INTRODUCTION
Lynch Syndrome is caused by mutations in one out of four mismatch repair (MMR) genes - MLH1, MSH2, MSH6 and PMS2 - and results in a 25% to 75% lifetime risk of colorectal cancer and 60% risk of endometrial cancer in women [1].
Genetic testing helps estimating the individual risk, plan appropriate care, screening and prophylactic treatments. Nevertheless, the risk to develop cancer conferred by a MMR mutation is modified by both the environment and genetic risk modifiers. The identification of such genetic risk modifiers can improve risk prediction and genetic counseling, through the individualization of surveillance programs and the evaluation of benefits versus burdens associated with prophylactic strategies [1, 2].
Searching for genetic risk modifiers is a challenge due to their expected small effect-size and their non-pathogenicity in the absence of a pathogenic mutation [2–4]. Genome-wide-association studies have identified loci and single nucleotide polymorphisms (SNPs) with risk modifying effects [5–8]. More recently, Next Generation Sequencing (NGS) of the whole genome or combined with gene-panels is emerging in genetic diagnostic to detect germ-line variants predisposing to cancer [4, 9–11].
Here, NGS combined with a 154 gene-panel was used to identify rare variants (minor allele frequency, MAF, <0.001) acting as gene-modifiers of Lynch syndrome MMR mutations. Unlike cancer somatic mutations, which map exclusively on tumor suppressors and oncogenes, the carcinogenic effect of germ-line variants can be indirect [2, 4, 11], hence they can map within but also outside the genes classically associated with cancer. A defect in the genome stability conferred by a Lynch syndrome MMR mutation can be aggravated by disturbed tissue homeostasis. Therefore, as proof of principle for Lynch syndrome related endometrial cancer, a panel that included genes controlling the endometrial physiology and homeostasis (i.e. hormone signaling) beside those associated with cancer was complied. A family based approach was used: the presence of candidate genetic risk modifiers was evaluated among 35 patients carrying already a MMR mutation and characterized by either a poor clinical phenotype (early age of diagnosis or the diagnosis of multiple cancers) or by a neutral clinical outcome (diagnosis with endometrial cancer only and late in life).
RESULTS
Patients
Table 1 shows the clinical features of the women enrolled. All were diagnosed with endometrial cancer between 31–81 years (mean: 53.1 ± 10.7). Subjects belonged to 29 families with a Lynch syndrome mismatch repair (MMR) pathogenic mutation, and all women carried the mutation. These mutations will be referred to as ‘familial MMR mutations’ (Table 1). One familial MMR mutation in MSH6 (c.3729_3732dupATTA – p.(Phe1245Ilefs*31)) was a founder mutation common to nine subjects belonging to six families.
Table 1: Overview of the study population
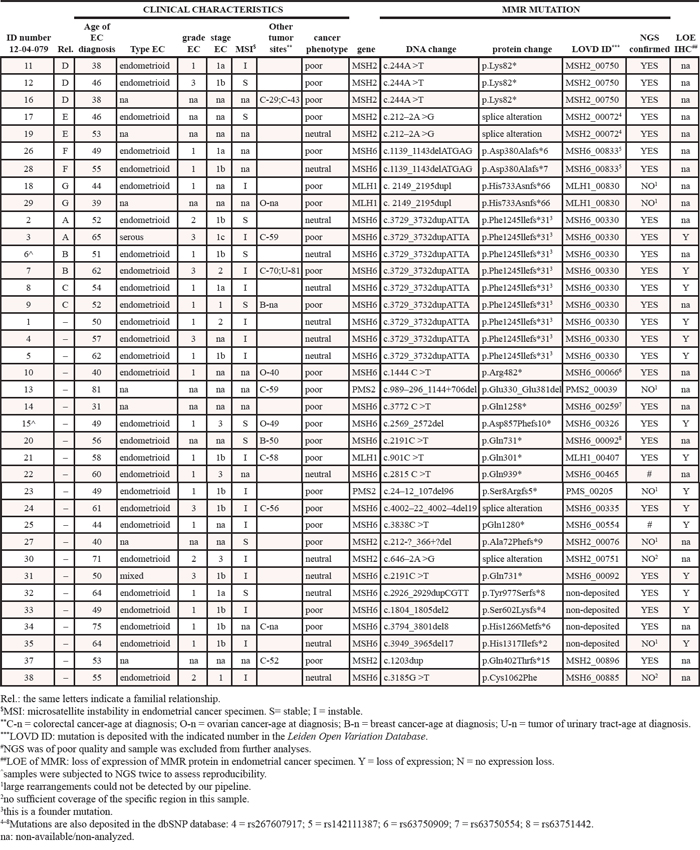
Eleven women were diagnosed with a second Lynch syndrome tumor (two in the breast, six in the intestine, three in the ovaries). One woman developed colorectal and urinary tract cancers and one was diagnosed twice with colorectal cancer (14 years apart). Additionally, 14 women developed endometrial cancer at the age of 49 or earlier. In the following analyses, women diagnosed with more than one cancer, or with endometrial cancer before 50 (49 or younger) are defined as patients with poor clinical phenotypes (or outcome, n = 23), whereas 14 patients who developed only endometrial cancer and after the age of 50 are defined as having a neutral clinical phenotype.
Estrogen and cancer related 154 gene-panel
The coding sequences and splice regions (10 non-coding nucleotides at each exon end) of 154 genes implied in endometrial physiology and carcinogenesis were explored by Next Generation Sequencing (NGS). Table 2 overviews the captured panel and Supplemental Table S1 gives the complete list of genes and the full set of related references.
Table 2: Overview of the panel design of 154 estrogen and cancer associated genes with the variants identified in 35 subjects analyzed
Capture plan |
Variants identified |
||||||||
|---|---|---|---|---|---|---|---|---|---|
Total |
dbSNP deposited* |
non-dbSNP deposited |
|||||||
No of genes |
nt (Kb) |
No |
Missense + non-sense |
Silent |
Intron-exon |
Missense + non-sense + ins/del |
Silent |
Intron-exon |
|
ESTROGEN |
47 |
101.407 |
25 |
9 |
4 |
0 |
11 |
0 |
1 |
ligand metabolism |
10 |
13.501 |
3 |
0 |
0 |
0 |
3 |
0 |
0 |
target genes |
24 |
46.847 |
12 |
6 |
2 |
0 |
3 |
0 |
1 |
responsive genes |
7 |
10.539 |
1 |
0 |
0 |
0 |
1 |
0 |
0 |
co-regulators |
6 |
30.52 |
9 |
3 |
2 |
0 |
4 |
0 |
0 |
ONCOGENES |
35 |
112.56 |
24 |
3 |
5 |
2 |
8 |
6 |
0 |
TUMOUR SUPPRESS |
63 |
162.306 |
40 |
17 |
9 |
0 |
8 |
4 |
2 |
OTHER |
9 |
19.395 |
9 |
0 |
3 |
0 |
5 |
0 |
1 |
TOTAL |
154 |
395.668 |
98 |
29 |
21 |
2 |
32 |
10 |
4 |
*variants deposited in the dbSNP database with a MAF <0.001.
The list of all variants is given in Supplemental Table S3.
Estrogens are important physiological regulators in the endometrium and risk factors for cancer [12]. Hence, the capture panel included 47 genes involved in estrogen (and progesterone) signaling, comprising genes controlling the metabolism and degradation of steroids [12], genes directly targeted by the hormone receptors [16–18], genes responsive to steroids [17, 19] and genes encoding for co-regulators controlling the hormone receptor transcriptional activity [16–18]. In addition, 35 oncogenes and 63 tumor suppressors were captured [10, 20–22]. NGS and Quality Control analyses are described as Supplemental Materials (Supplemental Methods, Supplemental Figures S1, 2 and Supplemental Table S2). Thirty-seven DNA samples were sequenced but two were excluded due to low quality (12–04-079_22 and 12–04-079_25, Supplemental Materials and Table 1). As control of NGS and raw data analyses, two samples (12–04-079_6 and 12–04-079_15) were randomly selected and subjected to one additional and independent library preparation and NGS run and they showed 100% concordance between called variants.
Variant analyses
Among the 35 patient’s DNA successfully sequenced, 865 independent variants deposited in dbSNP database were identified and each subject carried on average 245 single nucleotide polymorphisms (SNPs; range 207–270). Common SNPs were excluded from further analyses, but since rare dbSNP variants can be relevant in our search for genetic risk modifiers [23, 24], 52 variants with a minor allele frequency (MAF) lower than 0.001 were retrieved (Table 2). Of these, 28 were missense, one non-sense (stop), 21 were silent and two located in non-coding intron/exon boundaries. In addition, 46 identified variants were not reported in dbSNP, and 34 of these were never deposited before in any database (ExAC, Cosmic, LOVD and HGMD were checked; Table 2). Twenty-nine missense, one frame-shift non-sense, one in frame deletion and one in frame insertion of one amino-acid were identified plus 10 silent variants and four changes located in the non-coding regions flanking an exon (Table 2).
In total, 98 rare variants were identified (dbSNP with MAF <0.001 and non-dbSNP deposited). Considering only variants affecting the amino-acid sequence, there were 57 missense, one amino-acid insertion, one deletion (both in-frame) and two non-sense changes. All variants were heterozygous (Supplemental Table S3).
Putatively genetic risk modifiers: risk-variants
To predict the functional consequences and potential risk modifying effects, variants were curated using defined criteria [23–25], literature, controlling manually and in combination with bioinformatics tools such as: Alamut (version 2.4, Interactive Biosoftware, including SIFT and PolyPhen-2), Uniprot, PhosphoSitePlus® (for three-dimensional structure and post-translational modifications) [15], NCBI Clinical Variants, ExAC, NHLBI ESP, LOVD, HGMD, Cosmic.
Variants outside the MMR genes were analyzed first. The 98 rare variants reported earlier were considered plus three SNPs with a MAF ≥ 0.001 previously reported as pathogenic or disease related. These were: rs57865060, chromosome position Chr15:75012998:delT, with predicted protein change CYP1B1:p.(Glu229Lys); rs121918303, Chr13:32351535:A > C with predicted protein change RXFP2:p.(Thr222Pro); rs56378716, Chr17:56356502: A > G, with predicted protein change MPO:p.(Met251Thr).
The resulting 101 variants (Supplemental Table S3) were categorized in five groups according to the recommendations of the Dutch and British societies for clinical genetics [24]: categories 1 and 2 = no effect; 3 = variants of unknown significance – VUS; 4 = likely pathogenic; 5 = pathogenic. Silent and non-coding variants were categorized as 1 (none was predicted to affect splicing or other regulatory functions). Conservative amino acid changes were considered unlikely to affect the protein function (category 2), with the exclusion of four variants that had been associated with disease or were predicted to have damaging effects (Alamut prediction tool), which were categorized as 3. The remaining class 3 variants were all non-conservative protein changes. Eight variants were in category 4, four of which were pathogenic (class 5) for disorders other than endometrial cancer, hence they were considered as class 4 in the present study. Two independent investigators (RA and BMJ, of which BMJ is a Clinical Laboratory Geneticist with expertise in diagnostics and oncogenetics) performed this curation blindly for each other’s results. Supplemental Table S3 gives the curation details and the complete variant list.
Since genetic risk modifiers have modest effects (see discussion), all variants categorized as 3 (provided the presence of a non-conservative amino-acid substitution) or higher were considered and resulted in 54 candidate risk-modifiers. Such variants, referred to as ‘risk-variants’ from now on, were further characterized for the following features (Supplemental Table S3): i) the presence of cancer somatic (not silent) mutations in the same protein region (i.e. max +/−3 amino acids from the identified risk-variant; Cosmic database). This was considered as an indication that changes in the specific protein region could affect function and predisposition to cancer; ii) the interspecies protein domain conservation; iii) the presence of the same risk-variant among relatives; iv) the occurrence of the risk-variant in relevant protein domains; v) the predicted consequences based on Alamut, protein in silico analysis, literature mining or the occurrence of other pathogenic variants/SNPs in the same region; vi) we excluded that risk-variants could co-segregate with the familial MMR mutation by verifying their localization on separate chromosomes (Supplemental Table S3).
The majority of the patients carried one (or more) risk-variant in estrogen related genes (19 out of 35 patients, 54%) and 18 risk-variants were unique (i.e. the same variants occurring in relatives were counted only once: n = 5). Risk-variants in tumor suppressors were observed in 18 patients (49%), and 25 were unique. One (or more) oncogene contained nucleotide changes in nine patients (26%), and nine were unique.
MMR genes also contained three risk-variants beside the familial mutation and these have not been computed in the list of 101 variants given so far (Figure 1A). Two variants, one in PMS2 -rs116788608, Chr7:6035211:T > C, with predicted protein change PMS2:p.(Asp286Gly)- and the second in MSH2 - rs41295288, Chr2:47702191:A > G with predicted p.(Asp596Ser)- were on other chromosomes than the familial MMR mutation (MSH6) of these subjects. In contrast, MSH6 variant with predicted protein change p.(Ala25Ser) (rs267608026, Chr2:48010445:G > T) in patient 12–04-079_32 could co-segregate with the MSH6 familial mutation carried by this subject. Besides these three risk-variants, three additional variants in MMR genes with MAF < 0.001 were classified in category 2 (Figure 1A).
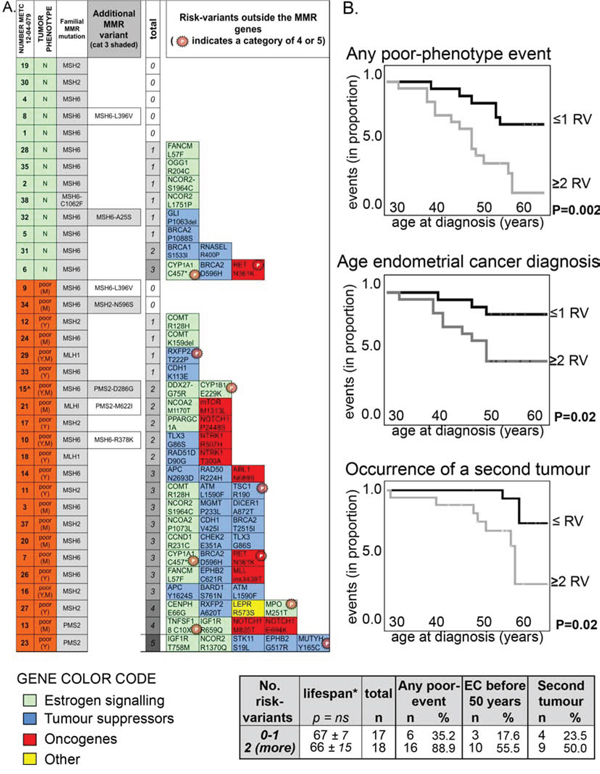
Figure 1: The number of class 3 (or more) risk-variants correlates with poor clinical phenotypes. A. Heat map showing the presence of class 3 (or more) risk-variants in each subject with neutral phenotype (green, N) or with a poor phenotype (orange, poor; Y = young age of endometrial cancer diagnosis; M = cancer at multiple sites). The familial MMR mutations (all non-sense but the one carried by subject 12–04-079_38) are indicated (3rd column). ‘Additional MRR variants’ (4th column) reports the six rare variants (MAF < 0.001) found in MMR genes among seven subjects. In particular, MSH6:p.(Arg378Lys) in subject 12–04-079_10 has been already described to be in linkage with the familial MSH6:p.(Arg482*) mutation detected in the patient [39]. ‘Total’ (5th column) refers to the total number of risk-variants outside the MMR genes identified in each patient. The specific risk-variants are reported at the right as gene name and predicted amino-acid change (for full chromosomal coordinates/nucleotide change, see Supplemental Table S3), together with a color code that indicates the gene function. Variants previously reported as (likely) pathogenic (categories 4 and 5) are labeled with a starred ‘P’. B. Kaplan-Meier curves showing the occurrence of any poor-clinical phenotype feature (Top chart; first events were only considered for statistic), of the diagnosis of endometrial cancer before 50 (Middle chart), or of the occurrence of a second tumor (Bottom chart; first events were only considered for statistic) in subjects carrying a maximum of one risk-variant (indicated as RV) compared to subjects carrying two or more risk-variants outside the MMR gene. The statistical p for significance is computed by Log Rank (Mantel-Cox, Test of equality of survival distributions for the different levels of risk-variants; SPSS).
Risk-variants are associated with poor clinical phenotypes
The distribution of MMR pathogenic mutations did not vary between women with poor and neutral clinical phenotypes (Table 3), but patients with poor clinical phenotypes carried more frequently one or more risk-variants outside the MMR genes compared with women with a neutral phenotype (Table 3 and Figure 1A).
Table 3: distribution of familial MMR mutations and risk-variants outside the MMR genes (class-3 or more, i.e. putatively risk modifiers) among patients with neutral and poor clinical phenotypes
Total subjects |
Neutral tumor phenotype |
Poor tumor phenotype |
||||
|---|---|---|---|---|---|---|
No. |
No. |
p-value |
||||
14** |
23** |
|||||
Lifespan* |
64.2 ± 6.0 |
66.8 ± 14.1 |
ns# |
|||
FAMILIAL MMR MUTATIONS |
No. |
% |
No. |
% |
p-value |
|
MLH1 |
0 |
0.0 |
3 |
13.6 |
ns##* |
|
MSH2 |
2 |
15.4 |
6 |
27.3 |
||
MSH6 |
12 |
92.3 |
12 |
54.5 |
||
PMS2 |
0 |
0.0 |
2 |
9.1 |
||
PRESENCE OF ADDITIONAL RISK-VARIANTS*** |
no variant |
5 |
38.5 |
2 |
9.1 |
0.0471##@ |
one variant |
6 |
46.2 |
4 |
18.2 |
||
two variants |
1 |
7.7 |
5 |
22.7 |
||
three variants |
1 |
7.7 |
8 |
36.4 |
||
four variants |
0 |
0.0 |
2 |
9.1 |
||
five variants |
0 |
0.0 |
1 |
4.5 |
||
0 or 1 variant |
11 |
84.6 |
6 |
27.3 |
0.0016### |
|
2 or more var. |
2 |
15.4 |
16 |
72.7 |
||
*age of subjects in 2013 was used as lifespan (mean age ± standard deviation in years).
**13 and 22 subjects were successfully analyzed by NGS.
***risk-variants, i.e. class-3 or higher, in genes other than the MMR were considered.
#T-test to compare mean values.
##*Chi square, with 3 degrees of freedom.
##@Chi square, with 5 degrees of freedom, Pearson’s: 11.225.
###Fisher exact test, two sided p-value.
ns = non-significant.
Cox-regression analysis confirmed that increasing number of risk-variants was associated with a poor clinical phenotype (Supplemental Figure S3). In these Kaplan-Meier analyses the curves of subjects with no or one risk-variant overlap with each other and are separated from the curves of subjects with two, three or more than four risk-variants. Table 3 (rows at the bottom) also shows a highest statistical significance by combining these two groups. Because of this net distinction between subjects carrying no or one risk-variant versus women carrying more than two risk-variants, in subsequent Kaplan-Meier cox-regression analyses we combined patients in these two groups (Figure 1B). The presence of two or more risk-variants beside the familial MMR mutation was associated with the occurrence of poor clinical features. As defined earlier, poor clinical features were either a diagnosis of endometrial cancer at a young age (<50), or the onset of multiple cancers. Subsequently, these two characteristics were assessed separately and the presence of two or more risk-variants was associated with both (Figure 1B). The lifespan among patients with neutral and poor clinical phenotypes, extrapolated at 2013 (the date of last update of our database), did not vary significantly between groups (Table 3, Figure 1B and Supplemental Figure S3).
To exclude that multiple risk-variants could co-occur because of co-segregation, we verified that all risk-variants in one subject were located on separate chromosomes. This was confirmed for 16 out of the 18 subjects. Subject 12–04-079_14 carried risk-variants Chr5:112179368:A > G (APC:p.(Asn2693Asp)) and Chr5:131915673:G > A (RAD50:p.(Arg224His)) both on chromosome 5 (at a distance of about 90 Mb). A second patient (12–04-079_13) carried two risk-variants in the same gene (NOTCH1). Results did not change by considering these four risk-variants as two co-segregating haplotypes.
Three possible risk-variants (category 3 or higher) were identified in MMR genes (grey shaded in Figure 1A), which were not considered in the aforementioned analyses because a linkage with the MMR familial mutation could not be excluded. Nevertheless, when these risk-variants were included, results did not change. In fact, two risk-variants that lied certainly in other chromosomes than the MMR familial mutation – Chr7:6035211:T > C, predicted protein change PMS2:p.(Asp286Gly) and Chr2:47702191:A > G with predicted MSH2:p(Asp596Ser)- occurred in patients with a poor clinical phenotype, whereas risk-variant Chr2:48010445:G > T, with predicted change MSH6:p.(Ala25Ser), which could be in linkage with the familial mutation, occurred in subject 12–04-079_32 having a neutral clinical phenotype.
Among 15 subjects there were some 1st and 2nd degree parental relationships and they belonged to 7 families. Among the five families in which members had distinct neutral or poor clinical phenotypes, there were three families (A, E and F) in which the woman with poor phenotype carried two or more risk-variants and carried more risk-variants than the relative with a neutral phenotype (Supplemental Figure S4). In family B, both members carried three risk-variants but the woman with neutral phenotype was significantly younger than the women with poor phenotype, and the occurrence of a poor phenotype later in life cannot be excluded.
Protein characterization in tumors
Immunohistochemistry was used to further characterize the protein expression in a limited number of endometrial tumor specimens carrying the risk-variants (formalin-fixed-paraffin-embedded, FFPE). Risk-variants and relative predicted protein change were: Chr10:43604498:C > A, with predicted protein change RET:p.(Asn361Lys); Chr15:75012998:delT, CYP1A1:p.(Cys457*); Chr15: 99465448:C > T, IGF1R:p.(Thr758Met), Chr12:124821523:G > C, NCOR2:p.(Ser1964Cys); Chr22: 19951274:delAAG, COMT:p.(Lys159del); Chr1:11264 625:T > G, mTOR:p.(Met1313Leu). We will refer from now on to these risk-variants using their predicted protein changes. Results on protein expression in the Lynch tumors were compared with a panel of sporadic endometrial cancers (n = 7) and post-menopausal healthy controls (n = 5). The presence of the risk-variant-allele in the Lynch tumors was confirmed by Sanger sequence analyses in 12 samples and none showed loss of heterozygosity (LOH, listed in Supplemental Table S4).
Since we found a substantial number or risk-variants mapping in the estrogen signaling, the expression levels of the estrogen receptor (ER-α) and the Ser-167-phosphorylated form, induced by growth factors (including mTOR, IGF1R and RET) and associated with ligand-independent activation (26–28), were assessed. ER-α and phospho-Ser-167-ER-α levels varied between samples, with highest expression in non-cancerous endometrium (both from healthy controls and normal tissues adjacent to cancer cells, i.e. peri-tumoural tissue), intermediate levels in well-differentiated cancers and lowest levels in high-grade lesions (Supplemental Figure S5).
The expression of NCOR2, CYP1A1 and COMT, involved in the estrogen signaling (see discussion), and the oncogenes mTOR, RET and IGF1R was high in normal and cancerous endometrium and varied between samples (Figure 2 and Supplemental Figures S5 and S6).
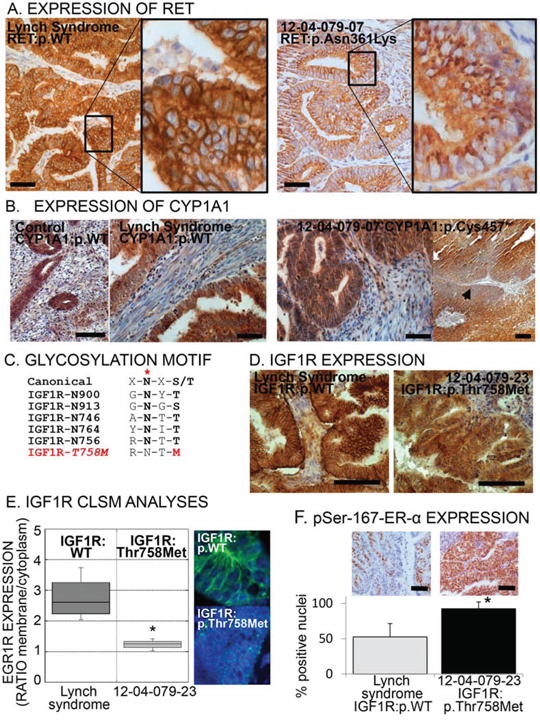
Figure 2: Aberrant expression or localization of RET, CYP1A1 and IGF1R in specimens carrying the respective risk-variants. A. Immunohistochemical images of tumors bearing the common allele (WT) of RET or the risk-variant RET:p.(Asn631Lys). Note the punctuated cytoplasmic pattern in the risk-variant carrying tumor indicative of trapping of the receptor inside the cell. Scale bar = 100 μm. B. Immunohistochemical images of endometrial specimens bearing the common allele (WT) of CYP1A1 (control post-menopausal endometrium and Lynch syndrome endometrial cancer: left images). Right images, CYP1A1 expression on tumor specimen bearing the p.(Cys457*) risk-variant, showing one area with loss of expression (arrowhead). Scale bar = 100 μm. C. Canonical consensus and the five glycosylation sequences (glycosylation site indicated by red asterisk) in the IGF1R plus the consequent disruption of this consensus caused by risk-variant IGF1R:p.(Thr768Met). D. Immunohistochemical images of tumor specimens with common allele (WT) of IGF1R or with risk-variant p.(Thr758Met). Scale bar = 100 μm. E. Representative confocal laser scanning microscopic (CLSM) images and fluorescence quantification of the membrane/cytoplasmic fractions of IGF1R in tumor with common allele or with p.(Thr758Met) IGF1R. The box plot represents the distribution of four independent samples for common allele and four areas of sample 12–04-079–23 [carrying the p.(Thr758Met) risk-variant]. Asterisk indicates a p-value < 0.05, Wilcoxon-Mann-Whitney Rank Sum Test. F. Representative immunohistochemical images (top) and quantification of the percentage of nuclei positive to phospho-Ser-167-ER-α among endometrial cancer with wild type IGF1R (the grey bar indicates the mean ± SD of seven Lynch syndrome tumor specimens) and with IFG1R:p.(Thr758Met) (black bar, mean ± SD of four independent areas of the tumor from subject 12–04-079–23, bearing the variant). Asterisk indicates a p-value < 0.05 compared to common allele (t-test). Scale bar = 100 μm.
The identified risk-variant p.(Asn361Lys) in the RET tyrosine kinase receptor has been previously described in Hirschsprung disease. Asn361 is a N-linked glycosylation site necessary for correct membrane localization of the receptor and mutated RET remains trapped in the endoplasmic reticulum (29). Immunohistochemistry on sample 12–04-079_7, carrying RET:p.(Asn361Lys), clearly shows an aberrant RET cytoplasmic localization compared with controls where RET is located at the plasma membrane (Figure 2A).
The frame-shift change in CYP1A1 at position 457 is predicted to replace the cysteine heme-binding site (30) with a stop codon thus disrupting the catalytic site and causing premature protein termination. Partial and localized loss of CYP1A1 expression was observed in the tumor carrying this risk-variant (Figure 2B).
Risk-variant p.(Thr758Met) in the tyrosine kinase receptor IGF1R is predicted to abolish the N-linked glycosylation at residue Asn756 (Figure 2C). Glycosylation is needed for correct receptor processing, membrane localization and signal transduction (31). Specimen 12–04-079_23 carrying this risk-variant showed a decreased membrane immuno-localization of the IGF1R (Figure 2D), which was confirmed with confocal laser scanning microscopy (CLSM; Figure 2E). This observation was concomitant with the highest percentage of phospho-Ser-167-ER-α staining (Figure 2F), one of the downstream target of the IGF1R kinase activity, suggestive of a sustained intracellular activation.
DISCUSSION
Genome-wide approaches [5, 6, 32] have identified modifiers of the cancer risk conferred by pathogenic Lynch syndrome mutations, but additional genetic modifiers are predicted to exist. For instance, genome-wide studies cannot detect rare variants [33] whose risk modifying contribution remains largely undetermined. To identify rare genetic modifiers of the risk of Lynch syndrome-related endometrial cancer, we have screened a panel of 154 genes among a cohort of 35 Lynch syndrome related endometrial cancer patients. Fifty-four variants with putative risk modifying action were selected (risk-variants), which occurred in 40 genes and the presence of two or more risk-variants was associated with poor clinical phenotypes (Figure 1). This result is in line with genome-wide studies that have shown that risk modifying alleles exert larger risks in carriers of multiple variants compared with single-variant carriers [6].
The rationale used to design the 154 gene-panel and the criteria used to select putative risk modifiers derived from recent understandings in cancer biology. Cancer somatic mutation landscapes have shown that while somatic driver mutations inactivate tumor suppressors or activate oncogenes, germ-line predisposing variants have more subtle effects and do not necessarily give a direct growth advantage or cancer phenotype [11]. For these reasons, the designed gene-panel included not only cancer associated-genes but also genes that are broadly involved in the endometrial physiology, like those encoding for proteins controlling the estrogen signaling. In line with this model suggesting a distinction between germ-line predisposing versus somatic driver mutations [11], no (germ-line) risk-variant was identified among the genes that are most frequently mutated at the somatic level in endometrial cancer, such as PTEN, PIK3CA, CTNNB1, KRAS (all included in the panel). When endometrial cancer somatic mutation landscape (COSMIC database [10]) was aligned to the risk modifiers identified in the present study, MTOR was the first common gene ranking 130th among somatic mutations. STK11 and ATM appeared as risk modifiers in our screen and are frequently mutated somatically [9] but not in endometrial cancer (COSMIC database [10]).
Well-established criteria were used to cluster variants in categories 1–5 [23–25, 34]. In addition, we considered that genetic modifiers are expected to have little or no influence in the general population (modest effect-size), while influencing the cancer-risk in predisposed subjects, i.e. in subjects carrying a penetrant cancer mutation [2]. Hence, we considered as putative risk modifiers all variants having a likely-pathogenic or pathogenic effect (category 4/5) but also those with ‘unclear significance’ (category 3), provided that the were missense leading to non-conservative amino-acid substitution. In this context, the recognition of the role of rare missense changes in cancer susceptibility is growing [35]. Lower categories (2 and 1, unlikely to have any effect at the protein level) were excluded.
Genes such as NOTCH1 (cell-fate determination), NCOR2 and NCOA2 (controlling the activity of ER-α), COMT (detoxification pathway and estrogen degradation), and classical oncogenes (the tyrosine kinase receptors NTRK1 and IGF1R) or tumor suppressors (APC, BRCA2, EPHB2 and CDH1, the latter being already identified as a risk modifier locus in genome-wide studies [6]) carried more than one independent risk-variant, indicating that they are hit frequently. In addition, the pathways where risk-variants were found underscored some signalings in which genetic modifier can be found: the STK11/TSC1/mTOR cascade; the estrogen signaling at ER-α transcriptional control (NCOR2, NCOA2 - both with multiple risk-variants - and PPARGC1A); or at ligand metabolism level, with risk-variants identified in CYP1A1, COMT and CYP1B1, phase I and II detoxifying enzymes degrading also estrogens via the formation of catechol-compounds. The CYP1A1 locus was reported previously as risk modifier for Lynch syndrome colorectal cancer, although data were not confirmed in independent studies [5] and common variants in CYP1A1 and in other genes controlling the estrogen metabolism have been associated with sporadic endometrial cancer [36].
Although it was not possible to assess the segregation of the identified risk-variants with the disease within the families in our cohort (DNA from unaffected relatives with matched life-span was not available), poorer phenotypes tended to segregate with a higher number of risk-variants among the small number of patient-relatives included in our cohort (Supplemental Figure S4).
Further immunohistochemical characterization of a limited number of risk-variants showed association with protein changes. The estrogen degrading protein CYP1A1 showed localized loss of expression in the tumor carrying CYP1A1:(p.Cys457*). Tyrosine receptors including RET and IGF1R have important crosstalk’s with the estrogen signaling and are relevant to the endometrial physiology [28, 37]. Variant RET:p.(Ans361Lys) impairs RET correct membrane localization [29], and was indeed associated with aberrant cytoplasmic immunoreactivity in this study. Glycosylation of IGF1R is reported to be important for cellular localization, response to anti-IGF therapy and signaling activation [31]. Variant IGF1R:p.Thr758Met, predicted to disrupt one N-linked glycosylation site, was associated with diminished membrane IGF1R localization and increased phosphorylation of ER-α, one of the downstream IGF1R kinase targets. The IGF gene locus (coding for the ligand of IGF1R) has been reported as a genetic risk modifier for Lynch syndrome [5].
In conclusion, we propose a gene-panel that contains potential genetic risk modifiers of Lynch syndrome related endometrial cancer in our population. Risk-variants that are likely pathogenic have been identified and assessing their segregation with the disease phenotypes can be relevant to the family members and the subjects assisted at our center.
For implications outside the families of the present investigation, the development of gene-panels in diagnostics is technically desirable compared to complete genome sequencing because it allows achieving sufficient coverage, cost effectiveness and simplicity in analyses [11, 23]. Therefore, it is intriguing to validate these data in independent populations and examine how this panel will translate to other ethnicities. The fact that a number of risk modifiers identified here correspond to loci that were previously characterized in genome-wide investigations is encouraging. Additionally, it is possible to improve our panel design by including genes recently identified as risk modifiers in genome-wide studies [6, 38].
MATERIALS AND METHODS
Ethical statement
Investigation has been conducted in accordance with the ethical standards, according to the Declaration of Helsinki and according to national and international guidelines. Protocols have been approved by the authors’ institutional medical ethical committee (see below for details).
Patient population
Thirty-seven Dutch Caucasian women from 29 families who developed hereditary endometrial cancer and carried a germ-line mismatch repair (MMR) gene mutation were enrolled (Table 1). Women were counseled at the Maastricht or Leiden Medical Centers (2011–2013) because of a Lynch syndrome family history, a Lynch syndrome mutation or a Lynch Syndrome diagnosis (Bethesda criteria). Genomic DNA was used for Next Generation Sequencing (NGS) analyses. All women had given consent to use their DNA for research. The local medical ethical committee approved the protocol (METC 12–04-079).
Archival formalin-fixed-paraffin-embedded (FFPE) tissues of 12 women’s tumor specimen were retrieved from the hospitals that performed the hysterectomy and they were used for DNA analyses, loss of heterozygosity (LOH; n = 12) and histology/immunohistochemistry (n = 8). Seven sporadic endometrial cancers and five post menopausal healthy controls were randomly selected from our tissue-bank [12] and used as control for immunohistochemistry (local medical ethical committee tissue-bank protocol approval: METC-14–04-003).
Gene-panel and NGS
Coding sequences plus intron/exon boundaries of 2012 regions from 154 genes (396kB; Table 2, Supplemental Table S1) and 57 off-target control regions (84kB) were designed and captured with Haloplex platform (Agilent Technologies, Ratingen, Germany). Illumina HiSeq NGS (Illumina, San Diego, USA) was used for sequencing and reads were aligned against the reference genome (GRCh37/hg19). Variants were called using NextGene (Softgenetics, State College, USA), SureCall software packages (Agilent Technologies, Ratingen, Germany) plus manual checking, which resulted in 0% false positives (confirmed by Sanger analyses) and in the successful detection of all re- sequenced pathogenic MMR mutations suitable to be detected (Supplemental Methods). Sanger sequencing was performed using BigDye Termination v.1.1. (Life Technologies, Bleiswijk, Netherlands). Detailed descriptions of the gene-panel design, capture, library preparation, NGS, variant calling and quality controls are given in Supplemental Methods. The dbSNP142 - The National Center for Biotechnology Information (NCBI) was used to identify deposited polymorphisms and variants.
Immunohistochemistry, confocal laser scanning microscopy (CLSM) and LOH
Standard protocols [12, 13] and antibody manufacturer’s instructions were used for immunohistochemistry. Antigens were retrieved with tris-EDTA buffer. Antibodies used: estrogen receptor-α (ERα; monoclonal D-5, 1:100; Dako, Glostrup, Denmark), IGF1R (1:400; monoclonal; Novus Biologicals, Littleton, USA), CYP1A1 (1:1000; polyclonal; Sigma-Aldrich, St. Louis, USA), COMT (1:100; polyclonal; Sigma-Aldrich, St. Louis, USA), Phospho-ERα ser167 (1:50; monoclonal; Cell Signaling Technology, Danvers, USA), RET (1:500; polyclonal; Sigma-Aldrich, St. Louis, USA). Chemate Envision and 3,3-diaminobenzidine (Dako, Glostrup, Denmark) were used to visualize antibody binding. Immunoscores were assessed by two independent investigators (DB and RA) as described [12]. Protocols for SMRT, Phospho-S6 Ribosomal protein-ser235/236 and mTOR staining are given in Supplemental Figure S6.
Confocal laser scanning microscopy (CLSM) was performed on 4 μm thick FFPE tissue sections. Anti IGF1R (1:250; monoclonal; Novus Biologicals, Littleton, U.S.A.) followed by rabbit-α-mouse IgG-FITC (1:100, Dako, Glostrup, Denmark) were used. CLSM was performed with Leica CTR 4000/CTC SPE and the Application Suite/advance fluorescence software (Leica Microsystems B.V., Rijswijk, Netherlands). Image-J program (v 1.48, National Institutes of Health, USA) was used for image analyses.
For LOH, an expert pathologist (VdVKK) identified the tumor regions from histological sections of FFPE materials, which were resected for genomic DNA isolation (QIAamp DNA FFPE tissue kit, Leusden, Netherlands) and Sanger sequencing. At least two separate tumor regions were analyzed.
Statistical analyses
Kaleidograph (v 4.1, Synergy Software, Reading, USA), SPSS (v 22, IBM Corporation, Armonk, USA) and the online Simple Interactive Statistical Analysis (SISA) were used as indicated in the text.
URLs
Gene browsers
Ensembl Genome Browser: http://www.ensembl.org; NCBI: http://ncbi.nlm.nih.gov; University of California Santa Cruz: UCSC Genome Browser: http://genome.ucsc.edu.
Variant analyses
dbSNP database Human Build 142: http://ncbi.nlm.nih.gov; Exome Aggregation Consortium: ExAC, v 0.3, (Cambridge, MA, USA; February 2015): http://exac.broadinstitute.org/; Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP, release ESP6500SI-V2, March-2015), Seattle, USA: http://evs.gs.washington.edu/EVS; Leiden Open Variation Database: LOVD 3.0: lovd.nl; The Human Gene Mutation Database: HGMD (Cardiff University): http://www.hgmd.cf.ac.uk; Genome of Netherlands project, release 5: http://www.nlgenome.nl [14]; Catalogue Of Somatic Mutations In Cancer: Cosmic (Sanger Institute): http://cancer.sanger.ac.uk; PhosphoSitePlus®: http://www.phosphosite.org [15].
Gene ontology, expression information
NCBI: http://ncbi.nlm.nih.gov; UniProt Knowledgebase: http://www.uniprot.org/uniprot; GeneCards (v 3.12.354 March 2015, Weizmann Institute of Science): http://www.genecards.org.
Statistics
SISA: http://www.quantitativeskills.com/sisa/.
ACKNOWLEDGMENTS AND FUNDING
The authors would like to thank the personnel from the pathology departments of the hospitals that kindly provided FFPE archival tissues from the patients: dr. S. Wouda (Viecuri Hospital, Venlo), dr. C. Huysentruyt (PAMM, Veldhoven), and the colleagues from the hospitals of Helmond (Elkerliek Ziekenhuis) and Nijmegen (Canisius-Wilhelmina Ziekenhuis). Special thanks go to G. Roemen, dr B. de Vries and L. Kooreman, dept. Pathology (MUMC), for helping with the archival materials and FFPE DNA isolation in our center; dr T. van Gorp and K. Cornel, dept. of Gynaecology (MUMC), for helping with the statistic analyses; dr. J. Broers and M. Kamps, dept. Molecular and Cell Biology (MUMC), for assistance with the CLSM. The authors would like to thank the NHLBI GO Exome Sequencing Project and its on-going studies, which produced and provided exome variant calls for comparison. The on-going studies are: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). The authors would like to thank also the Exome Aggregation Consortium and the groups that provided exome variant data for comparison (a full list of contributing groups can be found at http://exac.broadinstitute.org/about). Finally, we are grateful to all women who agreed to have their data and biological specimens used for research.
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. The study was sponsored by internal funds of the institute GROW - School for Oncology and Developmental Biology granted to BMJ, KRF, GGEB, RA.
CONFLICTS OF INTEREST
None of the authors has any conflict of interest to declare.
REFERENCES
1. Weissman SM, Burt R, Church J, Erdman S, Hampel H, Holter S, Jasperson K, Kalady MF, Haidle JL, Lynch HT, Palaniappan S, Wise PE and Senter L. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. Journal of genetic counseling. 2012; 21:484–493.
2. Antoniou AC and Chenevix-Trench G. Common genetic variants and cancer risk in Mendelian cancer syndromes. Curr Opin Genet Dev. 2010; 20:299–307.
3. Gaudet MM, Kuchenbaecker KB, Vijai J, Klein RJ, Kirchhoff T, McGuffog L, Barrowdale D, Dunning AM, Lee A, Dennis J, Healey S, Dicks E, Soucy P, Sinilnikova OM, Pankratz VS, Wang X, et al. Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS genetics. 2013; 9:e1003173.
4. Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, Devilee P, Meindl A, Couch FJ, Southey M, Goldgar DE, Evans DG, Chenevix-Trench G, Rahman N, Robson M, Domchek SM, et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N Engl J Med. 2015.
5. Houlle S, Charbonnier F, Houivet E, Tinat J, Buisine MP, Caron O, Benichou J, Baert-Desurmont S and Frebourg T. Evaluation of Lynch syndrome modifier genes in 748 MMR mutation carriers. European journal of human genetics: EJHG. 2011; 19:887–892.
6. Study C, Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I, Vijayakrishnan J, Sullivan K, Penegar S, Colorectal Cancer Association Study C, Carvajal-Carmona L, Howarth K, Jaeger E, Spain SL, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008; 40:1426–1435.
7. Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007; 39:984–988.
8. Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, Jaeger E, Fielding S, Rowan A, Vijayakrishnan J, Domingo E, Chandler I, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008; 40:623–630.
9. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153–158.
10. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES and Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501.
11. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr. and Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–1558.
12. Cornel KM, Kruitwagen RF, Delvoux B, Visconti L, Van de Vijver KK, Day JM, Van Gorp T, Hermans RJ, Dunselman GA and Romano A. Overexpression of 17beta-Hydroxysteroid Dehydrogenase Type 1 Increases the Exposure of Endometrial Cancer to 17beta-Estradiol. J Clin Endocrinol Metab. 2012; 97:E591–601.
13. Dassen H, Punyadeera C, Schulkens I, Delvoux B, Marchetti C, Kamps R, Klomp J, Dijks F, de Goeij T, D’Hooghe T, Kyama C, Ederveen A, Dunselman G, Groothuis P and Romano A. Olfactomedin-4 regulation by estrogen in the human endometrium requires epidermal growth factor signalling. Am J Pathol. 2010; 177:2495–2508.
14. Genome of the Netherlands C. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014; 46:818–825.
15. Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V and Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012; 40:D261–270.
16. Johnson AB and O’Malley BW. ERasing breast cancer resistance through the kinome. Nature medicine. 2011; 17:660–661.
17. Romano A, Adriaens M, Kuenen S, Delvoux B, Dunselman G, Evelo C and Groothuis P. Identification of novel ER-[alpha] target genes in breast cancer cells: Gene- and cellselective co-regulator recruitment at target promoters determines the response to 17[beta]-estradiol and tamoxifen. Mol Cell Endocrinol. 2010; 314:90–100.
18. Zwart W, Theodorou V, Kok M, Canisius S, Linn S and Carroll JS. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. Embo J. 2011; 30:4764–4776.
19. Groothuis PG, Dassen HH, Romano A and Punyadeera C. Estrogen and the endometrium: lessons learned from gene expression profiling in rodents and human. Hum Reprod Update. 2007; 13:405–417.
20. Pritchard CC, Smith C, Salipante SJ, Lee MK, Thornton AM, Nord AS, Gulden C, Kupfer SS, Swisher EM, Bennett RL, Novetsky AP, Jarvik GP, Olopade OI, Goodfellow PJ, King MC, Tait JF, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. The Journal of molecular diagnostics : JMD. 2012; 14:357–366.
21. Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L and Lopez-Bigas N. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Scientific reports. 2013; 3:2650.
22. Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM, Raeder MB, Sos ML, Engelsen IB, Trovik J, Wik E, Greulich H, Bo TH, Jonassen I, Thomas RK, Zander T, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009; 106:4834–4839.
23. Van Allen EM, Wagle N and Levy MA. Clinical analysis and interpretation of cancer genome data. J Clin Oncol. 2013; 31:1825–1833.
24. Wallis Y, Payne S, McAnulty C, Bodmer D, Sister-mans E, Robertson K, Moore D, Abbs S, Deans Z and Devereau A. (2013). Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics. (Association for Clinical Genetic Science & Dutch Society of Clinical Genetic Laboratory Specialists, pp. 1–16.
25. Cheon JY, Mozersky J and Cook-Deegan R. Variants of uncertain significance in BRCA: a harbinger of ethical and policy issues to comeΠGenome medicine. 2014; 6:121.
26. Borrello MG, Ardini E, Locati LD, Greco A, Licitra L and Pierotti MA. RET inhibition: implications in cancer therapy. Expert opinion on therapeutic targets. 2013; 17:403–419.
27. Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ and Holz MK. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem. 2009; 284:6361–6369.
28. Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, McLemore MS, Olivo SE and Stoica A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000; 141:4503–4511.
29. Kjaer S, Hanrahan S, Totty N and McDonald NQ. Mammal-restricted elements predispose human RET to folding impairment by HSCR mutations. Nature structural & molecular biology. 2010; 17:726–731.
30. Walsh AA, Szklarz GD and Scott EE. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J Biol Chem. 2013; 288:12932–12943.
31. Kim JG, Kang MJ, Yoon YK, Kim HP, Park J, Song SH, Han SW, Park JW, Kang GH, Kang KW, Oh do Y, Im SA, Bang YJ, Yi EC and Kim TY. Heterodimerization of glycosylated insulin-like growth factor-1 receptors and insulin receptors in cancer cells sensitive to anti-IGF1R antibody. PLoS One. 2012; 7:e33322.
32. Bellido F, Guino E, Jagmohan-Changur S, Segui N, Pineda M, Navarro M, Lazaro C, Blanco I, Vasen HF, Moreno V, Capella G, Wijnen JT and Valle L. Genetic variant in the telomerase gene modifies cancer risk in Lynch syndrome. European journal of human genetics: EJHG. 2013; 21:511–516.
33. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA and Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011; 12:745–755.
34. Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enns GM, David SP, Pakdaman N, Ormond KE, Caleshu C, Kingham K, Klein TE, Whirl-Carrillo M, et al. Clinical interpretation and implications of whole-genome sequencing. Jama. 2014; 311:1035–1045.
35. Tavtigian SV and Chenevix-Trench G. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomarkers in medicine. 2014; 8:589–603.
36. Yang HP, Gonzalez Bosquet J, Li Q, Platz EA, Brinton LA, Sherman ME, Lacey JV, Jr., Gaudet MM, Burdette LA, Figueroa JD, Ciampa JG, Lissowska J, Peplonska B, Chanock SJ and Garcia-Closas M. Common genetic variation in the sex hormone metabolic pathway and endometrial cancer risk: pathway-based evaluation of candidate genes. Carcinogenesis. 2010; 31:827–833.
37. Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, Wartmann M, Stumm M, Lane HA and Hynes NE. The Ret receptor tyrosine kinase pathway functionally interacts with the ERalpha pathway in breast cancer. Cancer Res. 2008; 68:3743–3751.
38. Wijnen JT, Brohet RM, van Eijk R, Jagmohan-Changur S, Middeldorp A, Tops CM, van Puijenbroek M, Ausems MG, Gomez Garcia E, Hes FJ, Hoogerbrugge N, Menko FH, van Os TA, Sijmons RH, Verhoef S, Wagner A, et al. Chromosome 8q23.3 and 11q23.1 variants modify colorectal cancer risk in Lynch syndrome. Gastroenterology. 2009; 136:131–137.
39. Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F, Sandkuijl L, Moller P, Genuardi M, Van Houwelingen H, Tops C, Van Puijenbroek M, Verkuijlen P, Kenter G, Van Mil A, Meijers-Heijboer H, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004; 127:17–25.