Introduction
Protein phosphorylation on tyrosine residues catalyzed by tyrosine kinases (TKs) has evolved as an important mechanism to coordinate cell communication in multicellular organisms [1]. This molecular process is highly regulated in vivo because only less than 1% of mammalian proteins are phosphorylated on Tyr residues. Tyr phosphorylation (pTyr)-dependent signal transduction results from the combination of three molecular actions: initiation of the pTyr signal induced by the TK (the writer), propagation of the signal through recognition of the phosphorylated protein by a SRC Homology 2 (SH2) domain-containing protein (the reader) and control of the signal by phosphatases that dephosphorylate the substrate (the eraser) [1]. The human genome encodes about 90 TKs, 100 SH2 domain-containing proteins and 40 tyrosine phosphatases [2]. The tyrosine kinome consists of receptor and non-receptor TK families [3]. The Receptor TK (RTK) family includes receptors for growth factors and factors involved in cell adhesion and motility, cell survival and metabolism [4]. The non-receptor TK family comprises cytoplasmic TKs (CTKs) that mainly mediate signals transduced by receptors devoid of TK activity [5]. These non-enzymatic receptors can be activated by a large variety of extracellular cues, such as hormones, neurotransmitters, cytokines and antigens as well as components of the extracellular matrix, to regulate cell activity. Deregulation of this pTyr-dependent signaling has a strong effect in cancer [6]. Over 50% of TKs display aberrant activities in human tumors due to overexpression or somatic mutation of the corresponding gene and these molecular alterations are thought to be the main cause of oncogenic induction driven by abnormal pTyr-dependent protein phosphorylation in human cells [6]. Hence, small inhibitors or antibodies that target this molecular process have become an attractive therapeutic strategy in oncology [7]. However, TK inhibitors have shown variable effects in the clinic, suggesting that TK deregulation alone may not be always sufficient to induce oncogenesis and to predict tumor response to TK inhibitors [7].
In light to the complexity of TK signaling, this mechanism has been reported to also be negatively controlled by a class of “readers” composed of small adaptor proteins, as originally reported by Yoshimura et al [8]. Interestingly, inactivation of this regulatory mechanism has recently emerged as an additional important mechanism of oncogenic induction driven by aberrant TK activities. This review outlines the role of this class of adaptors in the control of pTyr-dependent signaling in normal and tumorigenic conditions and discusses the potential therapeutic implications of these novel findings.
Negative regulation of TK signaling by small adaptors in non-transformed cells
Adaptor proteins define an important class of “readers” in the transmission of pTyr-dependent signaling. These proteins do not have enzymatic/catalytic or transcriptional activities, but act as molecular platforms that coordinate signaling events [9]. They mostly function as flexible molecular scaffolds that mediate protein–protein and protein–lipid interactions through interaction domains and binding motifs in their modular structure. These motifs allow specific interactions with effector proteins to regulate their localization and/or activities. Specifically, by binding to and bringing into proximity two or more signaling proteins, they can coordinate and regulate signaling events in space and time. Signaling proteins with “adaptor” function can be classified in three broad families: i) scaffold proteins that regulate a large number of effector proteins, ii) transmembrane proteins that dock signaling effectors at the plasma membrane, and iii) small cytoplasmic adaptors that bind two partners together [9]. This review will focus on this last class. The first small adaptors identified were either novel regulators of cell growth induced by growth factors or oncogenes (GRB2 and SHC) [10, 11], or transforming products of retroviruses (v-CRK) [12]. It is now established that this family of positive regulators includes members of the CRK, DAPP1, GRB2, NCK, SHB, SH3BP2, SHC, SH2D1-5, SLP76 and STAP families [2]. They can function through association with cognate effectors and subsequent targeting of the complex to the plasma membrane for activation. For example, in the cytoplasm, the adaptor GRB2 is constitutively associated with SOS, an activator of the small GTP-binding protein RAS, and upon growth factor stimulation, the complex is directed to the membrane by interaction via the SH2 domain with an RTK that enables RAS signaling [13].
More recently, a new group of small adaptors has emerged that inhibits TK-induced cell responses. One of the first examples was cytokine inducible SH2-domain containing protein (CIS) [8] that was discovered while searching for new immediate early response genes induced by cytokines. CIS is a small molecule that contains an SH2 domain and has some homology with the transcription factor STAT5. When overexpressed, the cytokine response is inhibited. Since then, several adaptors with similar properties have been identified, among which members of the SOCS, SLAP, SH2B, GRB7 and MIG6 families are included (Figure 1). Except for SOCS family members, these adaptors have emerged late during evolution to finely tune signaling and to prevent unwanted cellular responses. They mostly define feed-back loops, as revealed by loss of function experiments in mice combined with biochemical studies in cultured cells. They can negatively regulate TK signaling through direct inhibition of the TK catalytic activity, competitive inhibition of TK association with downstream signaling proteins, or destabilization of specific components of the pTyr signaling cascade via the recruitment of ubiquitination factors (Figure 2). A detailed analysis of their role in TK signaling is presented below.
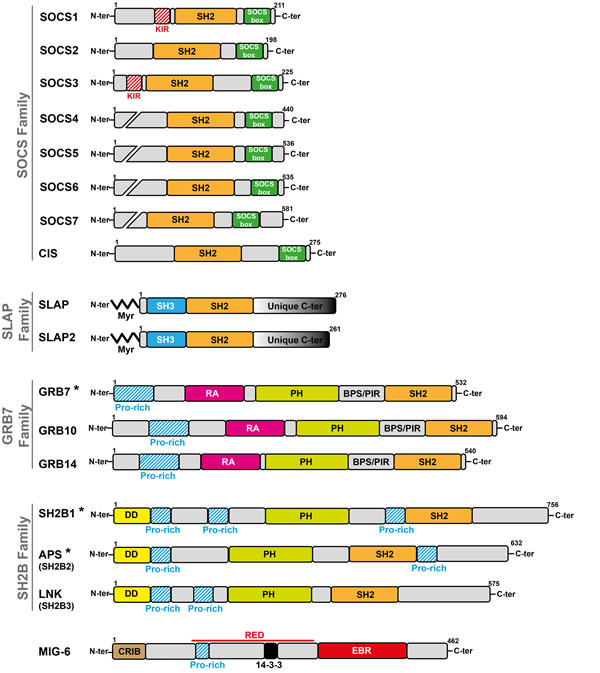
Figure 1: Modular structure of small adaptors that negatively regulate TK signaling. The size of adaptor proteins (number of amino acids), presence of specific homology domains, sequences and myristoylation sites (Myr) are indicated. (*) indicates that the adaptor of this subfamily positively regulates TK signaling.
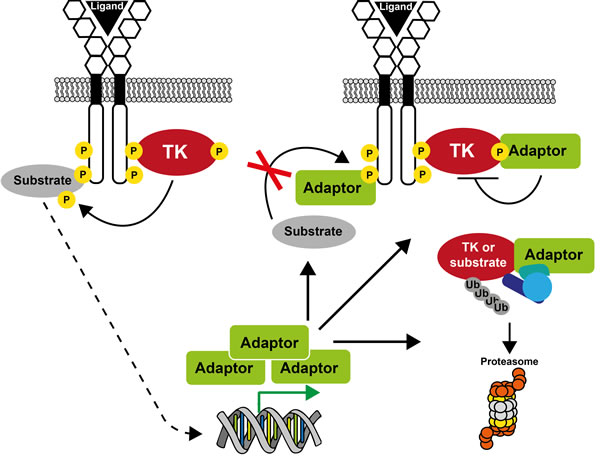
Figure 2: A unifying model on how small adaptors control TK signaling. Sustained receptor stimulation generally induces expression of small adaptor proteins. As a result, the adaptor protein can inhibit TK signaling by competing with effectors/substrates for receptor binding, by directly inhibiting TK activity, or by promoting substrate/TK degradation via its association with a specific ubiquitination factor.
SOCS family
The Suppressor of Cytokine Signaling (SOCS) family was originally identified based on the ability of its members to inhibit cytokine signaling and to bind to CTKs of the JAK family [14-16]. The mammalian SOCS family comprises eight members (SOCS1 to SOCS7 and CIS). They are composed of a variable N-terminal sequence, a central SH2 domain that shows homology with the STAT SH2, and a conserved sequence of about 40 amino acids, called “SOCS box” (Figure 1). This last domain is involved in the formation of Elongin/Cullin/SOCS box-type complexes that function as E3 ubiquitin ligases to promote the ubiquitination of targeted substrates. SOCS1 and SOCS3 additionally possess a short motif upstream of the SH2 domain that is called kinase-inhibitory region (KIR) and is critical for the inhibition of TK activity. SOCS proteins have emerged early in the animal kingdom. For instance, the C. elegans genome contains a single gene similar to SOCS6 and SOCS7, while the Drosophila genome already contains three SOCS genes (SOCS16D, SOCS36E and SOCS44A) that are homologous to SOCS4-7. Drosophila SOCS proteins share similar functions with their human homologues. SOCS36E is both a target and a negative regulator of JAK/STAT signaling, while SOCS44A and SOCS36E are regulators of Epidermal Growth Factor (EGF) receptor signaling [17]. SOCS family members are expressed in a wide range of tissues with variations and specificities [18]. Cytokine and growth factor receptors induce SOCS gene expression, mostly via the STAT pathway, which in turn counteracts their signaling activity [19, 20].
Most SOCS family members can link the associated substrates to the ubiquitination machinery via the SOCS box. A wide range of substrates are targeted for proteolytic degradation, including CTKs of the JAK family, JAK-associated cytokine receptors, RTKs [19, 21], MAL (a component of the inflammatory toll-like receptor signaling cascade) and IRS1/2 (components of the metabolic insulin signaling pathway) [22, 23]. However, SOCS-mediated ubiquitination can also promote internalization and routing of receptors, for instance growth hormone and granulocyte-colony stimulating factor (G-CSF) receptors [24, 25]. This results in the receptor and, possibly, JAK turnover. SOCS proteins can also suppress signaling by competing with downstream signal transducers for binding to shared phosphorylated motifs of the activated receptors. Particularly, SOCS proteins block STAT recruitment to cytokine receptors by masking the STAT binding sites of such receptors [26]. Finally, only SOCS1 and SOCS3 can bind to JAKs via the SH2 domain and directly inhibit JAK catalytic activity via the KIR, which acts as a pseudo-substrate that impairs substrate accessibility [27, 28]. SOCS1 can directly bind to the phosphorylated Tyr1007 residue in JAK2 activation loop [29]. SOCS3 shows weak affinity for JAK, but binds to the cytokine receptor in close proximity of the kinase. Recent structural and mechanistic analyses have revealed the molecular basis of JAK inhibition by SOCS3 [30, 31]. Upon binding to JAKs, the unstructured KIR domain of SOCS3 adopts an extended β-strand-like conformation that sits in the catalytic pocket of the kinase, resulting in prevention of substrate binding or phosphorylation. SOCS3 can inhibit JAK1, JAK2 and TYK2 via its KIR, but not JAK3. This is due to the absence of an evolutionarily conserved Gly-Gln-Met sequence (GQM motif) in JAK3 kinase domain. SOCS3 interacts with this motif through its SH2 and KIR domains. To date, there are no structural data on SOCS1, but high sequence conservation in the KIR domain with SOCS3 suggests that it may inhibit JAKs through the same mechanism.
Genetically manipulated mice were used to determine the physiological functions of SOCS proteins and demonstrated that they have a crucial role in immunological processes and in growth control in accordance with in vitro observations (for review see [18]). For example, Socs1-deficient mice die within three weeks of birth due to severe systemic inflammation, resulting from uncontrolled interferon-γ (IFN-γ) signaling [32, 33]. Mice lacking Socs3 die perinatally due to defective placental formation, whereas conditional Socs3 depletion induces inflammatory and metabolic disorders [34-36]. Leukemia Inhibitory Factor Receptor (LIFR) gene deficiency was able to rescue the Socs3 knockout placental defect and embryonic lethality, establishing SOCS3 as an essential regulator of LIFR signaling during placental formation [37]. Lethality of Socs1 or Socs3 deficient mice also revealed specific functions for these SOCS proteins that are not compensated by other family members. Socs2-deficient mice develop gigantism due to enhanced responses to growth hormone [38]. No Cis or Socs4 knockout mouse model has been reported by now, but Cis transgenic mice exhibit growth retardation [39]. Socs5 knockout mice showed no abnormalities, indicating possible redundancy between SOCS family members [40]. Socs6 deficient mice displayed an 8-10% reduction in body weight, but, despite the in vitro data, Socs6 knockout mice did not display any alterations in glucose metabolism [41]. Again, redundancy between SOCS family members may play a role in the absence of a phenotype in these mice. Finally, there have been conflicting reports regarding the in vivo function of SOCS7, probably due to differences in the genetic background of the respective mouse knockouts [42, 43]; however these observations suggested a role of SOCS7 in insulin signaling, consistent with the findings that SOCS7 can interact with the IR and their adaptor proteins.
SLAP family
SRC-Like Adaptor Protein (SLAP) was identified by yeast two-hybrid genetic screening using the cytoplasmic domain of the RTK EPHA2 as bait [44]. SLAP2 was then discovered using bioinformatic and functional screening approaches [45-47]. SLAP displays considerable structural homology with SRC, but lacks the kinase domain. It has a unique myristoylated N terminus for membrane localization, followed by the SH3 and SH2 domains with high homology to those of SRC family TKs (about 50% identity) and a unique C terminus involved in the interaction with downstream signaling proteins, such as the ubiquitination factor CBL (Figure 1) [48]. SLAP proteins have emerged during vertebrate evolution by SRC duplication [2]. SLAP is strongly expressed in the hematopoietic system, epithelial intestine, lung and brain and more weakly in other tissues. SLAP2 expression is more specific to the hematopoietic tissue and lungs [48]. SLAP is implicated in the negative regulation of RTK and immunoreceptor signaling. Surprisingly, the expression of SLAP proteins does not seem to be induced by receptor stimulation and, thus, it may not define a negative-feedback loop requiring transcriptional activation and novel protein synthesis, as reported for SOCS proteins. Their expression is rather induced during cell differentiation, for instance during thymocyte development [49].
SLAP negatively regulates SRC signaling by targeting SRC substrates for degradation or by competing with SRC for association with upstream receptors. Due to the homology with the SRC SH2 domain, SLAP SH2 competes with SRC SH2 for platelet-derived growth factor (PDGF) receptor interaction and thereby impairs SRC-mediated mitogenic signaling [50]. However, SLAP does not share the SRC-binding of FLT3 or EPHA2 suggesting that this competitive mechanism does not operate for all RTKs [51, 52]. SLAP proteins are also implicated in the degradation of SRC-like signaling components by facilitating the recruitment of ubiquitin ligases. For instance, they inhibit T cell receptor (TCR) and B cell receptor (BCR) activities by docking CBL to components of these receptor complexes and inducing their degradation [45, 53-55]. This mechanism may require phosphorylation by the TK LCK and the SLAP SH2 domain [53]. SLAP SH3 domain must also be intact for optimal attenuation of TCR signaling [56]. A recent structural analysis of SLAP2 revealed that the SH3 and SH2 domains directly interact through a beta-sheet formation and this may be important for SH2 binding activity [57]. Due to the high identity between SLAP proteins, this mechanism may be also operative in SLAP and could explain, at least in part, the important role of SLAP SH3 in TCR signaling. In addition to its role in lymphocytes, SLAP also controls F-actin assembly induced by PDGF in a CBL-dependent manner in fibroblasts, probably through destabilization of an upstream regulator of RAC GTPases [58]. Finally, SLAP also participates in CBL-dependent ubiquitination, internalization and/or degradation of many receptors, including GM-CSF receptor [59], FLT3 [51], KIT [60] and colony stimulating factor 1 (CSF-1) receptor [61]. SLAP also interacts with erythropoietin (EPO) receptor and negatively regulates erythroid terminal differentiation by unknown mechanisms [62].
Several in vivo SLAP functions have been revealed by Slap1 and 2 knockout experiments in mice (for reviews, see [48, 64, 65]). Surprisingly, these animals are healthy and without apparent physical defects [49, 55, 59] in contrast to the embryonic defects observed in Src, Fyn and Yes triple knockout mice [63]. These observations suggest that, in contrast to SFKs, SLAP proteins are not essential for embryonic development. SLAP and SLAP2 functions have been most intensely investigated in hematopoietic tissue and mainly in the context of lymphocyte signaling where they are strongly expressed. These in vivo analyses supported a model in which SLAP family members dampen immunoreceptor (TCR and BCR) signaling, thereby influencing lymphocytes development. For instance, disruption of the Slap1 gene showed that SLAP participates in a novel mechanism of TCR downregulation at the CD4+CD8+ stage and regulates positive selection. These in vivo analyses together with results obtained from cultured cells support a model in which SLAP regulates lymphocytes development by hindering immunoreceptor signaling [48, 64, 65]).
GRB7 family
The growth factor receptor bound protein-7/10/14 (GRB7/10/14) adaptors were originally identified as partners of activated EGFR [66-68]. Within the GRB7/10/14 family, GRB10 and GRB14 are major negative regulators of insulin and insulin growth factor 1 (IGF1) effects on metabolism and growth [69, 70]. Conversely, GRB7 is implicated in the transduction of FAK- and EPHB1-induced cell migration [71]. Therefore, in this review we will focus on GRB10 and GRB14. These proteins possess several signaling modules, including a poly-Pro region close to their N-terminus, a RAS-associating (RA) domain, followed by a Plekstrin Homology (PH) and a SH2 domain. They also include a region known as BPS (between the PH and SH2 domains) or PIR (phosphorylated insulin receptor-interacting region), which is unique to this adaptor family (Figure 1). Similarly to SLAP, GRB7/10/14 structure and function were acquired relatively late, in evolutionary terms [69]. GRB10 is an imprinted gene that is predominantly expressed from the maternally inherited allele in most human and mouse tissues, except brain [72]. GRB10 and 14 have similar patterns of expression in many tissues, but with some differences. Consistent with their role in insulin signaling, GRB10 and 14 are highly expressed in two major insulin target tissues: skeletal muscle and adipose tissue. GRB10 is also strongly expressed in pancreas, moderately in cardiac muscle and brain, and weakly in many other tissues. GRB14 is highly expressed in heart and liver, and is also detected in pancreas, kidney, gonads, brain and placenta [69]. Although it is unknown whether GRB10 and 14 are transcriptionally regulated by insulin, GRB14 can be up-regulated by insulin [73, 74]. Recently, a couple of studies revealed that mTORC1 directly phosphorylates GRB10, thereby enhancing its stability and, thus, acting as a negative regulator of insulin or IGF-1 signaling [75, 76].
GRB10 and 14 control TK-dependent signaling by a mechanism similar to the one described for SOCS proteins. The BPS domain of GRB10 and 14 specifically interacts with insulin and IGF-1 receptors (IR and IGF-1R) and inhibits their TK activity. Specifically, crystallographic studies showed that the BPS region of GRB14 acts as a pseudo-substrate that binds to the TK domain of IR and inhibits IR catalytic domain [77]. The SH2 domain potentiates the inhibitory effect of the BPS region by binding to the phosphorylated activation loop of the IR catalytic domain, while the RA and PH domains are involved in GRB14 membrane recruitment and participate in IR negative regulation [77, 78]. The interaction between GRB10/14 and IR/IGF-1R can also interfere with downstream partners. For example, GRB10 disrupts the association of insulin receptor substrate (IRS) proteins with IR [79]. In cells expressing IR and PTP1B, GRB14 co-expression maintains the pTyr of the IR activation loop, while favoring dephosphorylation of Tyr972 in the juxtamembrane domain [80]. As Tyr972 is the main docking site for IRS1, this may contribute to GRB14 ability to inhibit the association of IRS1 with IR. GRB10 might regulate the internalization and degradation of its target proteins, such as IGF-1R, by interacting with the ubiquitination factor NEDD4 [81-83]. GRB10 and 14 also regulate signaling initiated by other RTKs [70]. For example, GRB14 inhibits Fibroblasts Growth Factor (FGF) receptor-mediated signaling by altering FGF-induced PLCγ phosphorylation and activation [84]. Mechanistically, GRB14 SH2 binding to FGF receptor induces a conformational change that unmasks a PLCγ binding motif on GRB14, allowing PLCγ trapping and inactivation. On the other hand, GRB14 enhances RET-mediated signaling [85] and GRB10 promotes signaling mediated by PDGFR, vascular endothelial growth factor receptor (VEGFR) and KIT via not well known mechanisms [86-88].
In agreement with their role in insulin signaling, Grb10 and Grb14 knockout mice show improved insulin/IGF sensitivity [89-91]. The Grb10-/- phenotype also includes embryo and placenta overgrowth as well as increased skeletal muscle and pancreatic β-cell mass [92]. Conversely, transgenic mice overexpressing the Grb10 maternal allele show postnatal growth retardation, hyperinsulinemia, glucose intolerance and insulin resistance [93]. Accordingly with mouse phenotypes, modulations of GRB10/14 expression in cultured cells show that these adaptors inhibit insulin/IGF-induced receptor activation, revealed by the inhibition of endogenous substrate phosphorylation, such as IRS1, IRS2, SHC and p62DOK, and the inhibition of downstream signaling pathways such as PI3K/AKT and ERK1/2 (reviewed in [69, 70]).
SH2B family
The SH2B adaptor family includes SH2B1, APS (SH2B2) and LNK (SH2B3). LNK has a well-characterized negative function in JAK2 and TCR signaling [20, 94, 95], while the negative role of the other members remains controversial and may depend on the nature of the TK-dependent signaling. The molecular bases of these specificities are poorly understood, but they may involve alternative splicing of the SH2B1 and APS genes [70, 96]. SH2B family members contain an N-terminal dimerization domain, Pro-rich regions, a central PH domain and an SH2 domain (Figure 1). While highly homologous, the SH2 domains of these adaptors display distinct biochemical characteristics resulting in specific signaling functions. For example, the SH2B1 SH2 domain binds preferentially to JAK2, whereas the APS SH2 domain has higher affinity for the insulin receptor [97]. This specificity is attributable to the difference in the oligomeric states of the two SH2 domains: monomeric for SH2B1 and dimeric for APS [98]. Multiple consensus sites for Tyr and Ser/Thr phosphorylation are also found in SH2B adaptors. The SH2B family is evolutionarily conserved from insects to humans [99, 100]. Drosophila has only one dSH2B protein that is structurally similar to SH2B1. The core functions of dSH2B (e.g., growth, reproduction and metabolism) seem to be evolutionarily conserved, but the three mammalian SH2B family members also have new specific functions. Indeed, deletion of SH2B2 or SH2B3 does not alter growth or glucose metabolism in mice, in contrast to SH2B1 knockout [101]. LNK is mainly expressed in hematopoietic tissues, but also in testis, brain and muscle [102]. Similar to other negative regulators, LNK expression is induced following JAK/STAT or Tumor Necrosis Factor (TNF) α signaling, consistent with the existence of a transcription-dependent negative control loop [103, 104].
LNK negatively regulates TCR signaling in a LCK-dependent manner [95] and the activity of cytokine receptors, such as the EPO and thrombopoietin (TPO) receptors, in a JAK2-dependent manner [105, 106]. The role of LNK in TPO signaling has been especially well studied but the molecular mechanism of TPO-mediated signaling inhibition by LNK has not been fully elucidated. Mechanistically, the LNK SH2 domain binds to a JAK2 sequence located within the pseudo-kinase domain linker, upon JAK2 phosphorylation on Tyr813 induced by TPO [107]. The PH domain of LNK is also involved in this regulatory process [108]. As a result, LNK is a potent inhibitor of JAK2/STAT signaling during hematopoiesis. In hematopoietic stem cells, LNK/JAK2 interaction is further negatively regulated by 14-3-3 adaptor proteins. Binding of 14-3-3 to LNK requires phosphorylation of two serine residues in LNK. This binding abrogates LNK/JAK2 interaction, thereby affecting its inhibitory function [109]. LNK also controls signaling through RTKs, including KIT [110, 111], PDGFR [112], FLT3 [113] and CSF1 receptor [114]. For instance, the interaction of LNK SH2 with the phosphorylated Tyr568 present in KIT juxta-membrane domain [115] results in reduction of downstream signaling including the attenuation of GAB2 phosphorylation and MAPK activation [110]. The mechanism underlying this molecular process was not further elucidated in this study. Finally, LNK also interferes with TRKA signaling, possibly by competing with the positive-acting SH2B1 and APS for binding to TRKA [116].
Consistent with LNK negative role in cytokine receptor signaling reported in cell culture, Lnk-deficient mice are viable but display abnormal cytokine signaling and aberrations in hematopoiesis [102, 110]. Specifically, these studies revealed defects during lymphopoiesis, erythropoiesis, megakaryopoiesis, mast cell development and macrophages proliferation as well as in hematopoietic stem cell (HSC) expansion (for review, see [117, 118]). Moreover, Lnk and Tpo double-knockout mice revealed opposite physiological roles for LNK and TPO in HSC expansion [119] and LNK overexpression inhibits megakaryocyte development in mice, consistently with a role in TPO/JAK2 signaling regulation [120]. However, although high levels of LNK are present in non-hematopoietic tissues such as the testis, brain and muscle, no noticeable phenotypes of genetically modified mice were reported in these tissues, questioning about the physiological role of LNK in non-hematopoietic compartments.
MIG6
Mitogen-inducible gene 6 (MIG6, also known as receptor-associated late transducer, RALT, or ERBB feedback inhibitor 1, ERRFI1) was first cloned from a hydrocortisone-induced rat liver cDNA library [121]. MIG6 was then identified as a partner and an endogenous inhibitor of EGFR (also named ERBB1) and ERBB2, two members of the ERBB receptor family [122, 123]. MIG6 is a cytosolic protein composed of a CDC42/RAC interaction and binding (CRIB) domain, Pro-rich motifs, a 14-3-3 protein binding motif, the RALT endocytic domain (RED) and the ERBB-binding region (EBR) (Figure 1). MIG6 is present in higher order species, suggesting that its expression has been acquired during evolution for regulating more complex signaling circuits [124]. MIG6 is an immediate early response gene that is expressed in various tissues [124]. Its expression can be induced through a RAS/MAPK-dependent mechanism upon sustained stimulation by a broad spectrum of extracellular cues and as such, MIG6 defines a negative regulatory feedback loop that is tightly regulated. Indeed, the transcriptional activation of ERRFI1 (the gene encoding MIG6) appears to be transient [122] and MIG6 protein is degraded through a proteasome-dependent mechanism upon interaction with DNAJB1 [125, 126].
MIG6 inhibits ERBB kinase activities by direct binding of its EBR region to ERBB catalytic domain. Mechanistically, the N-terminal portion of EBR, called segment 1 (amino acids 336-364), interacts with ERBB C-lobe that overlaps with a site critical for forming the asymmetric dimer with the N-lobe of the other ERBB subunit. As a result, MIG6 locks ERBB in a catalytically inactive conformation and hinders its dimerization required for signal transduction [127]. Recently, it has been shown that phosphorylation on Tyr394 and Tyr395, which are located in segment 2 (amino acids 365-412) of EBR C-terminus, is critical for effective interaction of MIG6 with EGFR [128, 129]. A structural analysis suggests that EBR segment 1 binds across the base of the C lobe of ERBB and segment 2 forms a β-hairpin-like element that occupies the peptide-substrate binding site, once phosphorylated. Interestingly, Tyr394 and Tyr395 are phosphorylated by EGFR and SRC respectively [130], thus creating a forward feedback loop in the control of ERBB activity by MIG6. MIG6 also plays a role in ERBB receptor trafficking. Upon EBR docking onto the receptor, the RED domain interacts with the endocytic proteins AP-2 and intersectins to induce clathrin-mediated ERBB endocytosis [131]. Moreover, MIG6 mediates ERBB receptor sorting to late endosomes by binding to syntaxin 8, thus promoting ERBB lysosomal degradation [132]. MIG6 expression also negatively regulates MET signaling [133]. This activity requires an intact CRIB motif, suggesting that MIG6 acts, at least in part, distally from MET, possibly by inhibiting Rho-like GTPases. Surprisingly, MIG6 can also activate the CTK ABL by interacting with its kinase domain via the EBR domain. MIG6-dependent ABL activation occurs only when ERBB is inactive and leads to the induction of ABL apoptotic function [134]. This sophisticated mechanism is consistent with a switch-like mechanism, whereby MIG6 interacts with and attenuates EGFR activity, while in the absence of ERBB stimulation, it activates ABL pro-apoptotic function.
Gain and loss of function analyses in a wide range of cultured cells show that MIG6 inhibits downstream ERBB signaling, including activation of ERK and AKT, as well as biological responses regulated by ERBB receptors, such as cell proliferation and migration [123, 135-139]. Consistent with these data, Mig6 gene inactivation in mice induces sustained ERBB signaling, leading to overproliferation and impaired differentiation of epidermal keratinocytes and development of tumors in various tissues [140, 141].
Small adaptors in the control of oncogenic signaling driven by TK in human cancer
Most TKs targeted by these adaptor proteins display prominent transforming activity when deregulated. Hence, these negative regulators could also control aberrant signaling driven by oncogenic TKs. As they mostly function by activating a feedback loop, their suppressive activity could be exacerbated by aberrant TK activity in transformed cells. Consequently, they could acquire a potent tumor suppressor function in human cancer. Curiously, their role during malignant cell transformation is much less documented than that of adaptors with positive regulatory functions. In addition, the underlying mechanism of their tumor suppressive role is poorly characterized. Like in normal cells, these adaptors may control TK-dependent oncogenic signaling by directly affecting TK catalytic activity and interaction with components of the signaling cascade, and/or by promoting the destruction of specific elements of this oncogenic signaling, resulting in a restriction of pTyr-dependent cell transformation (Figures 3, 4, 5). Their inactivation by genetic or epigenetic mechanisms could promote TK-driven oncogenic signaling, thus enhancing malignant cell transformation (Table 1). Intriguingly, the tumor suppressive activity of some adaptors appears to be highly context-dependent, because they seem also to participate in the formation of some tumors (Table 1). The molecular cause of these variable effects is unclear. They might target additional processes involved in the control of tumor formation, such as tissue integrity, tumor cell differentiation or tumor immune suppression, resulting in aggravation of the tumor phenotype. A short analysis of the role of these adaptors in human cancer is presented below.
Table 1: Status of small adaptors that negatively regulate TK signaling in human cancers.
Adaptor |
Status in tumors |
Type of tumor |
SOCS1 |
Mutation |
|
Hypermethylation |
AML [148, 149], CML [150], MPN [151], uterine cervical cancer [152], Barrett’s adenocarcinoma [154], ovarian cancer [155], esophageal squamous cell carcinoma [153], glioblastoma multiforme [157], breast cancer [155, 158] |
|
Hypermethylation/Gene loss |
hepatocellular carcinoma [156] |
|
Down-regulation |
Colorectal [159], prostate and pancreatic cancer, myeloma, laryngeal carcinoma [194] |
|
Up-regulation |
Melanoma [168] |
|
SOCS2 |
Hypermethylation |
Ovarian cancer [155] |
Hypermethylation/Gene loss |
||
Down-regulation |
||
Up-regulation |
Acromegaly associated colonic polyps [200], CML [201, 202], AML, glioblastoma and myeloma [203] |
|
SOCS3 |
Mutation |
MPN [161] |
Hypermethylation |
MPN [204], hepatocellular carcinoma [162], glioma [163], cholangiocarcinoma [164], breast [165], lung [205], prostate [206], head and neck cancer [207]; Barrett’s adenocarcinoma [154] |
|
Up-regulation |
Follicular lymphoma [169] |
|
Hyperphosphorylation |
MPN [170] |
|
SOCS4 |
Hypermethylation |
Gastric cancer [208] |
SOCS5 |
Down-regulation |
Thyroid gland cancer [209] |
SOCS6 |
Hypermethylation/Gene loss |
Gastric cancer [210] |
Gene loss |
Colorectal cancer [211] |
|
Down-regulation |
Primary lung squamous cell carcinoma [212], prostate cancer [198], hepatocellular carcinoma [197], liver and thyroid gland cancer [209] |
|
SOCS7 |
Down-regulation |
Breast cancer [158] |
SLAP |
Down-regulation |
|
Up-regulation |
CML, chronic lymphocytic leukemia, glioblastoma, prostate cancer [51] |
|
GRB10 |
Down-regulation |
Myeloma, bladder, brain, breast, prostate and pancreatic cancer [75] |
Up-regulation |
||
GRB14 |
Mutation |
Colorectal cancer [177] |
Up-regulation |
Thyroid cancer [85] |
|
LNK |
Mutation |
MPN, leukemia [117] |
Up-regulation |
Skin, kidney, ovarian cancer [182] |
|
MIG6 |
LOH; Gene deletion |
|
Hypermethylation |
Papillary thyroid carcinoma [186] |
|
Down-regulation |
Hepatocellular carcinoma [139], breast [183], lung [184], skin, pancreatic and ovarian cancer [140] |
Types of tumors where adaptors clearly participate in tumor progression are indicated in italic. LOH, loss of heterozygosity; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MPN, myleoproliferative neoplasms.
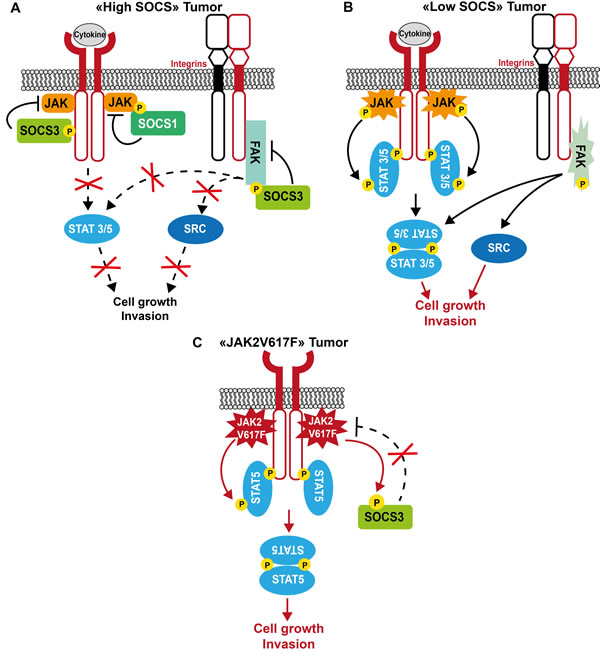
Figure 3: Model of SOCS tumor suppressor function in human cancer. A. In tumor cells with high SOCS expression, these adaptor proteins inhibit tumor cell growth by controlling JAK/STAT-dependent cytokine signaling, and restrict integrin-dependent cell invasion by inhibiting FAK/SRC signaling. B. Upon SOCS inactivation in tumor cells, cytokine and integrin signaling are exacerbated, thus contributing to tumor progression. C. Upon expression of the JAK2V617F oncogene, SOCS inhibitory function is inactivated by tyrosine phosphorylation, which results in increased JAK2V617F oncogenic activity.
SOCS family
SOCS proteins regulate inflammation, hematopoiesis, cell growth and metabolism. They might also play important tumor suppressor roles in many cancers [18, 142] where they are frequently inactivated. SOCS1 and 3 anti-oncogenic activities are particularly well documented. For example, SOCS1 can suppress STAT-dependent signaling induced by oncogenic KIT, TEL-JAK2 and BCR-ABL [143]. However, SOCS1 is frequently inactivated in human lymphoma by gene inactivating mutations leading to increased STAT5 and 6 signaling [144-147]. SOCS1 is commonly silenced by hypermethylation and occasionally mutated in acute myeloid leukemia (AML) [148, 149]. In patients with chronic myeloid leukemia (CML), SOCS1 is often hypermethylated, but can revert to the unmethylated state during remission [150]. Some BCR-ABL-negative myeloproliferative neoplasms (MPN) also exhibit SOCS1 hypermethylation, which may complement other mutations, such as the hyperactive JAK2V617F mutation [151]. SOCS1 hypermethylation is commonly reported in solid tumors [152-155], and combined hypermethylation/gene loss have been observed in hepatocellular carcinoma [156]. However, the functional consequence of SOCS1 inactivation has not been fully elucidated yet. SOCS1 hypermethylation is associated with enhanced radio-resistance in glioblastoma multiforme, indicative of a pro-apoptotic function [157]. Conversely, higher SOCS1 expression has been observed in early stage tumors and has been linked to better clinical outcome in breast and colorectal cancers (CRC) [158, 159]. A functional analysis suggested that SOCS1 may control CRC metastatic progression, possibly through destabilization of metastatic inducers [159]. SOCS1 is also important for preventing chronic inflammation-mediated carcinogenesis. For instance, Socs1 knockout mice spontaneously develop intestinal tumors in an IFNγ/STAT1-dependent manner, suggesting that chronic inflammation is a critical determinant for CRC development [160]. Therefore, SOCS1 is a unique anti-oncogene that prevents carcinogenesis by suppressing chronic inflammation.
Although mutations in SOCS3 are rare events, a loss of function mutation within the SH2 domain of SOCS3 (F136L) was recently described in a cohort of Japanese patients with MPN [161]. SOCS3 hypermethylation also has been observed in this type of cancer and in solid tumors. For instance, SOCS3 inactivation increases JAK/STAT and FAK signaling, promoting growth and migration of hepatocellular carcinoma cells [162], invasion of glioma cells [163], enhanced anti-apoptotic IL-6/STAT3 signaling in cholangiocarcinoma cells [164] and, possibly, increased dissemination of breast cancer cells (Figure 3) [165]. SOCS3 inactivation is also associated with STAT5-dependent CRC metastatic progression [166]. Finally, in mice, SOCS3 can limit inflammation-associated tumorigenesis in colon, by inactivating STAT3 and NFκB [167].
Opposing functions for SOCS proteins have been also reported in some tumors. For example, SOCS1, which is not expressed in normal skin or melanocytic nevi, is up-regulated in melanoma. Moreover, high SOCS1 expression level correlates with the metastatic stage of the disease, suggesting that SOCS1 might be an additional marker of human melanoma progression. Mechanistically, SOCS1 may reduce the tumor cell response to endogenous and/or therapeutically administered cytokines [168]. Similarly, SOCS3 up-regulation was associated with decreased survival in a cohort of patients with de novo follicular lymphoma [169]. Moreover, aberrantly active TKs may find a way to escape SOCS3 negative regulation. For example, JAK2V617F inhibits SOCS3 inhibitory function by promoting SOCS3 hyperphosphorylation in MPN. Additionally, JAK2 diverts SOCS3 to sustain a transformed phenotype by a still not well understood mechanism (Figure 3) [170]. Similarly, truncated G-CSF receptor variants expressed in AML lack the sequences required for SOCS3-mediated control of STAT5 activation [171].
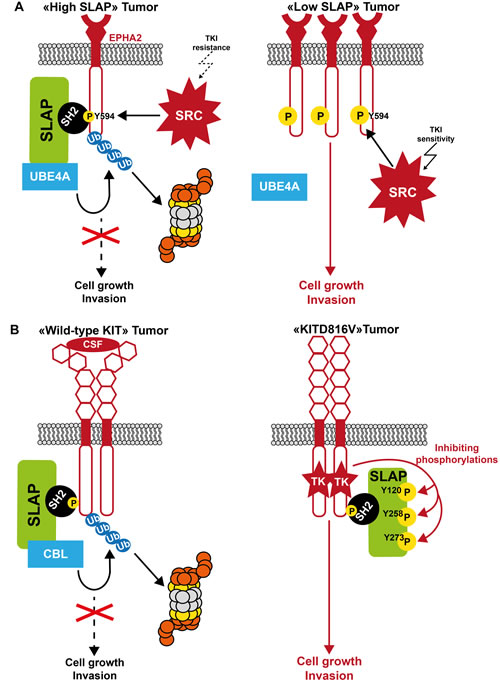
Figure 4: Model of SLAP tumor suppressor function in human cancer. A. Control of SRC oncogenic signaling by SLAP. In tumor cells with high SLAP expression, SLAP inhibits SRC oncogenic signaling by promoting destabilization of SRC oncogenic substrates, including the cell adhesive receptor EPHA2. This results in the restriction of tumor cell growth and invasion. When SLAP is inactivated, EPHA2 protein level is abnormally increased and SRC oncogenic signaling exacerbated, thus enabling metastatic progression. Consequently, tumor cells may be more sensitive to SRC-like inhibitors. B. Control of KIT oncogenic signaling by SLAP. SLAP regulates KIT-driven oncogenic signaling by promoting ubiquitination-dependent KIT degradation. However, upon expression of oncogenic KITD816V, this SLAP-mediated inhibitory mechanism is impaired through tyrosine phosphorylation, thus alleviating SLAP control on KITD816V oncogenic signaling.
SLAP family
Little was known about their role in human cancer until recently, mainly because SLAP functions were thought to be restricted to the immune system. Nevertheless, we previously reported that SLAP has a strong capacity to counteract SRC oncogenic activity in fibroblasts through a SH3-dependent mechanism, in agreement with a potential anti-oncogenic activity [58]. More recently, we observed that SLAP is abundantly expressed in colon epithelium, but frequently down-regulated in the associated tumor [52]. SLAP inactivation is not mediated by a gene methylation-dependent mechanism, but in rare cases could be caused by SLAP inactivating mutations located in the SH2 and SH3 domains. SLAP silencing promotes tumor initiation, progression and metastasis formation, while SLAP overexpression inhibits tumor growth and invasion. SLAP promotes the degradation of EPHA2, an important adhesive receptor and key substrate for SRC function in cell tumorigenesis and invasiveness [172, 173]. This novel activity is CBL-independent, but requires SLAP interaction with the ubiquitination factor UBE4A. Mechanistically, SRC phosphorylates EPHA2 on Tyr594, resulting in the promotion of a UBE4A/SLAP/EPHA2 complex for EPHA2 proteasomal degradation, thus limiting the SRC metastatic potential (Figure 4). Consistently, SLAP inactivation in CRC dramatically increases EPHA2 protein level and amplifies the SRC/EPHA2/AKT signaling cascade that promotes tissue invasion of tumor cells. Thus, inactivation of SLAP-mediated degradation of specific SRC substrates defines an additional and important mechanism of SRC-mediated oncogenic induction in CRC. It is not known whether SLAP targets additional SRC oncogenic substrates to mediate its tumor suppressive activity. SLAP expression might exert a similar tumor suppressor function in AML and myeloma [51].
However, deregulated RTKs may evade SLAP negative regulation in pathological conditions. For instance, it was recently reported that wild type KIT, but not the oncogenic KIT-D816V mutant, is degraded through a SLAP-dependent pathway. SLAP can associate with KIT and KIT-D816V, but only KIT-D816V phosphorylates SLAP on Tyr120, Tyr258 and Tyr273, leading to inhibition of SLAP activity and sustained KIT oncogenic signaling (Figure 4) [60].
Finally, SLAP may also participate in cell transformation in specific situations, as reported for SOCS proteins. For instance, SLAP could be involved in the blockade of cell differentiation required for the induction of erythroleukemia by the FLI-1 oncoprotein [62]. Mechanistically, SLAP inhibits several components of EPO receptor signaling needed for late survival and terminal differentiation of erythroblasts. Likewise, SLAP may also be induced in acute promyelocytic leukemia subtypes that harbor FLT3-ITD mutations, suggesting that this adaptor protein might also participate in this transforming process [51].
GRB7 family
GRB10 and GRB14 roles in human cancer are poorly documented and not clear. GRB10 may participate in progression of some human tumors, as suggested for primary cervical squamous carcinoma [174] and FLT3-ITD positive AML [175], and also in BCR-ABL-mediated leukemogenesis in mice [176]. Similarly GRB14 increases thyroid cancer cell growth by promoting RET signaling and its expression is correlated with human thyroid cancer invasiveness [85]. However, these adaptors may also acquire tumor suppressive functions, as suggested by a recent transcriptomic meta-analysis showing that GRB10 expression is down-regulated in many human tumors [75]. Moreover, the GRB14 gene is frequently mutated in human CRC with microsatellite instability [177].
LNK
LNK may have tumor suppressor function as suggested by the phenotype of Lnk−/− mice that resembles the myeloproliferative abnormalities found in human MPN [178]. LNK ectopic expression inhibits proliferation of leukemic cells through binding to and catalytic inhibition of transforming TKs [103, 112, 113, 179-181]. Surprisingly, analysis of LNK expression in a large panel of hematological malignancies revealed that LNK is strongly expressed in nearly half of the patient samples, possibly as a consequence of aberrant cytokine signaling activation [103, 180]. How JAK2 can cope with high LNK level in these cells is unclear. LNK can be mutated in MPN (3-5%) and in some leukemias [117]. Somatic mutations mostly target a hot spot in LNK PH domain, resulting in aberrant JAK/STAT signaling, even in MPN harboring wild type JAK [108]. Consistently, cells co-expressing the TPO receptor and mutated LNK are hyper-responsive to TPO and display aberrant growth and enhanced JAK2/STAT signaling.
Surprisingly, LNK is up-regulated in some solid tumors, including the mesenchymal subtype of serous ovarian cancer, and this is associated with poorer outcome [182]. Functionally, LNK participates in AKT- and MAPK-dependent tumor cell growth and survival.
MIG6
MIG6 tumor suppressive role is supported by the finding that upon deletion of ERRFI1, mice frequently develop spontaneous or chemical-induced tumors [140]. Additionally, MIG6 expression is frequently reduced in various human cancer types [139, 140, 183, 184] and this is correlated with poor survival in patients with breast or lung cancer [183, 184]. MIG6 inactivation can be the result of loss of heterozygosity or focal deletion of ERRFI1, as reported in EGFR-amplified glioblastoma [129, 132, 185], or promoter methylation, as found in papillary thyroid carcinoma [186]. MIG6 inactivation may affect RTK aberrant signaling (Figure 5). Accordingly, MIG6 down-regulation in thyroid cancer cells and papillary carcinoma enhances EGFR, ERBB2 and MET activity.
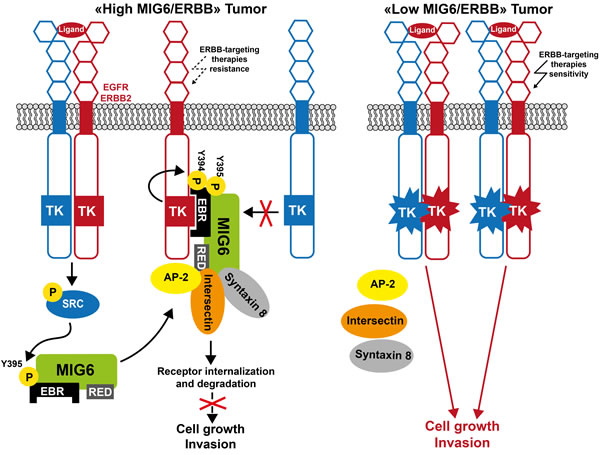
Figure 5: Model of MIG6 tumor suppressor function in human cancer. In tumor cells with a high ratio of MIG6/ERBB receptors, MIG6 inhibits the receptor kinase activity and promotes their internalization for lysosomal degradation, resulting in a dramatic reduction of ERBB oncogenic signaling. In tumor cells with a low MIG6/ERBB ratio, MIG6 activity is reduced and ERBB oncogenic signaling is restored. Consequently, tumor cells may be more sensitive to ERBB-like inhibitors.
Therapeutic consequences in oncology
All this data indicates that loss of adaptor-mediated regulation of oncogenic TKs contributes to tumor formation. These novel findings may have significant consequences for cancer treatment/management, such as the discovery/development of novel biomarkers and the improvement of therapy targeting TK-dependent signaling.
Adaptor expression levels as novel biomarkers
The expression status of some of these adaptors in transformed cells may predict the tumor responses to drug that target oncogenic TK signaling. For instance, low expression in tumor cells of adaptors that control TK catalytic activity, such as MIG6, might predict heightened oncogenic signaling dependent of deregulated TKs, such as ERBB receptors, and therefore may be a good predicator of tumor cell response to TK inhibitors (TKIs) (Figure 5). Indeed, carcinogen-induced tumors in Errfi1-/- mice are highly sensitive to the EGFR TKI gefitinib [140]. MIG6 silencing also increases bladder cancer cell sensitivity to the therapeutic anti-EGFR antibody cetuximab [187]. A low MIG6/EGFR ratio, predicting high EGFR activity, is highly correlated with erlotinib sensitivity in cancer cell lines derived from different tissues [188]. Analysis of a cohort of patients with lung cancer treated with gefitinib alone demonstrated higher response rates and a marked increase in progression-free survival in patients with a low MIG6/EGFR ratio [188]. Overall, these studies highlight a mechanism of resistance to EGFR-targeted therapies in tumors with high MIG6/EGFR ratio. They also suggest that this ratio could represent a novel biomarker for guiding the decision to incorporate these drugs into chemotherapeutic regimens.
Inactivation in tumor cells of adaptors that control substrate stability, such as SLAP, would also predict heightened oncogenic signaling dependent of the deregulated TK, such as SRC, and therefore may be a good predictor of tumor cell response to TKIs. Conversely, high SLAP expression level might indicate that although SRC activity is aberrant in these tumors, its transforming activity could be hampered by SLAP-mediated destruction of important SRC substrates. This would render tumor cells less dependent on SRC oncogenic signaling and, consequently, rather resistant to a SRC-based therapy (Figure 4). This model has been experimentally verified in CRC cells, where response to SRC inhibitors was greatly enhanced in cells where SLA was silenced [52]. Therefore, SLAP down-regulation may be a key predictor of SRC inhibitory response, and may explain, at least in part, why SRC inhibitors failed to generate promising results in solid tumors so far. Overall, these findings support the idea that the level of these regulators may be used as valuable biomarkers to select patients who could respond to TK-targeted therapy.
Adaptors as potential therapeutic targets
Due to their prominent tumor suppressor function, forced expression of these adaptors in tumor cells could represent an alternative therapeutic strategy to target oncogenic TK signaling in human cancer. Restoration of adaptor expression in tumor cells can reduce tumor growth and/or metastasis formation. Nevertheless, gene therapy is still a technical challenge and alternative methods to modulate adaptor activity or expression might be considered. These adaptor proteins are frequently inactivated in tumors via a methylation-dependent mechanism. Accordingly, demethylation drugs, such as 5-aza-2’-deoxycytidine that exhibits clear tumor suppressive roles, could restore adaptor expression in tumor cells and may represent a potential anti-tumor therapeutic strategy. Forced expression of many of these adaptors, such as LNK [181] and SOCS proteins [18], was used to test their capacity to overcome TK-induced cell transformation. While promising, this strategy may be limited by the capacity of oncogenic TKs to escape negative regulation by phosphorylation-dependent mechanisms, as reported for SOCS and SLAP. Besides, modulation of their expression in tumor cells for anti-tumor therapy is highly context-dependent due to their opposite function in different cancers, as described above. Another potential therapeutic strategy, although not experimentally validated, would arise from the development of small molecules that mimic their actions on their capacity to inhibit TK catalytic activity as demonstrated for the targeting of JAKs by the SOCS3-KIR domain.
Finally, because of their prominent role in immune response, their level of expression may dictate the tumor cell response to cytokine-based therapy. For instance, SOCS1 and SOCS3 up-regulation seem to be responsible for the unresponsiveness to IFN therapy in patients with leukemia. Indeed, SOCS1 constitutive expression has been observed in patients with CML [189], in agreement with the reported hypomethylation of this gene [190], and was correlated with poor response to IFNα treatment, possibly due to a direct effect on receptor signaling [189]. SOCS3 expression is also elevated in CML and confers resistance to IFNα treatment [191]. SOCS1 expression is also higher in IFN-resistant neuroendocrine tumor cells and siRNA inhibition of SOCS1 expression enhances their IFN-responsiveness [192]. Likewise, siRNA-mediated inhibition of SOCS1 and SOCS3 expression in melanoma cells enhances their responsiveness to IFN [193]. This supports the idea that siRNA-mediated reduction of SOCS level could be a promising new approach to enhance IFN therapeutic effectiveness.
Concluding remarks and perspectives
Since the discovery of CIS twenty years ago, this small class of adaptor proteins has emerged as an important player in the mechanisms to finely tune TK-mediated signal transduction. While their functions have been mostly investigated in hematopoietic cells due to their high expression levels and to the essential role of TK signaling in hematopoiesis, more recent data revealed additional important roles in non-haematopoietic cells, as exemplified by the SLAP tumor suppressive role in the colon. Therefore, one important perspective will be to uncover such physio-pathological activities in non-hematopoietic tissues using novel relevant in vitro and animal models. Similarly, while underlying mechanisms of adaptor-mediated negative feedback regulation was thought to be rather well established, recent molecular analyses challenged this idea and suggest the existence of broader mechanisms involved in their activities. For example, CBL has been reported as the main SLAP effector in the control of TK signaling through ubiquitin-mediated degradation via the proteasome or the lysosome [45, 53-55] but our recent study revealed that SLAP recruits distinct ubiquitination factors such as UBE4A in the colon to control transforming substrates stability, despite a high expression level of CBL in this tissue [52]. Additionally, proteomics identified a dozen of ubiquitination factors as specific SLAP interactors in CRC suggesting that these adaptors share common mechanisms to control signaling, but recruit a large repertoire of effectors to target a wide range of TK-dependent signaling. Surprisingly, proteomics also identified a hundred of additional SLAP binders unrelated to the control of receptor signaling or protein stability, predicting a much broader role of this adaptor in signal transduction and raising the question whether its functions are restricted to TK-dependent signaling [52]. We thus anticipate novel functions for these adaptors to be identified in the future and proteomic methods combined with functional analyses are warranted to give a comprehensive view on the role of these adaptors in cell biology.
Due to their feedback role in the control of TK signaling, the negative function of these adaptors is expected to be exacerbated upon aberrant expression of TK activities, uncovering novel tumor suppressor functions for these adaptors. This idea has been experimentally validated in myeloproliferative malignancies (e.g. LNK) but tumor suppressor and anti-metastatic activity for these adaptors have been now reported also in solid tumors (e.g. SLAP and SOCS1). Therefore, a broader tumor suppressor role for these adaptors is expected in human cancer, which will deserve more investigation in the future. Besides, we suspect that oncogenic TKs may find a way to overcome this control mechanism and in some situations divert this regulatory process to sustain tumor progression. How oncogenic TKs cope with this control mechanism is an additional important issue to be addressed in the future. Intriguingly, the function of some adaptors in cancer is more complex because their activity seems to be context-dependent. The reason for such variable activity is quite obscure, but we suggest that these molecules target additional important signals that are involved in the control of tumor development/progression, such as the immune response. It will be thus important to elucidate such tumor promoting function in order to better understand their general role in this human disease and improve TK-based therapeutic strategy according to their activities. In conclusion, this rather neglected mechanism in the control of TK signaling is emerging as an additional important mechanism for the control of human malignancy and a better understanding on the role of this adaptor family in human cancer should ultimately improve TK-based tumor therapies.
Acknowledgments
We thank our colleagues for critically reading the manuscript.
Funding
CN and CC were supported by “La Ligue Nationale contre le Cancer” and SR is an INSERM investigator. This work was also supported by “La Ligue Nationale contre le Cancer” (équipe labellisée LIGUE), CNRS and Montpellier University.
conflicts of interest
The authors declare no conflict of interest.
References
1. Lim WA and Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010; 142(5):661-667.
2. Liu BA and Nash PD. Evolution of SH2 domains and phosphotyrosine signalling networks. Philos Trans R Soc Lond B Biol Sci. 2012; 367(1602):2556-2573.
3. Manning G, Whyte DB, Martinez R, Hunter T and Sudarsanam S. The protein kinase complement of the human genome. Science. 2002; 298(5600):1912-1934.
4. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000; 103(2):211-225.
5. Tsygankov AY. Non-receptor protein tyrosine kinases. Front Biosci. 2003; 8:s595-635.
6. Blume-Jensen P and Hunter T. Oncogenic kinase signalling. Nature. 2001; 411(6835):355-365.
7. Krause DS and Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005; 353(2):172-187.
8. Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, Hara T and Miyajima A. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995; 14(12):2816-2826.
9. Pawson T and Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997; 278(5346):2075-2080.
10. Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D and Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992; 70(3):431-442.
11. Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T and Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992; 70(1):93-104.
12. Mayer BJ, Hamaguchi M and Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988; 332(6161):272-275.
13. van der Geer P, Hunter T and Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol. 1994; 10:251-337.
14. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA and Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997; 387(6636):917-921.
15. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997; 387(6636):921-924.
16. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S and Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997; 387(6636):924-929.
17. Stec WJ and Zeidler MP. Drosophila SOCS Proteins. J Signal Transduct. 2011; 2011:894510.
18. Trengove MC and Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol. 2013; 2(1):1-29.
19. Kazi JU, Kabir NN, Flores-Morales A and Ronnstrand L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cellular and molecular life sciences. 2014; 71(17):3297-3310.
20. Babon JJ, Lucet IS, Murphy JM, Nicola NA and Varghese LN. The molecular regulation of Janus kinase (JAK) activation. Biochem J. 2014; 462(1):1-13.
21. Piessevaux J, Lavens D, Peelman F and Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev. 2008; 19(5-6):371-381.
22. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O’Neill LA and Hertzog PJ. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006; 7(2):148-155.
23. Rui L, Yuan M, Frantz D, Shoelson S and White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002; 277(44):42394-42398.
24. Landsman T and Waxman DJ. Role of the cytokine-induced SH2 domain-containing protein CIS in growth hormone receptor internalization. J Biol Chem. 2005; 280(45):37471-37480.
25. Irandoust MI, Aarts LH, Roovers O, Gits J, Erkeland SJ and Touw IP. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J. 2007; 26(7):1782-1793.
26. Ram PA and Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. 1999; 274(50):35553-35561.
27. Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN and Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999; 18(5):1309-1320.
28. Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, Sasaki M, Johnston JA and Yoshimura A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells. 1999; 4(6):339-351.
29. Giordanetto F and Kroemer RT. A three-dimensional model of Suppressor Of Cytokine Signalling 1 (SOCS-1). Protein Eng. 2003; 16(2):115-124.
30. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS and Nicola NA. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012; 36(2):239-250.
31. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA and Babon JJ. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nature structural & molecular biology. 2013; 20(4):469-476.
32. Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ and Alexander WS. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95(24):14395-14399.
33. Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D and Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999; 98(5):597-608.
34. Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ and Alexander WS. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98(16):9324-9329.
35. Jo D, Liu D, Yao S, Collins RD and Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med. 2005; 11(8):892-898.
36. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H and Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004; 10(7):739-743.
37. Robb L, Boyle K, Rakar S, Hartley L, Lochland J, Roberts AW, Alexander WS and Metcalf D. Genetic reduction of embryonic leukemia-inhibitory factor production rescues placentation in SOCS3-null embryos but does not prevent inflammatory disease. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(45):16333-16338.
38. Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ and Alexander WS. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000; 405(6790):1069-1073.
39. Matsumoto A, Seki Y, Kubo M, Ohtsuka S, Suzuki A, Hayashi I, Tsuji K, Nakahata T, Okabe M, Yamada S and Yoshimura A. Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine-inducible SH2-containing protein 1 transgenic mice. Molecular and cellular biology. 1999; 19(9):6396-6407.
40. Brender C, Columbus R, Metcalf D, Handman E, Starr R, Huntington N, Tarlinton D, Odum N, Nicholson SE, Nicola NA, Hilton DJ and Alexander WS. SOCS5 is expressed in primary B and T lymphoid cells but is dispensable for lymphocyte production and function. Molecular and cellular biology. 2004; 24(13):6094-6103.
41. Krebs DL, Uren RT, Metcalf D, Rakar S, Zhang JG, Starr R, De Souza DP, Hanzinikolas K, Eyles J, Connolly LM, Simpson RJ, Nicola NA, Nicholson SE, Baca M, Hilton DJ and Alexander WS. SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Molecular and cellular biology. 2002; 22(13):4567-4578.
42. Krebs DL, Metcalf D, Merson TD, Voss AK, Thomas T, Zhang JG, Rakar S, O’Bryan M K, Willson TA, Viney EM, Mielke LA, Nicola NA, Hilton DJ and Alexander WS. Development of hydrocephalus in mice lacking SOCS7. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(43):15446-15451.
43. Banks AS, Li J, McKeag L, Hribal ML, Kashiwada M, Accili D and Rothman PB. Deletion of SOCS7 leads to enhanced insulin action and enlarged islets of Langerhans. The Journal of clinical investigation. 2005; 115(9):2462-2471.
44. Pandey A, Duan H and Dixit VM. Characterization of a novel Src-like adapter protein that associates with the Eck receptor tyrosine kinase. J Biol Chem. 1995; 270(33):19201-19204.
45. Loreto MP, Berry DM and McGlade CJ. Functional cooperation between c-Cbl and Src-like adaptor protein 2 in the negative regulation of T-cell receptor signaling. Molecular and cellular biology. 2002; 22(12):4241-4255.
46. Pandey A, Ibarrola N, Kratchmarova I, Fernandez MM, Constantinescu SN, Ohara O, Sawasdikosol S, Lodish HF and Mann M. A novel Src homology 2 domain-containing molecule, Src-like adapter protein-2 (SLAP-2), which negatively regulates T cell receptor signaling. J Biol Chem. 2002; 277(21):19131-19138.
47. Holland SJ, Liao XC, Mendenhall MK, Zhou X, Pardo J, Chu P, Spencer C, Fu A, Sheng N, Yu P, Pali E, Nagin A, Shen M, Yu S, Chan E, Wu X, et al. Functional cloning of Src-like adapter protein-2 (SLAP-2), a novel inhibitor of antigen receptor signaling. J Exp Med. 2001; 194(9):1263-1276.
48. Dragone LL, Shaw LA, Myers MD and Weiss A. SLAP, a regulator of immunoreceptor ubiquitination, signaling, and trafficking. Immunol Rev. 2009; 232(1):218-228.
49. Sosinowski T, Killeen N and Weiss A. The Src-like adaptor protein downregulates the T cell receptor on CD4+CD8+ thymocytes and regulates positive selection. Immunity. 2001; 15(3):457-466.
50. Roche S, Alonso G, Kazlauskas A, Dixit VM, Courtneidge SA and Pandey A. Src-like adaptor protein (Slap) is a negative regulator of mitogenesis. Curr Biol. 1998; 8(17):975-978.
51. Kazi JU and Ronnstrand L. Src-Like adaptor protein (SLAP) binds to the receptor tyrosine kinase Flt3 and modulates receptor stability and downstream signaling. PLoS One. 2012; 7(12):e53509.
52. Naudin C, Sirvent A, Leroy C, Larive R, Simon V, Pannequin J, Bourgaux JF, Pierre J, Robert B, Hollande F and Roche S. SLAP displays tumour suppressor functions in colorectal cancer via destabilization of the SRC substrate EPHA2. Nat Commun. 2014; 5:3159.
53. Myers MD, Sosinowski T, Dragone LL, White C, Band H, Gu H and Weiss A. Src-like adaptor protein regulates TCR expression on thymocytes by linking the ubiquitin ligase c-Cbl to the TCR complex. Nat Immunol. 2006; 7(1):57-66.
54. Myers MD, Dragone LL and Weiss A. Src-like adaptor protein down-regulates T cell receptor (TCR)-CD3 expression by targeting TCRzeta for degradation. J Cell Biol. 2005; 170(2):285-294.
55. Dragone LL, Myers MD, White C, Sosinowski T and Weiss A. SRC-like adaptor protein regulates B cell development and function. J Immunol. 2006; 176(1):335-345.
56. Sosinowski T, Pandey A, Dixit VM and Weiss A. Src-like adaptor protein (SLAP) is a negative regulator of T cell receptor signaling. J Exp Med. 2000; 191(3):463-474.
57. Wybenga-Groot LE and McGlade CJ. Crystal structure of Src-like adaptor protein 2 reveals close association of SH3 and SH2 domains through beta-sheet formation. Cell Signal. 2013; 25(12):2702-2708.
58. Sirvent A, Leroy C, Boureux A, Simon V and Roche S. The Src-like adaptor protein regulates PDGF-induced actin dorsal ruffles in a c-Cbl-dependent manner. Oncogene. 2008; 27(24):3494-3500.
59. Liontos LM, Dissanayake D, Ohashi PS, Weiss A, Dragone LL and McGlade CJ. The Src-like adaptor protein regulates GM-CSFR signaling and monocytic dendritic cell maturation. J Immunol. 2011; 186(4):1923-1933.
60. Kazi JU, Agarwal S, Sun J, Bracco E and Ronnstrand L. Src-like-adaptor protein (SLAP) differentially regulates normal and oncogenic c-Kit signaling. J Cell Sci. 2014; 127(Pt 3):653-662.
61. Pakuts B, Debonneville C, Liontos LM, Loreto MP and McGlade CJ. The Src-like adaptor protein 2 regulates colony-stimulating factor-1 receptor signaling and down-regulation. J Biol Chem. 2007; 282(25):17953-17963.
62. Lebigot I, Gardellin P, Lefebvre L, Beug H, Ghysdael J and Quang CT. Up-regulation of SLAP in FLI-1-transformed erythroblasts interferes with EpoR signaling. Blood. 2003; 102(13):4555-4562.
63. Klinghoffer RA, Sachsenmaier C, Cooper JA and Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. Embo J. 1999; 18(9):2459-2471.
64. Kazi JU, Kabir NN and Ronnstrand L. Role of SRC-like adaptor protein (SLAP) in immune and malignant cell signaling. Cellular and molecular life sciences. 2015; 72(13):2535-2544.
65. Marton N, Baricza E, Ersek B, Buzas EI and Nagy G. The Emerging and Diverse Roles of Src-Like Adaptor Proteins in Health and Disease. Mediators of inflammation. 2015; 2015:952536.
66. Margolis B, Silvennoinen O, Comoglio F, Roonprapunt C, Skolnik E, Ullrich A and Schlessinger J. High-efficiency expression/cloning of epidermal growth factor-receptor-binding proteins with Src homology 2 domains. Proceedings of the National Academy of Sciences of the United States of America. 1992; 89(19):8894-8898.
67. Ooi J, Yajnik V, Immanuel D, Gordon M, Moskow JJ, Buchberg AM and Margolis B. The cloning of Grb10 reveals a new family of SH2 domain proteins. Oncogene. 1995; 10(8):1621-1630.
68. Daly RJ, Sanderson GM, Janes PW and Sutherland RL. Cloning and characterization of GRB14, a novel member of the GRB7 gene family. J Biol Chem. 1996; 271(21):12502-12510.
69. Holt LJ and Siddle K. Grb10 and Grb14: enigmatic regulators of insulin action--and more? Biochem J. 2005; 388(Pt 2):393-406.
70. Desbuquois B, Carre N and Burnol AF. Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J. 2013; 280(3):794-816.
71. Shen TL and Guan JL. Grb7 in intracellular signaling and its role in cell regulation. Front Biosci. 2004; 9:192-200.
72. Lucas-Fernandez E, Garcia-Palmero I and Villalobo A. Genomic organization and control of the grb7 gene family. Curr Genomics. 2008; 9(1):60-68.
73. Cariou B, Capitaine N, Le Marcis V, Vega N, Bereziat V, Kergoat M, Laville M, Girard J, Vidal H and Burnol AF. Increased adipose tissue expression of Grb14 in several models of insulin resistance. FASEB J. 2004; 18(9):965-967.
74. Kairouz R, Parmar J, Lyons RJ, Swarbrick A, Musgrove EA and Daly RJ. Hormonal regulation of the Grb14 signal modulator and its role in cell cycle progression of MCF-7 human breast cancer cells. J Cell Physiol. 2005; 203(1):85-93.
75. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP and Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011; 332(6035):1322-1326.
76. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA and Sabatini DM. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011; 332(6035):1317-1322.
77. Depetris RS, Hu J, Gimpelevich I, Holt LJ, Daly RJ and Hubbard SR. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Molecular cell. 2005; 20(2):325-333.
78. Depetris RS, Wu J and Hubbard SR. Structural and functional studies of the Ras-associating and pleckstrin-homology domains of Grb10 and Grb14. Nature structural & molecular biology. 2009; 16(8):833-839.
79. Wick KR, Werner ED, Langlais P, Ramos FJ, Dong LQ, Shoelson SE and Liu F. Grb10 inhibits insulin-stimulated insulin receptor substrate (IRS)-phosphatidylinositol 3-kinase/Akt signaling pathway by disrupting the association of IRS-1/IRS-2 with the insulin receptor. J Biol Chem. 2003; 278(10):8460-8467.
80. Nouaille S, Blanquart C, Zilberfarb V, Boute N, Perdereau D, Roix J, Burnol AF and Issad T. Interaction with Grb14 results in site-specific regulation of tyrosine phosphorylation of the insulin receptor. EMBO Rep. 2006; 7(5):512-518.
81. Morrione A, Plant P, Valentinis B, Staub O, Kumar S, Rotin D and Baserga R. mGrb10 interacts with Nedd4. J Biol Chem. 1999; 274(34):24094-24099.
82. Vecchione A, Marchese A, Henry P, Rotin D and Morrione A. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Molecular and cellular biology. 2003; 23(9):3363-3372.
83. Monami G, Emiliozzi V and Morrione A. Grb10/Nedd4-mediated multiubiquitination of the insulin-like growth factor receptor regulates receptor internalization. J Cell Physiol. 2008; 216(2):426-437.
84. Browaeys-Poly E, Blanquart C, Perdereau D, Antoine AF, Goenaga D, Luzy JP, Chen H, Garbay C, Issad T, Cailliau K and Burnol AF. Grb14 inhibits FGF receptor signaling through the regulation of PLCgamma recruitment and activation. FEBS Lett. 2010; 584(21):4383-4388.
85. Balogh K, Asa SL, Zheng L, Cassol C, Cheng S and Ezzat S. The insulin resistance Grb14 adaptor protein promotes thyroid cancer ret signaling and progression. Oncogene. 2012; 31(36):4012-4021.
86. Wang J, Dai H, Yousaf N, Moussaif M, Deng Y, Boufelliga A, Swamy OR, Leone ME and Riedel H. Grb10, a positive, stimulatory signaling adapter in platelet-derived growth factor BB-, insulin-like growth factor I-, and insulin-mediated mitogenesis. Molecular and cellular biology. 1999; 19(9):6217-6228.
87. Giorgetti-Peraldi S, Murdaca J, Mas JC and Van Obberghen E. The adapter protein, Grb10, is a positive regulator of vascular endothelial growth factor signaling. Oncogene. 2001; 20(30):3959-3968.
88. Jahn T, Seipel P, Urschel S, Peschel C and Duyster J. Role for the adaptor protein Grb10 in the activation of Akt. Molecular and cellular biology. 2002; 22(4):979-991.
89. Smith FM, Holt LJ, Garfield AS, Charalambous M, Koumanov F, Perry M, Bazzani R, Sheardown SA, Hegarty BD, Lyons RJ, Cooney GJ, Daly RJ and Ward A. Mice with a disruption of the imprinted Grb10 gene exhibit altered body composition, glucose homeostasis, and insulin signaling during postnatal life. Molecular and cellular biology. 2007; 27(16):5871-5886.
90. Wang L, Balas B, Christ-Roberts CY, Kim RY, Ramos FJ, Kikani CK, Li C, Deng C, Reyna S, Musi N, Dong LQ, DeFronzo RA and Liu F. Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo. Molecular and cellular biology. 2007; 27(18):6497-6505.
91. Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, Biden TJ, Ormandy CJ, James DE and Daly RJ. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J. 2004; 23(3):582-593.
92. Charalambous M, Smith FM, Bennett WR, Crew TE, Mackenzie F and Ward A. Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100(14):8292-8297.
93. Shiura H, Miyoshi N, Konishi A, Wakisaka-Saito N, Suzuki R, Muguruma K, Kohda T, Wakana S, Yokoyama M, Ishino F and Kaneko-Ishino T. Meg1/Grb10 overexpression causes postnatal growth retardation and insulin resistance via negative modulation of the IGF1R and IR cascades. Biochem Biophys Res Commun. 2005; 329(3):909-916.
94. Huang X, Li Y, Tanaka K, Moore KG and Hayashi JI. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase C gamma 1, Grb2, and phosphatidylinositol 3-kinase. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92(25):11618-11622.
95. Li Y, He X, Schembri-King J, Jakes S and Hayashi J. Cloning and characterization of human Lnk, an adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J Immunol. 2000; 164(10):5199-5206.
96. Li M, Li Z, Morris DL and Rui L. Identification of SH2B2beta as an inhibitor for SH2B1- and SH2B2alpha-promoted Janus kinase-2 activation and insulin signaling. Endocrinology. 2007; 148(4):1615-1621.
97. Hu J and Hubbard SR. Structural basis for phosphotyrosine recognition by the Src homology-2 domains of the adapter proteins SH2-B and APS. Journal of molecular biology. 2006; 361(1):69-79.
98. Hu J, Liu J, Ghirlando R, Saltiel AR and Hubbard SR. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Molecular cell. 2003; 12(6):1379-1389.
99. Song W, Ren D, Li W, Jiang L, Cho KW, Huang P, Fan C, Song Y, Liu Y and Rui L. SH2B regulation of growth, metabolism, and longevity in both insects and mammals. Cell Metab. 2010; 11(5):427-437.
100. Slack C, Werz C, Wieser D, Alic N, Foley A, Stocker H, Withers DJ, Thornton JM, Hafen E and Partridge L. Regulation of lifespan, metabolism, and stress responses by the Drosophila SH2B protein, Lnk. PLoS Genet. 2010; 6(3):e1000881.
101. Rui L. SH2B1 regulation of energy balance, body weight, and glucose metabolism. World J Diabetes. 2014; 5(4):511-526.
102. Velazquez L, Cheng AM, Fleming HE, Furlonger C, Vesely S, Bernstein A, Paige CJ and Pawson T. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med. 2002; 195(12):1599-1611.
103. Baran-Marszak F, Magdoud H, Desterke C, Alvarado A, Roger C, Harel S, Mazoyer E, Cassinat B, Chevret S, Tonetti C, Giraudier S, Fenaux P, Cymbalista F, Varin-Blank N, Le Bousse-Kerdiles MC, Kiladjian JJ, et al. Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood. 2010; 116(26):5961-5971.
104. Boulday G, Coulon F, Fraser CC, Soulillou JP and Charreau B. Transcriptional up-regulation of the signaling regulatory protein LNK in activated endothelial cells. Transplantation. 2002; 74(9):1352-1354.
105. Tong W, Zhang J and Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood. 2005; 105(12):4604-4612.
106. Seita J, Ema H, Ooehara J, Yamazaki S, Tadokoro Y, Yamasaki A, Eto K, Takaki S, Takatsu K and Nakauchi H. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(7):2349-2354.
107. Bersenev A, Wu C, Balcerek J and Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. The Journal of clinical investigation. 2008; 118(8):2832-2844.
108. Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs KD, Jr., Merker JD, Zehnder JL, Nolan GP and Gotlib J. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010; 116(6):988-992.
109. Jiang J, Balcerek J, Rozenova K, Cheng Y, Bersenev A, Wu C, Song Y and Tong W. 14-3-3 regulates the LNK/JAK2 pathway in mouse hematopoietic stem and progenitor cells. The Journal of clinical investigation. 2012; 122(6):2079-2091.
110. Takaki S, Morita H, Tezuka Y and Takatsu K. Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J Exp Med. 2002; 195(2):151-160.
111. Simon C, Dondi E, Chaix A, de Sepulveda P, Kubiseski TJ, Varin-Blank N and Velazquez L. Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood. 2008; 112(10):4039-4047.
112. Gueller S, Hehn S, Nowak V, Gery S, Serve H, Brandts CH and Koeffler HP. Adaptor protein Lnk binds to PDGF receptor and inhibits PDGF-dependent signaling. Exp Hematol. 2011; 39(5):591-600.
113. Lin DC, Yin T, Koren-Michowitz M, Ding LW, Gueller S, Gery S, Tabayashi T, Bergholz U, Kazi JU, Ronnstrand L, Stocking C and Koeffler HP. Adaptor protein Lnk binds to and inhibits normal and leukemic FLT3. Blood. 2012; 120(16):3310-3317.
114. Gueller S, Goodridge HS, Niebuhr B, Xing H, Koren-Michowitz M, Serve H, Underhill DM, Brandts CH and Koeffler HP. Adaptor protein Lnk inhibits c-Fms-mediated macrophage function. J Leukoc Biol. 2010; 88(4):699-706.
115. Gueller S, Gery S, Nowak V, Liu L, Serve H and Koeffler HP. Adaptor protein Lnk associates with Tyr(568) in c-Kit. Biochem J. 2008; 415(2):241-245.
116. Wang TC, Chiu H, Chang YJ, Hsu TY, Chiu IM and Chen L. The adaptor protein SH2B3 (Lnk) negatively regulates neurite outgrowth of PC12 cells and cortical neurons. PLoS One. 2011; 6(10):e26433.
117. Gery S and Koeffler HP. Role of the adaptor protein LNK in normal and malignant hematopoiesis. Oncogene. 2013; 32(26):3111-3118.
118. Devalliere J and Charreau B. The adaptor Lnk (SH2B3): an emerging regulator in vascular cells and a link between immune and inflammatory signaling. Biochem Pharmacol. 2011; 82(10):1391-1402.
119. Buza-Vidas N, Antonchuk J, Qian H, Mansson R, Luc S, Zandi S, Anderson K, Takaki S, Nygren JM, Jensen CT and Jacobsen SE. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev. 2006; 20(15):2018-2023.
120. Tong W and Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004; 200(5):569-580.
121. Lee KL, Isham KR, Stringfellow L, Rothrock R and Kenney FT. Molecular cloning of cDNAs cognate to genes sensitive to hormonal control in rat liver. J Biol Chem. 1985; 260(30):16433-16438.
122. Fiorentino L, Pertica C, Fiorini M, Talora C, Crescenzi M, Castellani L, Alema S, Benedetti P and Segatto O. Inhibition of ErbB-2 mitogenic and transforming activity by RALT, a mitogen-induced signal transducer which binds to the ErbB-2 kinase domain. Molecular and cellular biology. 2000; 20(20):7735-7750.
123. Hackel PO, Gishizky M and Ullrich A. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol Chem. 2001; 382(12):1649-1662.
124. Zhang YW and Vande Woude GF. Mig-6, signal transduction, stress response and cancer. Cell Cycle. 2007; 6(5):507-513.
125. Fiorini M, Ballaro C, Sala G, Falcone G, Alema S and Segatto O. Expression of RALT, a feedback inhibitor of ErbB receptors, is subjected to an integrated transcriptional and post-translational control. Oncogene. 2002; 21(42):6530-6539.
126. Park SY, Choi HK, Seo JS, Yoo JY, Jeong JW, Choi Y, Choi KC and Yoon HG. DNAJB1 negatively regulates MIG6 to promote epidermal growth factor receptor signaling. Biochim Biophys Acta. 2015; 1853(10 Pt A):2722-2730.
127. Zhang X, Pickin KA, Bose R, Jura N, Cole PA and Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007; 450(7170):741-744.
128. Maity TK, Venugopalan A, Linnoila I, Cultraro CM, Giannakou A, Nemati R, Zhang X, Webster JD, Ritt D, Ghosal S, Hoschuetzky H, Simpson RM, Biswas R, Politi K, Morrison DK, Varmus HE, et al. Loss of MIG6 Accelerates Initiation and Progression of Mutant Epidermal Growth Factor Receptor-Driven Lung Adenocarcinoma. Cancer Discov. 5(5):534-549.
129. Park E, Kim N, Ficarro SB, Zhang Y, Lee BI, Cho A, Kim K, Park AK, Park WY, Murray B, Meyerson M, Beroukhim R, Marto JA, Cho J and Eck MJ. Structure and mechanism of activity-based inhibition of the EGF receptor by Mig6. Nature structural & molecular biology. 22(9):703-711.
130. Begley MJ, Yun CH, Gewinner CA, Asara JM, Johnson JL, Coyle AJ, Eck MJ, Apostolou I and Cantley LC. EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nature structural & molecular biology. 2015; Nov 9. doi: 10.1038/nsmb.3117
131. Frosi Y, Anastasi S, Ballaro C, Varsano G, Castellani L, Maspero E, Polo S, Alema S and Segatto O. A two-tiered mechanism of EGFR inhibition by RALT/MIG6 via kinase suppression and receptor degradation. J Cell Biol. 2010; 189(3):557-571.
132. Ying H, Zheng H, Scott K, Wiedemeyer R, Yan H, Lim C, Huang J, Dhakal S, Ivanova E, Xiao Y, Zhang H, Hu J, Stommel JM, Lee MA, Chen AJ, Paik JH, et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(15):6912-6917.
133. Pante G, Thompson J, Lamballe F, Iwata T, Ferby I, Barr FA, Davies AM, Maina F and Klein R. Mitogen-inducible gene 6 is an endogenous inhibitor of HGF/Met-induced cell migration and neurite growth. J Cell Biol. 2005; 171(2):337-348.
134. Hopkins S, Linderoth E, Hantschel O, Suarez-Henriques P, Pilia G, Kendrick H, Smalley MJ, Superti-Furga G and Ferby I. Mig6 is a sensor of EGF receptor inactivation that directly activates c-Abl to induce apoptosis during epithelial homeostasis. Dev Cell. 2012; 23(3):547-559.
135. Ballaro C, Ceccarelli S, Tiveron C, Tatangelo L, Salvatore AM, Segatto O and Alema S. Targeted expression of RALT in mouse skin inhibits epidermal growth factor receptor signalling and generates a Waved-like phenotype. EMBO Rep. 2005; 6(8):755-761.
136. Anastasi S, Sala G, Huiping C, Caprini E, Russo G, Iacovelli S, Lucini F, Ingvarsson S and Segatto O. Loss of RALT/MIG-6 expression in ERBB2-amplified breast carcinomas enhances ErbB-2 oncogenic potency and favors resistance to Herceptin. Oncogene. 2005; 24(28):4540-4548.
137. Anastasi S, Fiorentino L, Fiorini M, Fraioli R, Sala G, Castellani L, Alema S, Alimandi M and Segatto O. Feedback inhibition by RALT controls signal output by the ErbB network. Oncogene. 2003; 22(27):4221-4234.
138. Xu D, Makkinje A and Kyriakis JM. Gene 33 is an endogenous inhibitor of epidermal growth factor (EGF) receptor signaling and mediates dexamethasone-induced suppression of EGF function. J Biol Chem. 2005; 280(4):2924-2933.
139. Reschke M, Ferby I, Stepniak E, Seitzer N, Horst D, Wagner EF and Ullrich A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology. 2010; 51(4):1383-1390.
140. Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, Sommergruber W, Kraut N, Ullrich A, Fassler R and Klein R. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006; 12(5):568-573.
141. Zhang YW, Staal B, Su Y, Swiatek P, Zhao P, Cao B, Resau J, Sigler R, Bronson R and Vande Woude GF. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene. 2007; 26(2):269-276.
142. Inagaki-Ohara K, Kondo T, Ito M and Yoshimura A. SOCS, inflammation, and cancer. JAKSTAT. 2013; 2(3):e24053.
143. Rottapel R, Ilangumaran S, Neale C, La Rose J, Ho JM, Nguyen MH, Barber D, Dubreuil P and de Sepulveda P. The tumor suppressor activity of SOCS-1. Oncogene. 2002; 21(28):4351-4362.
144. Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K, Mattfeldt T, Barth TF and Moller P. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene. 2006; 25(18):2679-2684.
145. Mottok A, Renne C, Willenbrock K, Hansmann ML and Brauninger A. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood. 2007; 110(9):3387-3390.
146. Mottok A, Renne C, Seifert M, Oppermann E, Bechstein W, Hansmann ML, Kuppers R and Brauninger A. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood. 2009; 114(20):4503-4506.
147. Melzner I, Bucur AJ, Bruderlein S, Dorsch K, Hasel C, Barth TF, Leithauser F and Moller P. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood. 2005; 105(6):2535-2542.
148. Chen CY, Tsay W, Tang JL, Shen HL, Lin SW, Huang SY, Yao M, Chen YC, Shen MC, Wang CH and Tien HF. SOCS1 methylation in patients with newly diagnosed acute myeloid leukemia. Genes Chromosomes Cancer. 2003; 37(3):300-305.
149. Watanabe D, Ezoe S, Fujimoto M, Kimura A, Saito Y, Nagai H, Tachibana I, Matsumura I, Tanaka T, Kanegane H, Miyawaki T, Emi M, Kanakura Y, Kawase I, Naka T and Kishimoto T. Suppressor of cytokine signalling-1 gene silencing in acute myeloid leukaemia and human haematopoietic cell lines. Br J Haematol. 2004; 126(5):726-735.
150. Liu TC, Lin SF, Chang JG, Yang MY, Hung SY and Chang CS. Epigenetic alteration of the SOCS1 gene in chronic myeloid leukaemia. Br J Haematol. 2003; 123(4):654-661.
151. Jost E, do ON, Dahl E, Maintz CE, Jousten P, Habets L, Wilop S, Herman JG, Osieka R and Galm O. Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia. 2007; 21(3):505-510.
152. Sobti RC, Singh N, Hussain S, Suri V, Nijhawan R, Bharti AC, Bharadwaj M and Das BC. Aberrant promoter methylation and loss of suppressor of cytokine signalling-1 gene expression in the development of uterine cervical carcinogenesis. Cell Oncol (Dordr). 2011; 34(6):533-543.
153. Hussain S, Singh N, Salam I, Bandil K, Yuvaraj M, Akbar Bhat M, Muzaffar Mir M, Siddiqi MA, Sobti RC, Bharadwaj M and Das BC. Methylation-mediated gene silencing of suppressor of cytokine signaling-1 (SOCS-1) gene in esophageal squamous cell carcinoma patients of Kashmir valley. J Recept Signal Transduct Res. 2011; 31(2):147-156.
154. Tischoff I, Hengge UR, Vieth M, Ell C, Stolte M, Weber A, Schmidt WE and Tannapfel A. Methylation of SOCS-3 and SOCS-1 in the carcinogenesis of Barrett’s adenocarcinoma. Gut. 2007; 56(8):1047-1053.
155. Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, Campbell IG and Visvader JE. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 2004; 23(46):7726-7733.
156. Nagai H, Kim YS, Konishi N, Baba M, Kubota T, Yoshimura A and Emi M. Combined hypermethylation and chromosome loss associated with inactivation of SSI-1/SOCS-1/JAB gene in human hepatocellular carcinomas. Cancer Lett. 2002; 186(1):59-65.
157. Zhou H, Miki R, Eeva M, Fike FM, Seligson D, Yang L, Yoshimura A, Teitell MA, Jamieson CA and Cacalano NA. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res. 2007; 13(8):2344-2353.
158. Sasi W, Jiang WG, Sharma A and Mokbel K. Higher expression levels of SOCS 1,3,4,7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer. 2010; 10:178.
159. David M, Naudin C, Letourneur M, Polrot M, Renoir JM, Lazar V, Dessen P, Roche S, Bertoglio J and Pierre J. Suppressor of cytokine signaling 1 modulates invasion and metastatic potential of colorectal cancer cells. Mol Oncol. 2014; 8(5):942-955.
160. Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H, Kato S and Yoshimura A. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006; 203(6):1391-1397.
161. Suessmuth Y, Elliott J, Percy MJ, Inami M, Attal H, Harrison CN, Inokuchi K, McMullin MF and Johnston JA. A new polycythaemia vera-associated SOCS3 SH2 mutant (SOCS3F136L) cannot regulate erythropoietin responses. Br J Haematol. 2009; 147(4):450-458.
162. Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005; 24(42):6406-6417.
163. Lindemann C, Hackmann O, Delic S, Schmidt N, Reifenberger G and Riemenschneider MJ. SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol. 2011; 122(2):241-251.
164. Isomoto H, Mott JL, Kobayashi S, Werneburg NW, Bronk SF, Haan S and Gores GJ. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007; 132(1):384-396.
165. Nakagawa T, Iida S, Osanai T, Uetake H, Aruga T, Toriya Y, Takagi Y, Kawachi H and Sugihara K. Decreased expression of SOCS-3 mRNA in breast cancer with lymph node metastasis. Oncol Rep. 2008; 19(1):33-39.
166. Zhang L, Li J, Li L, Zhang J, Wang X, Yang C, Li Y, Lan F and Lin P. IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis. 2014; 35(6):1330-1340.
167. Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS and Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007; 26(33):4833-4841.
168. Li Z, Metze D, Nashan D, Muller-Tidow C, Serve HL, Poremba C, Luger TA and Bohm M. Expression of SOCS-1, suppressor of cytokine signalling-1, in human melanoma. J Invest Dermatol. 2004; 123(4):737-745.
169. Krishnadasan R, Bifulco C, Kim J, Rodov S, Zieske AW and Vanasse GJ. Overexpression of SOCS3 is associated with decreased survival in a cohort of patients with de novo follicular lymphoma. Br J Haematol. 2006; 135(1):72-75.
170. Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W, Percy MJ, McMullin MF, Constantinescu SN and Johnston JA. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood. 2007; 109(11):4924-4929.
171. van de Geijn GJ, Gits J, Aarts LH, Heijmans-Antonissen C and Touw IP. G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood. 2004; 104(3):667-674.
172. Leroy C, Fialin C, Sirvent A, Simon V, Urbach S, Poncet J, Robert B, Jouin P and Roche S. Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Res. 2009; 69(6):2279-2286.
173. Sirvent A, Benistant C and Roche S. Oncogenic signaling by tyrosine kinases of the SRC family in advanced colorectal cancer. Am J Cancer Res. 2012; 2(4):357-371.
174. Okino K, Konishi H, Doi D, Yoneyama K, Ota Y, Jin E, Kawanami O and Takeshita T. Up-regulation of growth factor receptor-bound protein 10 in cervical squamous cell carcinoma. Oncol Rep. 2005; 13(6):1069-1074.
175. Kazi JU and Ronnstrand L. FLT3 signals via the adapter protein Grb10 and overexpression of Grb10 leads to aberrant cell proliferation in acute myeloid leukemia. Mol Oncol. 2013; 7(3):402-418.
176. Illert AL, Albers C, Kreutmair S, Leischner H, Peschel C, Miething C and Duyster J. Grb10 is involved in BCR-ABL-positive leukemia in mice. Leukemia. 2014.
177. Duval A, Rolland S, Compoint A, Tubacher E, Iacopetta B, Thomas G and Hamelin R. Evolution of instability at coding and non-coding repeat sequences in human MSI-H colorectal cancers. Hum Mol Genet. 2001; 10(5):513-518.
178. Bersenev A, Wu C, Balcerek J, Jing J, Kundu M, Blobel GA, Chikwava KR and Tong W. Lnk constrains myeloproliferative diseases in mice. The Journal of clinical investigation. 2010; 120(6):2058-2069.
179. Gery S, Gueller S, Chumakova K, Kawamata N, Liu L and Koeffler HP. Adaptor protein Lnk negatively regulates the mutant MPL, MPLW515L associated with myeloproliferative disorders. Blood. 2007; 110(9):3360-3364.
180. Gery S, Gueller S, Nowak V, Sohn J, Hofmann WK and Koeffler HP. Expression of the adaptor protein Lnk in leukemia cells. Exp Hematol. 2009; 37(5):585-592 e582.
181. Gery S, Cao Q, Gueller S, Xing H, Tefferi A and Koeffler HP. Lnk inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J Leukoc Biol. 2009; 85(6):957-965.
182. Ding LW, Sun QY, Lin DC, Chien W, Hattori N, Dong XM, Gery S, Garg M, Doan NB, Said JW, Xiao JF, Yang H, Liu LZ, Meng X, Huang RY, Tang K, et al. LNK (SH2B3): paradoxical effects in ovarian cancer. Oncogene. 2015; 34(11):1463-1474.
183. Amatschek S, Koenig U, Auer H, Steinlein P, Pacher M, Gruenfelder A, Dekan G, Vogl S, Kubista E, Heider KH, Stratowa C, Schreiber M and Sommergruber W. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res. 2004; 64(3):844-856.
184. Li Z, Qu L, Zhong H, Xu K, Qiu X and Wang E. Low expression of Mig-6 is associated with poor survival outcome in NSCLC and inhibits cell apoptosis via ERK-mediated upregulation of Bcl-2. Oncol Rep. 2014; 31(4):1707-1714.
185. Duncan CG, Killela PJ, Payne CA, Lampson B, Chen WC, Liu J, Solomon D, Waldman T, Towers AJ, Gregory SG, McDonald KL, McLendon RE, Bigner DD and Yan H. Integrated genomic analyses identify ERRFI1 and TACC3 as glioblastoma-targeted genes. Oncotarget. 2010; 1(4):265-277. doi: 10.18632/oncotarget.137.
186. Lin CI, Du J, Shen WT, Whang EE, Donner DB, Griff N, He F, Moore FD, Jr., Clark OH and Ruan DT. Mitogen-inducible gene-6 is a multifunctional adaptor protein with tumor suppressor-like activity in papillary thyroid cancer. J Clin Endocrinol Metab. 2011; 96(3):E554-565.
187. Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, Calin G, Wang H, Siefker-Radtke A, McConkey D, Bar-Eli M and Dinney C. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009; 15(16):5060-5072.
188. Chang X, Izumchenko E, Solis LM, Kim MS, Chatterjee A, Ling S, Monitto CL, Harari PM, Hidalgo M, Goodman SN, Wistuba, II, Bedi A and Sidransky D. The relative expression of Mig6 and EGFR is associated with resistance to EGFR kinase inhibitors. PLoS One. 2013; 8(7):e68966.
189. Roman-Gomez J, Jimenez-Velasco A, Castillejo JA, Cervantes F, Barrios M, Colomer D, Heiniger A and Torres A. The suppressor of cytokine signaling-1 is constitutively expressed in chronic myeloid leukemia and correlates with poor cytogenetic response to interferon-alpha. Haematologica. 2004; 89(1):42-48.
190. Hatirnaz O, Ure U, Ar C, Akyerli C, Soysal T, Ferhanoglu B, Ozcelik T and Ozbek U. The SOCS-1 gene methylation in chronic myeloid leukemia patients. Am J Hematol. 2007; 82(8):729-730.
191. Sakai I, Takeuchi K, Yamauchi H, Narumi H and Fujita S. Constitutive expression of SOCS3 confers resistance to IFN-alpha in chronic myelogenous leukemia cells. Blood. 2002; 100(8):2926-2931.
192. Zitzmann K, Brand S, De Toni EN, Baehs S, Goke B, Meinecke J, Spottl G, Meyer HH and Auernhammer CJ. SOCS1 silencing enhances antitumor activity of type I IFNs by regulating apoptosis in neuroendocrine tumor cells. Cancer Res. 2007; 67(10):5025-5032.
193. Lesinski GB, Zimmerer JM, Kreiner M, Trefry J, Bill MA, Young GS, Becknell B and Carson WE, 3rd. Modulation of SOCS protein expression influences the interferon responsiveness of human melanoma cells. BMC Cancer. 2010; 10:142.
194. Zhang J, Li H, Yu JP, Wang SE and Ren XB. Role of SOCS1 in tumor progression and therapeutic application. Int J Cancer. 2012; 130(9):1971-1980.
195. Quentmeier H, Geffers R, Jost E, Macleod RA, Nagel S, Rohrs S, Romani J, Scherr M, Zaborski M and Drexler HG. SOCS2: inhibitor of JAK2V617F-mediated signal transduction. Leukemia. 2008; 22(12):2169-2175.
196. Etienne A, Carbuccia N, Adelaide J, Bekhouche I, Remy V, Sohn C, Sainty D, Gastaut JA, Olschwang S, Birnbaum D, Mozziconacci MJ and Chaffanet M. Rearrangements involving 12q in myeloproliferative disorders: possible role of HMGA2 and SOCS2 genes. Cancer Genet Cytogenet. 2007; 176(1):80-88.
197. Qiu X, Zheng J, Guo X, Gao X, Liu H, Tu Y and Zhang Y. Reduced expression of SOCS2 and SOCS6 in hepatocellular carcinoma correlates with aggressive tumor progression and poor prognosis. Mol Cell Biochem. 2013; 378(1-2):99-106.
198. Zhu JG, Dai QS, Han ZD, He HC, Mo RJ, Chen G, Chen YF, Wu YD, Yang SB, Jiang FN, Chen WH, Sun ZL and Zhong WD. Expression of SOCSs in human prostate cancer and their association in prognosis. Mol Cell Biochem. 2013; 381(1-2):51-59.
199. Iglesias-Gato D, Chuan YC, Wikstrom P, Augsten S, Jiang N, Niu Y, Seipel A, Danneman D, Vermeij M, Fernandez-Perez L, Jenster G, Egevad L, Norstedt G and Flores-Morales A. SOCS2 mediates the cross talk between androgen and growth hormone signaling in prostate cancer. Carcinogenesis. 2014; 35(1):24-33.
200. Bogazzi F, Ultimieri F, Raggi F, Russo D, Costa A, Marciano E, Bartalena L and Martino E. Changes in the expression of suppressor of cytokine signalling (SOCS) 2 in the colonic mucosa of acromegalic patients are associated with hyperplastic polyps. Clin Endocrinol (Oxf). 2009; 70(6):898-906.
201. Zheng C, Li L, Haak M, Brors B, Frank O, Giehl M, Fabarius A, Schatz M, Weisser A, Lorentz C, Gretz N, Hehlmann R, Hochhaus A and Seifarth W. Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia. 2006; 20(6):1028-1034.
202. Schultheis B, Carapeti-Marootian M, Hochhaus A, Weisser A, Goldman JM and Melo JV. Overexpression of SOCS-2 in advanced stages of chronic myeloid leukemia: possible inadequacy of a negative feedback mechanism. Blood. 2002; 99(5):1766-1775.
203. Kazi JU and Ronnstrand L. Suppressor of cytokine signaling 2 (SOCS2) associates with FLT3 and negatively regulates downstream signaling. Mol Oncol. 2013; 7(3):693-703.
204. Fourouclas N, Li J, Gilby DC, Campbell PJ, Beer PA, Boyd EM, Goodeve AC, Bareford D, Harrison CN, Reilly JT, Green AR and Bench AJ. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica. 2008; 93(11):1635-1644.
205. He B, You L, Uematsu K, Zang K, Xu Z, Lee AY, Costello JF, McCormick F and Jablons DM. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100(24):14133-14138.
206. Pierconti F, Martini M, Pinto F, Cenci T, Capodimonti S, Calarco A, Bassi PF and Larocca LM. Epigenetic silencing of SOCS3 identifies a subset of prostate cancer with an aggressive behavior. Prostate. 2011; 71(3):318-325.
207. Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, Dietz A, Wittekind C and Tannapfel A. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005; 24(44):6699-6708.
208. Kobayashi D, Nomoto S, Kodera Y, Fujiwara M, Koike M, Nakayama G, Ohashi N and Nakao A. Suppressor of cytokine signaling 4 detected as a novel gastric cancer suppressor gene using double combination array analysis. World J Surg. 2012; 36(2):362-372.
209. Yoon S, Yi YS, Kim SS, Kim JH, Park WS and Nam SW. SOCS5 and SOCS6 have similar expression patterns in normal and cancer tissues. Tumour Biol. 2012; 33(1):215-221.
210. Lai RH, Hsiao YW, Wang MJ, Lin HY, Wu CW, Chi CW, Li AF, Jou YS and Chen JY. SOCS6, down-regulated in gastric cancer, inhibits cell proliferation and colony formation. Cancer Lett. 2010; 288(1):75-85.
211. Storojeva I, Boulay JL, Ballabeni P, Buess M, Terracciano L, Laffer U, Mild G, Herrmann R and Rochlitz C. Prognostic and predictive relevance of DNAM-1, SOCS6 and CADH-7 genes on chromosome 18q in colorectal cancer. Oncology. 2005; 68(2-3):246-255.
212. Sriram KB, Larsen JE, Savarimuthu Francis SM, Wright CM, Clarke BE, Duhig EE, Brown KM, Hayward NK, Yang IA, Bowman RV and Fong KM. Array-comparative genomic hybridization reveals loss of SOCS6 is associated with poor prognosis in primary lung squamous cell carcinoma. PLoS One. 2012; 7(2):e30398.