our results show a high frequency of mutations in SRSF2, SETBP1, ASXL1 and NRAS genes. In cases where isolated i(17q) abnormality was acquired during the disease course, SRSF2 and ASXL1 mutations preceded i(17q) detection, whereas SETBP1 mutations were associated with i(17q). The acquisition of i(17q) abnormality and the presence of these adverse molecular markers may contribute to the poorer outcome of these patients. SRSF2 and RAS pathway gene mutations may be potential therapeutic targets for aggressive management of these neoplasms.
MATERIALS AND METHODS
Patient cohort
This study was approved by the institutional review board. Informed consent was obtained from all patients. We retrieved all cases of myeloid neoplasm with i(17q) as the primary cytogenetic abnormality from our database from 1998–2014. Clinical data was collected from the medical records. All relevant pathology material including peripheral blood, bone marrow smears and biopsy sections, and immunocytochemical stains were reviewed. All cases were classified according to the 2008 WHO classification.
Cytogenetic analysis
Conventional cytogenetic studies were performed on metaphase cells prepared from bone marrow aspirate smears using standard techniques. The results were reported using the International System for Human Cytogenetic Nomenclature (2009 and 2013).
Genetic analysis
We performed amplicon-based next-generation sequencing (NGS) targeting the coding regions of a panel of 28 genes implicated in myeloid neoplasms using MiSeq platform (Illumina, San Diego, CA) on archived DNA extracted from fresh bone marrow aspirate samples. We used 250 ng of DNA to prepare the genomic library. The genes included in this panel are as follows: ABL1, ASXL1, BRAF, DNMT3A, FGFR, EZH2, FLT3, GATA1, GATA2, HRAS, IDH1, IDH2, IKZF2, JAK2, KIT, KRAS, MDM2, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PTPN11, RUNX1, TET2, TP53, and WT1. Following successful library generation and purification, equal quantities of DNA from each sample were used for multiplex paired-end sequencing on MiSeq personal genome sequencer using the MiSeq Reagent Kit v2 (500 cycles). Human genome build 19 (hg19) was used as the reference for sequence alignment. MiSeq Reporter Software 2.2 and Integrative Genomics Viewer (IGV) were used for variant calling and visualization respectively. Successful sequencing was indicated by a Q30 score of more than 85%. For this study, variants with more than 5% allelic frequency with a minimum coverage of 250X in both directions were included. Since matched normal tissue was not tested, data from literature and COSMIC database was used to infer somatic nature. Single nucleotide polymorphisms listed in dbSNP 137 and 138 and 10K genome project were excluded. The functional significance of all variants was analyzed using PolyPhen-2 website (http://genetics.bwh.harvard.edu/pph2/); only variants that resulted in “probably damaging” were included. Selected variants were confirmed using an alternate platform, such as Sanger sequencing, pyrosequencing or capillary electrophoresis (data not shown). Mutation analyses of 5 additional genes that are not included in the NGS panel (SETBP1, CEBPA, MDM4, and splicing factor genes SRSF2, SF3B1 and U2AF1) were performed by Sanger sequencing. In 25 cases, FLT3 mutational analysis for internal tandem duplication was also performed by capillary electrophoresis.
Statistical analysis
Frequencies and percentages were calculated for categorical variables, and means and standard deviation were calculated for continuous variables. Fisher’s exact test was used to assess the association between categorical variables and mutation rate (not corrected for multiple testing). All statistical analyses were performed using SAS 9.3 for Windows.
ACKNOWLEDGMENTS
We thank Dr. Gary Lu for his support.
GRANT SUPPORT
This work was supported, in part, by The University of Texas MD Anderson Cancer Center Faculty Startup Fund (to RK-S), The University of Texas MD Anderson Cancer Center Specialized Program of Research Excellence (SPORE) in Leukemia CA100632-11 (to CB-R), The University of Texas MD Anderson Moon Shots Program and the MD Anderson Cancer Center Support Grant (CCSG) CA016672 and the Cancer Center Support Grant (NCI P30 CA016672).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Kanagal-Shamanna R, Bueso-Ramos CE, Barkoh B, Lu G, Wang S, Garcia-Manero G, Vadhan-Raj S, Hoehn D, Medeiros LJ, Yin CC. Myeloid neoplasms with isolated isochromosome 17q represent a clinicopathologic entity associated with myelodysplastic/myeloproliferative features, a high risk of leukemic transformation, and wild-type TP53. Cancer. 2012; 118:2879–2888.
2. McClure RF, Dewald GW, Hoyer JD, Hanson CA. Isolated isochromosome 17q: a distinct type of mixed myeloproliferative disorder/myelodysplastic syndrome with an aggressive clinical course. Br J Haematol. 1999; 106:445–454.
3. Visconte V, Tabarroki A, Zhang L, Hasrouni E, Gerace C, Frum R, Ai J, Advani AS, Duong HK, Kalaycio M, Saunthararajah Y, Sekeres MA, His ED, et al. Clinicopathologic and molecular characterization of myeloid neoplasms harboring isochromosome 17(q10). Am J Haematol. 2014; 89:862.
4. Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, Granada I, Hildebrandt B, Slovak ML, Ohyashiki K, Steidl C, Fonatsch C, Pfeilstocker M, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012; 30:820–829.
5. Sánchez-Castro J, Marco-Betés V, Gómez-Arbonés X, Arenillas L, Valcarcel D, Vallespí T, Costa D, Nomdedeu B, Jimenez MJ, Granada I. Characterization and prognostic implication of 17 chromosome abnormalities in myelodysplastic syndrome. Leuk Res. 2013; 37:769–776.
6. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK, National Cancer Research Institute Adult Leukaemia Working G. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010; 116:354–365.
7. Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Hoglund M, Johansson B, Hagemeijer A, Park SS, Mitelman F, Lupski JR, Fioretos T. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet. 2004; 74:1–10.
8. Fioretos T, Strombeck B, Sandberg T, Johansson B, Billstrom R, Borg A, Nilsson PG, Van Den Berghe H, Hagemeijer A, Mitelman F, Hoglund M. Isochromosome 17q in blast crisis of chronic myeloid leukemia and in other hematologic malignancies is the result of clustered breakpoints in 17p11 and is not associated with coding TP53 mutations. Blood. 1999; 94:225–232.
9. Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, Haferlach T, Schnittger S. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013; 27:1852–1860.
10. Adema V, Larrayoz MJ, Calasanz MJ, Palomo L, Patino-Garcia A, Agirre X, Hernandez-Rivas JM, Lumbreras E, Buno I, Martinez-Laperche C, Mallo M, Garcia O, Alvarez S, et al. Correlation of myelodysplastic syndromes with i(17) (q10) and TP53 and SETBP1 mutations. Br J Haematol. 2015; 171:137–141.
11. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Engl J Med. 2013; 368:2059–2074.
12. Meggendorfer M HC, Kern W, Schnittger S, Haferlach T. The Landscape of Myeloid Neoplasms with Isochromosome 17q Discloses a Specific Mutation Profile and Is Characterized By an Accumulation of Prognostically Adverse Molecular Markers. Blood. 2015; 126.
13. Lindsley R, Mar B, Mazzola E, Grauman P, Shareef S, Allen S, Pigneux A, Wetzler M, Stuart R, Erba H. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2014; 125:1367–76.
14. Meggendorfer MHT, Jeromin S, Haferlach C, Kern W, Schnittger S. Molecular Analyses of MDS/MPN Overlap Entities According to WHO Classification Reveal a Distinct Molecular Pattern for MDS/MPN, Unclassifiable. Blood. 2014; 124.
15. Savona MR, Malcovati L, Komrokji R, Tiu RV, Mughal TI, Orazi A, Kiladjian JJ, Padron E, Solary E, Tibes R, Itzykson R, Cazzola M, Mesa R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015; 125:1857–1865.
16. Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, Berthon C, Ades L, Fenaux P, Beyne-Rauzy O, Vey N, Braun T, Haferlach T, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013; 31:2428–2436.
17. Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, Kohlmann A, Alpermann T, Yoshida K, Ogawa S. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012; 120:3080–3088.
18. Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W, Schnittger S. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica. 2014; 99:e244–246.
19. Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, Knudson RA, Ketterling RP, Tefferi A, Solary E. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014; 28:2206–2212.
20. Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, Ng KP, Gudmundsson KO, Vishwakarma BA, Jerez A, Gomez-Segui I, Takahashi M, Shiraishi Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013; 45:942–946.
21. Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, Ketterling RP, Pardanani A, Tefferi A. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013; 27:2100–2102.
22. Kohlmann A, Grossmann V, Klein H-U, Schindela S, Weiss T, Kazak B, Dicker F, Schnittger S, Dugas M, Kern W. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010; 28:3858–3865.
23. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, Antolini L, Mologni L, Donadoni C, Papaemmanuil E, Schnittger S, Kim DW, Boultwood J, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013; 45:18–24.
24. Wang SA, Hasserjian RP, Fox PS, Rogers HJ, Geyer JT, Chabot-Richards D, Weinzierl E, Hatem J, Jaso J, Kanagal-Shamanna R, Stingo FC, Patel KP, Mehrotra M, et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood. 2014; 123:2645–2651.
25. Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013; 122:1707–1711.
26. Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci USA. 2015; 112:E4726–4734.