
INTRODUCTION
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of gastrointestinal (GI) tract, but they are relatively uncommon ~10–20 per GISTs per million people [1]. The major mechanisms of tumorigenesis of GISTs are oncogenic mutations of the KIT or PDGFRA genes and these account for 80–90% of GISTs [2]. According to the National Comprehensive Cancer Network guidelines, GISTs with no mutations in exons 9, 11, 13, and 17 of the KIT gene and exons 12, and 18 of the PDGFRA gene are defined as KIT/PDGFRA wild-type GISTs, and they represent 15–20% of GISTs [3]. However, pathogenic mechanisms and molecular characteristics of KIT/PDGFRA wild-type GISTs are poorly understood. Recently, frequent succinate dehydrogenase (SDH) mutations were identified in KIT/PDGFRA wild-type GISTs, especially in pediatric patients, which was considered a subtype of GISTs [4, 5].
Imatinib mesylate (imatinib) is the only first-line drug for GIST treatment and efficacy depends on KIT or PDGFRA genotypes [6, 7]. Drug response in KIT/PDGFRA wild-type patients is poor and data show that 70% of these patients are resistant to imatinib. Thus, ~30% of KIT/PDGFRA wild-type patients may benefit from imatinib, suggesting susceptible factors in wild-type individuals or that some mutations are not detected with current sequencing methods.
Several studies have explored possible mechanisms of imatinib resistance and GIST pathogenesis. Miranda’s group reported that KRAS and BRAF mutations existed in GIST patients that these predicted imatinib resistant in in vitro experiments [8]; however, no KRAS mutation was found in a cohort of 514 cases [9]. PTEN-deficient expression and PI3K/AKT pathway activation were shown to be important to imatinib resistance [10, 11]. A subset GISTs tested in vitro had KIT mutations in exon 8, and these cells were sensitive to imatinib [12]. To explore unknown mutations and possible pathogenic mechanisms of KIT/PDGFRA wild-type GISTs, we sequenced KIT and PDGFRA genes and critical molecules downstream of these genes using targeted next-generation sequencing (NGS).
RESULTS
Patient characteristics
We studied 146 KIT/PDGFRA wild-type patients and these data appear in Table 1. All patients had records of primary tumor sites, tumor sizes and mitosis, however, CD117, DOG-1, and CD34 expressions were collected from 139, 93, and 132 patients, respectively. Among 146 patients, 12 patients received imatinib palliative treatment after diagnosis, 2 patients received imatinib neoadjuvant therapy followed by surgery, 18 patients received imatinib adjuvant therapy after surgery, 2 patients received sunitinib palliative treatment when diagnosis, and the rest 112 patients received surgery alone or no any treatment when diagnosis.
Table 1: Characteristics of patients
Characteristics |
No. of patients (%) |
|---|---|
Sex |
|
Male |
69 (47.3) |
Female |
77 (52.7) |
Age (years) |
|
Median |
52 |
Range |
16–78 |
Primary sites |
|
Stomach |
56 (38.3) |
Small bowel |
41 (28.1) |
Abdominal/pelvic cavity/omentum |
27 (18.5) |
Others* |
22 (15.1) |
Long diameter of tumor (cm) |
|
≤ 2 |
12 (8.2) |
2–5 |
37 (25.3) |
5–10 |
63 (43.2) |
> 10 |
34 (23.3) |
Mitosis |
|
≤ 5/50HPF |
66 (45.2) |
6–10/50HPF |
50 (34.2) |
> 10/50HPF |
30 (20.5) |
CD117 expression |
|
Positive |
108 (74.0) |
Negative |
31 (21.2) |
NA |
7 (4.8) |
DOG-1 expression |
|
Positive |
66 (45.2) |
Negative |
27 (18.5) |
NA |
53 (36.3) |
CD34 expression |
|
Positive |
96 (65.8) |
Negative |
36 (24.6) |
NA |
14 (9.6) |
Note: *including colon, rectum, renal, etc. NA: none available.
Quality control of next-generation sequencing
Average coverage of next-generation sequencing was > 200× and sequence content of four bases T, C, A, G was well called and balanced. The actual GC distribution over all sequences was similar with theoretical distributions. Furthermore, the proportion of N appearing in a sequence was low and the distribution of fragment sizes was uniform (chiefly 100 bp; range: 99–101 bp). This guaranteed accuracy of sequencing and established a foundation for data elucidation (Supplementary Figure S2).
Variants of 48 genes in 146 KIT/PDGFRA wild-type GISTs
Among the 146 patients, 119 had at least one nonsynonymous or deletion variant in the captured gene set with a median variant of 2 (range: 0–34; Supplementary Figure S3). For 48 captured genes, TP53 (43.15%), ROS1 (20.55%), NF1 (19.86%), ATRX (19.86%), and KIT (19.18%) were the five most frequently variants. Moreover, other variants with prevalence > 10% were identified including BRCA2 (17.12%), BRAF (16.44%), TSC1 (15.07%), MET (15.07%), F5 (13.01%), DEPDC5 (13.01%), PDGFRA (12.33%), SLTM (11.64%), KDR (11.64%), ALK (11.64%), and DDR2 (10.27%) (Supplementary Figure S4). Whether these variants participated in tumorigenesis of GISTs is unclear.
Variation profile of 48 genes based on clinicopathological features
The heat maps of 48 genes based on different features were analyzed using R software (Supplementary Figure S5–S13). Data from hierarchical cluster analysis for all patients (Figure S5) suggested the following stratified cluster analysis based on sex (Figure S6), age (Figure S7), tumor sites (Figure S8), tumor sizes (Figure S9), mitosis (Figure S10), CD117 (Figure S11), DOG-1 (Figure S12), and CD34 (Figure S13) expression. No obvious differences in variation profile were noted among different features.
KIT/PDGFRA mutations in KIT/PDGFRA wild-type GISTs and correlations to imatinib sensitivity
For hot spots for KIT (exons 9, 11, 13, and 17) and PDGFRA (exons 12, and 18) genes, 19 (13.0%) and 4 (2.7%) of 146 KIT/PDGFRA wild-type GISTs patients carried KIT and PDGFRA mutations, respectively, with a mutation ratio (mutratio, mutcount/coverage) less than 25%. The mutation types contained W557G (n = 1), W557R (n = 2), V559D (n = 1), Del 557–558 (n = 3), L576P (n = 6), Del 579 (n = 1) in exon 11 of KIT gene, A814S (n = 1), N822K (n = 4) in exon 17 of KIT gene, and R585K (n = 1) in exon 12 of PDGFRA gene, D842V (n = 2), D842Y (n = 1) in exon 18 of PDGFRA gene (Table 2). These mutations were mutually exclusive, and all mutation types in exon 11, N822K in exon 17 of the KIT gene, and D842V in exon 18 of the PDGFRA gene were frequently reported in GISTs. Based on our previous large scale analysis [13], only one mutation type codon (a 502–503 duplication) was found in exon 9 of the KIT gene. No mutation in exon 9 was found in this study and mutations were not found in exon 13 of the KIT gene.
Table 2: Hot spots mutations found by next-generation sequencing in KIT/PDGFRA wild-type GISTs
Case |
Exon of gene |
Mutation type |
MutRatio* |
|---|---|---|---|
2014-BZ0157 |
11 of KIT |
Del 557–558 |
12.1% |
2014-BZ0027 |
11 of KIT |
L576P |
20.4% |
2014-BZ0129 |
11 of KIT |
L576P |
17.7% |
2014-BZ0132 |
11 of KIT |
W557R |
22.8% |
2014-BZ0069 |
11 of KIT |
Del 557–558 |
16.9% |
2014-BZ0184 |
11 of KIT |
L576P |
11.8% |
2014-BZ0020 |
11 of KIT |
W557R |
24.1% |
2014-BZ0075 |
11 of KIT |
Del 579 |
14.4% |
2014-BZ0093 |
11 of KIT |
W557G |
18.4% |
2014-BZ0019 |
11 of KIT |
V559D |
23.5% |
2014-BZ0021 |
11 of KIT |
L576P |
10.5% |
2014-BZ0128 |
11 of KIT |
L576P |
11.7% |
2014-BZ0166 |
11 of KIT |
L576P |
13.4% |
2014-BZ0017 |
11 of KIT |
Del 557–558 |
11.0% |
2014-BZ0028 |
17 of KIT |
N822K |
11.9% |
2014-BZ0162 |
17 of KIT |
N822K |
19.8% |
2014-BZ0096 |
17 of KIT |
A814S |
10.8% |
2014-BZ0024 |
17 of KIT |
N822K |
22.8% |
2014-BZ0135 |
17 of KIT |
N822K |
10.1% |
2014-BZ0127 |
12 of PDGFRA |
R585K |
22.9% |
2014-BZ0015 |
18 of PDGFRA |
D842V |
19.1% |
2014-BZ0114 |
18 of PDGFRA |
D842Y |
10.3% |
2014-BZ0038 |
18 of PDGFRA |
D842V |
18.1% |
Note: *MutRatio = MutCount/Coverage ×100%.
Among 146 patients, 12 received imatinib palliative therapy and 11 patients (91.7%) were evaluable for clinical response. Two of 3 patients (66.7%) with KIT mutations identified by NGS achieved partial response (PR), while only 1 of 8 patients (12.5%) without KIT mutations reached PR, suggesting NGS could identify a portion of patients eligible for imatinib therapy.
In addition to hot spots, other exons of the KIT or PDGFRA genes were confirmed to carry missense or deletion mutations (Figure 1), which could co-exist with each other and with mutations in hot spots. A total of 19 patients were confirmed to carry 33 types of mutations in other exons of the KIT or PDGFRA genes. Six patients carried mutations both in hot spots and in other exons of the KIT or PDGFRA genes but the potential functions of these mutations are unclear.
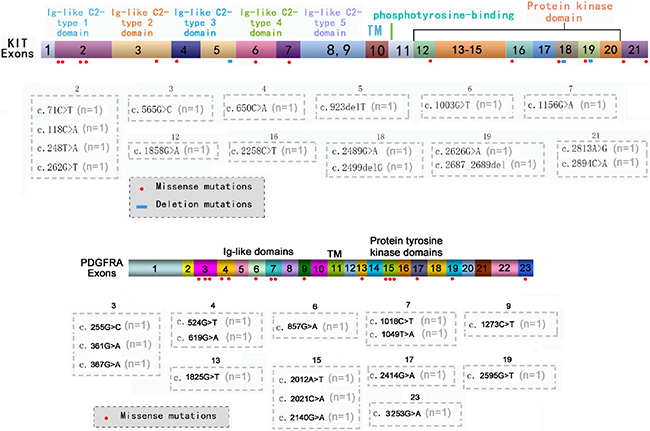
Figure 1: Mutations located in other exons of KIT/PDGFRA genes. The distribution of missense or deletion mutations identified by NGS in other exons of KIT (A) or PDGFRA (B) genes.
Intratumoral KIT mutational heterogeneity
Data show that 19 KIT/PDGFRA wild-type patients carried mutations in hot spots of the KIT gene, which were not identified using Sanger sequencing. Polyclonal features of KIT mutations have been reported previously, and consequently, formalin-fixed paraffin-embedded sections of each patient were macrodissected into four regions based on H & E staining. Genomic DNA was extracted from macrodissected samples followed by PCR amplification and Sanger sequencing. Data show that 4 of 19 patients had intratumoral KIT mutational heterogeneity (Figure 2). In addition to wild-type cells, four patients carried mutant cells with different mutation types containing W557G, W557R, L576P, and N822K, data consisted with Table 2 data. The mutational heterogeneity potentially triggered mechanisms of polyclonal evolution and metastasis, as well as different therapeutic sensitivities.
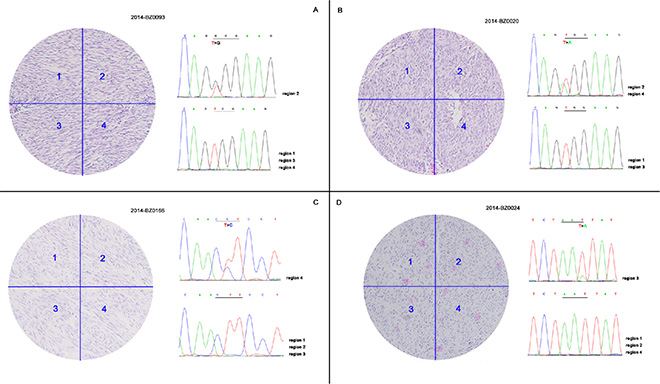
Figure 2: Intratumoral KIT mutational heterogeneity of 4 patients. FFPE sections of 19 patients identified to carry hot spots mutations of KIT by NGS were macrodissected into four regions followed by PCR amplification and Sanger sequencing. Four of 19 patients demonstrated intratumoral KIT mutational heterogeneity with concurrent wild-type and mutant tumor cells.
KRAS and BRAF mutations in KIT/PDGFRA wild-type GISTs
Important downstream molecules of KIT/PDGFRA, KRAS and BRAF mutations have been reported in different studies. For common mutations of KRAS (exon 2) and BRAF (exon 15), one patient carried a G13D mutation in exon 2 of the KRAS gene, and six carried mutations (5 carried V600E mutations and one carried the A598D mutation) in exon 15 of the BRAF gene in this study, and this excluded hot spots of KIT/PDGFRA genes. Various mutations were also observed in other exons of KRAS and BRAF genes, which could co-exist with each other and with common mutations (Supplementary Table S2).
DISCUSSION
Oncogenic mutations of KIT/PDGFRA genes are the major mechanisms of tumorigenesis of GISTs, and KIT/PDGFRA genotypes are correlated to imatinib efficacy [3]. At present, patients with KIT/PDGFRA wild-type GISTs are usually not treated with imatinib but 30% of these patients may benefit from imatinib, likely due to unidentified susceptibility factors or incomplete sequencing data with Sanger sequencing. Here, we sequenced whole exomes of 48 genes containing KIT/PDGFRA with targeted NGS in a sample of KIT/PDGFRA wild-type GISTs.
For hot spots in the KIT and PDGFRA genes, 19 KIT mutations and 4 PDGFRA mutations were identified in KIT/PDGFRA wild-type cases according NGS. These mutations were mutually exclusive and most mutations (n = 20) were point mutations. Mutations in exons 9 and 13 of the KIT gene were not identified. NGS may not be suitable for identifying deletion mutations in large fragments located in exon 11 of the KIT gene. Xu and colleagues reported that KIT and PDGFRA mutations in 121 samples by NGS were 49.6% and 0.8%, respectively [14], significantly fewer mutations than previously reported [7, 13]. However, Gleeson FC and colleagues reported that targeted NGS of cytology samples from 19 patients with GISTs is clinically feasible [15]. Our data show that NGS did not identify deletion mutations in exon 11 of the KIT gene in several samples. Thus, NGS should be optimized for clinical practice for assessing GISTs.
The five most frequently mutated genes were TP53, ROS1, NF1, ATRX, and KIT, but whether these mutation genes participate in tumorigenesis of GISTs warrants more study. Hechtman’s group analyzed 8 patients with wild-type KIT/PDGFRA, and 8 cases had loss of SDHB expression and carried ARIDIA, TP53, and other gene alterations [16]. Pantaleo and colleagues reported that SDH mutations were frequently observed in patients with KIT/PDGFRA wild-type GISTs [5], especially in pediatric patients [4]. Patients with wild-type KIT/PDGFRA had high expression of IGF pathway family members [17, 18], which offered an alternative therapeutic strategy for treating KIT/PDGFRA wild-type GISTs. Studies with small samples indicated that genomic profiles between KIT/PDGFRA wild-type and mutant GISTs were different [19, 20]. Until now, a pathogenic mechanism to explain KIT/PDGFRA wild-type GISTs was not known and comparative analyses of whole genomic sequencing of large samples of KIT/PDGFRA wild-type and mutant GISTs should provide insights about KIT/PDGFRA wild-type GISTs.
Hot spots in KIT/PDGFRA genes were mutually exclusive and mutations in other exons of the KIT/PDGFRA gene co-existed with each other and with hot spot mutations. Mechanisms or reasons for exclusive hot spot mutations were similar to KRAS/BRAF exclusive mutations in colorectal cancer [21]. For GISTs, only primary hot spot mutations in KIT/PDGFRA genes were excluded, however, primary and secondary mutations were concomitantly located in hot spots of the KIT/PDGFRA genes [22].
Previous research suggests that KIT mutations have polyclonal features [2, 23], so FFPE sections of 19 patients identified to carry hot spots mutations of the KIT gene were macrodissected into four regions followed by PCR amplification and Sanger sequencing. Four of the 19 cases had intratumoral KIT mutational heterogeneity with concurrent wild-type and mutant tumor cells, which may have triggered polyclonal evolution and metastasis and unique therapeutic sensitivity.
Downstream pathways of KIT/PDGFRA genes may be important sources for explaining drug sensitivity. KRAS and BRAF mutations have been reported but data were inconsistent [8, 24]. Miranda C and colleagues reported that KRAS (5%) and BRAF (2%) mutations were identified in GISTs carrying KIT/PDGFRA mutations, not in KIT/PDGFRA wild-type GISTs [8]. Agaimy A and colleagues demonstrated BRAF mutations (7%) were detected in KIT/PDGFRA wild-type GISTs not in mutant GISTs [25]. Our previous results presented that KRAS (1.7%) and BRAF (1.7%) mutations were detected in KIT/PDGFRA wild-type GISTs not in mutant GISTs (unpublished data). Whether KRAS/BRAF mutations can predict imatinib resistance in GISTs requires validation in a larger sample size.
Nannini and coworkers described KIT/PDGFRA wild-type GIST as a set of different diseases sustained by specific molecular alterations not yet known [25]. Although our results were somewhat superficial, we identified patients eligible for imatinib therapy by NGS. Intratumoral KIT mutational heterogeneity may be monitored to evaluate imatinib efficacy.
MATERIALS AND METHODS
Patients and samples
From October 2001 to January 2014, a total of 1,080 individuals with GISTs were screened for KIT or PDGFRA mutations at Peking University Cancer Hospital. We identified 185 KIT/PDGFRA wild-type patients and among these 39 were excluded due to lack of genomic DNA data or failure of library preparation for NGS (Figure 3). All clinicopathological features and treatments were retrospectively assessed from medical records, and samples were taken prior to imatinib or sunitinib treatment. Written informed consent was obtained from all patients for sample study, and the study was approved by the Medical Ethics Committee of Peking University Cancer Hospital.
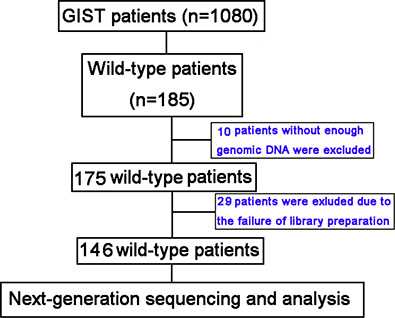
Figure 3: Patient screening flow chart. From 1,080 patients studied for KIT/PDGFRA mutations, 185 were KIT/PDGFRA wild-type and 146 were analyzed using targeted next-generation sequencing (NGS). There was insufficient genomic DNA or the library preparation failed for 39 patients.
DNA extraction and mutation detection of KIT and PDGFRA genes
Genomic DNA was extracted from formalin-mixed paraffin-embedded tumor specimens with tumor cells > 50% based on H & E staining using QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany), and stored at −80°C for future use after quantification with Nanodrop 2000. Mutational analysis in exons 9, 11, 13, and 17 of KIT gene and exons 12, and 18 of PDGFRA gene was performed with PCR amplification and Sanger sequencing according to published procedures [7]. Each sample was sequenced at least twice.
Library preparation and targeted NGS
Genomic DNA (3 µg) was used for library preparation according to the manufacturer’s instruction (MyGenostics, Beijing, China), and the final library size of 350–450 bp, containing adapter sequences was used in the following experiment.
A panel of 48 genes (Supplementary Table S1) including KIT and PDGFRA genes was captured with OncoCap Enrichment System (MyGenostics, Beijing, China) based on previously published methods [26]. After enrichment, libraries were sequenced on an Illumina Solexa HiSeq 2000 sequencer for paired reads of 100 bp followed by data retrieval using Solexa QA package and a cutadapt program (http://code.google.com/p/cutadapt/).
Bioinformatic analysis
Supplementary Figure S1 shows the experimental design. Briefly, illumina clean reads (the sequencing quality > 20 and read length > 80 bp) were aligned to each human reference genome (hg19) using the BWA program and quality scores were recalibrated and realigned to references using GATK software. Duplicated reads were removed using Sequence Alignment/Map tools (SAMtools) and only uniquely mapping reads were used for variation assessment. Low frequency variants were identified on the basis of the bam file. The SAMtools mpileup command was used to generate pileup files. VarScan was performed to assess pileup files from tumor samples to heuristically call for a genotype at positions achieving certain thresholds of coverage and quality.
SNVs were detected and genotyped with the GATK UnifiedGenotyper in single-sample mode, and variants were filtered with GATK VariantFiltration module (with filters “QUAL < 50.0 & QD < 5.0 & HRun > 10 & DP < 4” and parameters –cluster 3 -window 10). Indels were detected with GATK IndelGenotyperV2 and filtered with a custom python module that removed sites with amax_cons_av ≥ 1.9 (maximum average number of mismatches across reads supporting the indel) or max_cons_nqs_av_mm ≥ 0.2 (maximum average mismatch rate in the 5-bp NQS window around the indel, across indel-supporting reads).
Low frequency variants were identified with Fisher’s exact test. Post-calling filters are based on read depth, sequencing quality, mismatches, and overlap with indels. Variation annotations such as locations (exonic, intronic and intergenic region) and effects on protein coding (synonymous, missense, nonsense, frameshift), were performed with an in-house developed bioinformatics tool with RefSeq (hg19, from UCSC) and UCSC annotation (http://www.ncbi.nlm.nih.gov/refseq/). Variants with the following conditions could be analyzed: (1) located within an exonic or splicing region; (2) nonsynonymous; (3) MAF < 0.05 in the European 1,000 genomes variant database; (4) reads supporting the variation ≥ 5; and (5) variation frequency > 0.01.
Experimental validation
Macrodissection of tumor sections and subsequent PCR amplification and Sanger sequencing was used to validate detected missense mutations by NGS. PCR products were sequenced with a 3730XL genetic analyzer and Chromas software was used to analyze sequencing results.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by Novartis, the National Natural Science Foundation of China (No. 81172110, 81301853), and the National High Technology Research and Development Program (No. 2012AA 02A 504). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Joensuu H, Fletcher C, Dimitrijevic S, Silberman S, Roberts P, Demetri G. Management of malignant gastrointestinal stromal tumours. Lancet Oncol. 2002; 3:655–64.
2. Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, Speidel N, Pietsch T, Buettner R, Pink D, Reichardt P, Hohenberger P. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006; 12:1743–9.
3. von Mehren M, Benjamin RS, Bui MM, Casper ES, Conrad EU 3rd, DeLaney TF, Ganjoo KN, George S, Gonzalez R, Heslin MJ, Kane JM 3rd, Mayerson J, McGarry SV, et al. Soft tissue sarcoma, version 2. 2012: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2012; 10:951–60.
4. Oudijk L, Gaal J, Korpershoek E, van Nederveen FH, Kelly L, Schiavon G, Verweij J, Mathijssen RH, den Bakker MA, Oldenburg RA, van Loon RL, O’Sullivan MJ, de Krijger RR, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013; 26:456–63.
5. Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, Saponara M, Formica S, Ceccarelli C, Casadio R, Rossi G, Bertolini F, Santini D, et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet. 2014; 22:32–9.
6. Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008; 26:5360–7.
7. Gao J, Dang Y, Sun N, Li J, Shen L. C-KIT mutations were closely associated with the response to Imatinib in Chinese advanced gastrointestinal stromal tumor patients. Med Oncol. 2012; 29:3039–45.
8. Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, Saletti P, Mazzucchelli L, Pilotti S, Pierotti MA, Tamborini E, Greco A, Frattini M. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012; 18:1769–76.
9. Lasota J, Xi L, Coates T, Dennis R, Evbuomwan MO, Wang ZF, Raffeld M, Miettinen M. No KRAS mutations found in gastrointestinal stromal tumors (GISTs): molecular genetic study of 514 cases. Mod Pathol. 2013; 26:1488–91.
10. Quattrone A, Wozniak A, Dewaele B, Floris G, Vanspauwen V, Van Looy T, Schöffski P, Rutkowski P, Sciot R, Debiec-Rychter M. Frequent mono-allelic loss associated with deficient PTEN expression in imatinib-resistant gastrointestinal stromaltumors. Mod Pathol. 2014; 27:1510–20.
11. Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007; 26:7560–8.
12. Huss S, Künstlinger H, Wardelmann E, Kleine MA, Binot E, Merkelbach-Bruse S, Rüdiger T, Mittler J, Hartmann W, Büttner R, Schildhaus HU. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del). Mod Pathol. 2013; 26:1004–12.
13. Li Y, Gao J, Tian Y, Li J, Shen L. Mutation profiles of c-kit/PDGFRα and its associations with clinicopathological characteristics in Chinese gastrointestinal stromal tumors: analysis of 827 cases. Zhonghua Wei Chang Wai Ke Za Zhi. 2015; 18:332–7.
14. Xu Z, Huo X, Tang C, Ye H, Nandakumar V, Lou F, Zhang D, Jiang S, Sun H, Dong H, Zhang G, Liu Z, Dong Z, et al. Frequent KIT mutations in human gastrointestinal stromal tumors. Sci Rep. 2014; 4:5907.
15. Gleeson FC, Kipp BR, Kerr SE, Voss JS, Graham RP, Campion MB, Minot DM, Tu ZJ, Klee EW, Lazaridis KN, Henry MR, Levy MJ. Kinase genotype analysis of gastric gastrointestinal stromal tumor cytology samples using targeted next-generation sequencing. Clin Gastroenterol Hepatol. 2015; 13:202–6.
16. Hechtman JF, Zehir A, Mitchell T, Borsu L, Singer S, Tap W, Oultache A, Ladanyi M, Nafa K. Novel oncogene and tumor suppressor mutations in KIT and PDGFRA wild type gastrointestinal stromal tumor revealed by next generation sequencing. Genes Chromosomes Cancer. 2015; 54:177–84.
17. Beadling C, Patterson J, Justusson E, Nelson D, Pantaleo MA, Hornick JL, Chacón M, Corless CL, Heinrich MC. Gene expression of the IGF pathway family distinguishes subsets of gastrointestinal stromal tumors wild type forKIT and PDGFRA. Cancer Med. 2013; 2:21–31.
18. Nannini M, Astolfi A, Paterini P, Urbini M, Santini D, Catena F, Indio V, Casadio R, Pinna AD, Biasco G, Pantaleo MA. Expression of IGF-1 receptor in KIT/PDGF receptor-α wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol. 2013; 9:121–6.
19. Nannini M, Astolfi A, Urbini M, Indio V, Santini D, Heinrich MC, Corless CL, Ceccarelli C, Saponara M, Mandrioli A, Lolli C, Ercolani G, Brandi G, et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer. 2014; 14:685.
20. Astolfi A, Nannini M, Pantaleo MA, Di Battista M, Heinrich MC, Santini D, Catena F, Corless CL, Maleddu A, Saponara M, Lolli C, Di Scioscio V, Formica S, et al. A molecular portrait of gastrointestinal stromal tumors: an integrative analysis of gene expression profiling and high-resolution genomic copy number. Lab Invest. 2010; 90:1285–94.
21. Gao J, Sun ZW, Li YY, Shen L. Mutations of KRAS and BRAF in Chinese patients with colorectal carcinoma: analyses of 966 cases. Zhonghua Bing Li Xue Za Zhi. 2012; 41:579–83.
22. Gao J, Tian Y, Li J, Sun N, Yuan J, Shen L. Secondary mutations of c-KIT contribute to to acquired resistance to imatinib and decrease efficacy of sunitinib in Chinese patients with gastrointestinal stromal tumors. Med Oncol. 2013; 30:522.
23. Chen H, Hirota S, Isozaki K, Sun H, Ohashi A, Kinoshita K, O’Brien P, Kapusta L, Dardick I, Obayashi T, Okazaki T, Shinomura Y, Matsuzawa Y, et al. Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut. 2002; 51:793–6.
24. Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foerster A, Hartmann A, Bihl MP. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol. 2009; 62:613–6.
25. Nannini M, Biasco G, Astolfi A, Pantaleo MA. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet. 2013; 50:653–61.
26. Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, Wolfgang CL, Klein AP, Diaz LA Jr, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011; 3:92ra66.