
INTRODUCTION
Prostate cancer is the number one incidence of cancer with 233,000 new cases and the second leading cause of cancer-related deaths in men in the United States [1]. Androgen-deprivation therapy is initially effective, but this therapy eventually fails and the disease progresses to castration-resistant prostate cancer (CRPC) [2–4]. Patients with CRPC have poor survival rates due to limited effective therapies. Despite recent phase III trials showing a survival advantage for abiraterone acetate (17α-hydroxylase/C17, 20-lyase inhibitor) and enzalutamide (AR inhibitor), the median duration of response is < 1 year [5, 6]. Several mechanisms including androgen receptor (AR) mutations, AR or its co-regulator proteins overexpression, intraprostatic androgen synthesis up-regulation and gene fusions have been proposed to result in CRPC progression, which suggests AR signaling remains a critical factor for the growth and survival of the hormone-refractory prostate cancer [7–15].
In addition to AR signaling, the PI3K/Akt/mTOR pathway is associated with prostate cancer. Uncontrolled activation of PI3K-signaling pathway through loss of PTEN has been found in about 40% of primary and 70% of metastatic prostate cancers [16–18]. Recently it was demonstrated that there is a bidirectional crosstalk between PI3K and AR survival pathways in PTEN-negative prostate cancers [19]. Inhibition of the PI3K pathway stimulates up-stream HER2/3 resulting in activation of the AR, whereas blockade of AR reduces FKBP5 levels impairing PHLPP function leading to upregulation of pAKT. Inhibition of the PI3K pathway alone causes growth arrest but not significant tumor regression in Pten-negative prostate cancers, however, combined PI3K and AR pathway inhibition gives profound tumor regressions. It has been reported that AZD5363, an AKT inhibitor potently inhibited proliferation and induced apoptosis in prostate cancer cell lines expressing the AR and had anticancer activity in vivo in androgen-sensitive and castration-resistant phases of a LNCaP xenograft model [20]. However, the effect of castration-resistant tumor growth inhibition and prostate-specific antigen (PSA) stabilization was transient and resistance occurred after approximately 30 days of treatment. Mechanistically, they found that single agent AZD5363 induced increase of AR binding to androgen response element, AR transcriptional activity, and AR-dependent genes such as PSA and NKX3.1 expression. These effects were overcome by the combination of AZD5363 with the antiandrogen bicalutamide, resulting in synergistic inhibition of cell proliferation and induction of apoptosis in vitro, and prolongation of tumor growth inhibition and PSA stabilization in CRPC in vivo.
In this study, we demonstrated that androgen-depletion increases AR expression and Akt activity. Further, we generated resistance cell lines of LNCaP to Enzalutamide (MDV3100) (LNCaP ‘MDV-R’), an AR inhibitor and PF-04691502 (LNCaP ‘PF-R’), a potent, selective pan-PI3K/mTOR inhibitor. MTS analysis showed that LNCaP ‘PF-R’ was strongly resistant to Enzalutamide however LNCaP ‘MDV-R’ was 6-fold resistant to PF-04691502. Mechanistically, we observed PI3K/Akt and AR pathway is activated in LNCaP ‘MDV-R’ and LNCaP ‘PF-R’, respectively. Combination targeting of PI3K/mTOR and AR pathways with a variety of small molecular inhibitors led to synergistic suppression of proliferation in both androgen-dependent LNCaP and independent 22Rv1 cell lines with increased apoptosis and cell cycle arrest. Together, this study provided a preclinical proof-of-concept that combination of a PI3K/mTOR inhibitor with an AR inhibitor resulted in an anti-tumor activity in both non-CRPC and CRPC models.
RESULTS
Androgen depletion increases AR protein level and Akt phosphorylation
AR inhibitor resistance acquired in prostate cancer is associated with increased AR expression and PI3K/Akt activity. To further evaluate this observation, we grew LNCaP, an androgen-dependent cell line in androgen-depleted medium, phenol red-free RPMI 1640 supplemented with 10% charcoal/dextran-treated FBS for 10 days. Compared to LNCaP cells cultured in regular medium and serum, androgen depletion significantly resulted in increased AR protein and Akt phosphorylation at Ser473 and Thr308 (Fig. 1). These data suggest AR over-expression and Akt activity play important roles in resistance to androgen-deprivation therapy in prostate cancer.
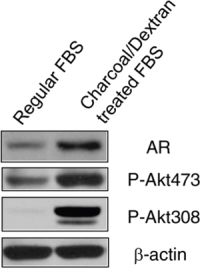
Figure 1: Depletion of androgen increased AR expression and Akt activity. LNCaP cells were grown in regular RPMI 1640 medium with 10% FBS and androgen-depleted medium, phenol red-free RPMI 1640 supplemented with 10% charcoal/dextran-treated FBS for 10 days. Cells were lyzed and 50 μg total protein was resolved by electrophoresis on a 10% SDS-PAGE gel. Immunoblotting was performed using AR and phospho-Akt antibodies, respectively. β-actin protein was used as a loading control.
Acquired reciprocal resistance to PI3K/mTOR inhibition or AR inhibition in LNCaP cells
To further examine the role of AR and PI3K/Akt/mTOR signaling, we generated both AR and PI3K/mTOR inhibitor resistant LNCaP cell lines. Parental LNCaP cells were exposed intermittently and incrementally to various concentrations Enzalutamide (MDV3100) (5–10 μM) or PI3K/mTOR inhibitor PF-04691502 (0.1–0.3 μM) for 12 months. The LNCaP cells finally became stable and can grow in 10 μM of Enzalutamide or 0.3 μM of PF-04691502 which were designated LNCaP ‘MDV-R’ or LNCaP ‘PF-R’, respectively. The LNCaP ‘MDV-R’, LNCaP ‘PF-R’ and parental LNCaP cell lines were evaluated by MTS assays for cell viability at Enzalutamide and PF-04691502 concentrations varying from 0.001 to 100 μM. As expected, LNCaP ‘MDV-R’ cells were completely resistant to Enzalutamide and 25% LNCaP ‘PF-R’ cells were resistant to PF-04691502 (Fig. 2A). Interestingly, LNCaP ‘PF-R’ cells were totally resistant to Enzalutamide and ~50% LNCaP ‘MDV-R’ cells were resistant to PF-04691502 (Fig. 2A). Consistent with this observation, loss of AR expression, loss of PSA and activation of Akt signaling were identified in LNCaP ‘MDV-R’ but the opposite effect was seen in LNCaP ‘PF-R’ (Fig. 2B). These results demonstrate the existence of a reciprocal feedback activation pathway between the AR and the PI3K/mTOR pathway in prostate cancer.
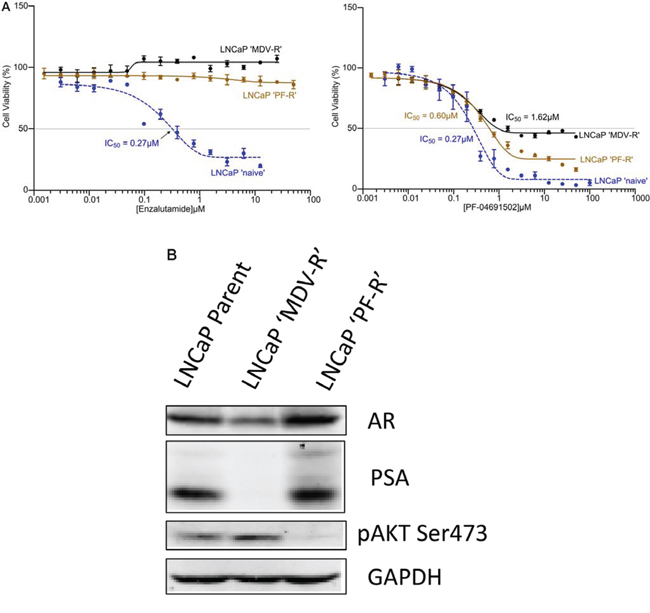
Figure 2: Acquired resistance to AR inhibitor and PI3K/mTOR inhibitor in LNCaP cells. A. The AR or PI3K/mTOR inhibitor-resistant cell lines were developed by intermittent, incremental exposure of the LNCaP cells to various concentrations (5–10 μM) of Enzalutamide or 0.1–0.3 μM of PF-04691502 for 12 months designated as LNCaP ‘MDV-R’ and LNCaP ‘PF-R’ respectively. The LNCaP ‘MDV-R’, LNCaP ‘PF-R’ and parental LNCaP cell lines were evaluated in an MTS assay for cell viability at Enzalutamide and PF-04691502 concentrations varying from 0.001 to 100 μM. B. The parent LNCaP, LNCaP ‘MDV-R’ and LNCaP ‘PF-R’ cells were lysed and immunoblotting was performed to detect AR, PSA and phosphorylation of Akt. GAPDH was used as a loading control.
Suppression of PI3K/mTOR inhibits cell proliferation in AR inhibitor sensitive and resistant prostate cancer cell lines
The PI3K pathway plays a central role in cancer cell survival and proliferation across a variety of malignancies and has been validated as a drug target for cancer therapy [21, 22]. To examine whether suppression of PI3K pathway inhibits prostate cancer cells, MTS assays were performed to evaluate the growth in LNCaP, 22Rv1 and VCaP cell lines. First, we evaluated two drugs, Enzalutamide and Abiraterone and found that the IC50 of Enzalutamide and Abiraterone were 0.27 μM and 10.49 μM in LNCaP cells, 20.27 μM and 25.47 μM in 22Rv1 cells, and unreached IC50 and 30.32 μM in VCaP cells, indicating LNCaP cells were sensitive to AR inhibition but 22Rv1 and VCaP were not (Fig. 3A and Table 1). However, except LY294002, the other three PI3K/mTOR inhibitors GDC-0980, GDC-0941 and PF-04691502 effectively inhibited the growth of these three cell lines with IC50 values of 0.06 μM to 0.43 μM (Fig. 3A and Table 1). Moreover, to determine whether these inhibitors specifically inhibited the PI3K pathway, we treated LNCaP cells with a representative drug PF-04691502. Immunoblotting showed complete suppression downstream of PI3K/mTOR, including Akt, S6 and 4E-BP1 (Fig. 3B). Together, the data demonstrate that blockage of PI3K/mTOR pathway successfully suppress cell proliferation in both AR sensitive and non-sensitive prostate cancer cell lines.
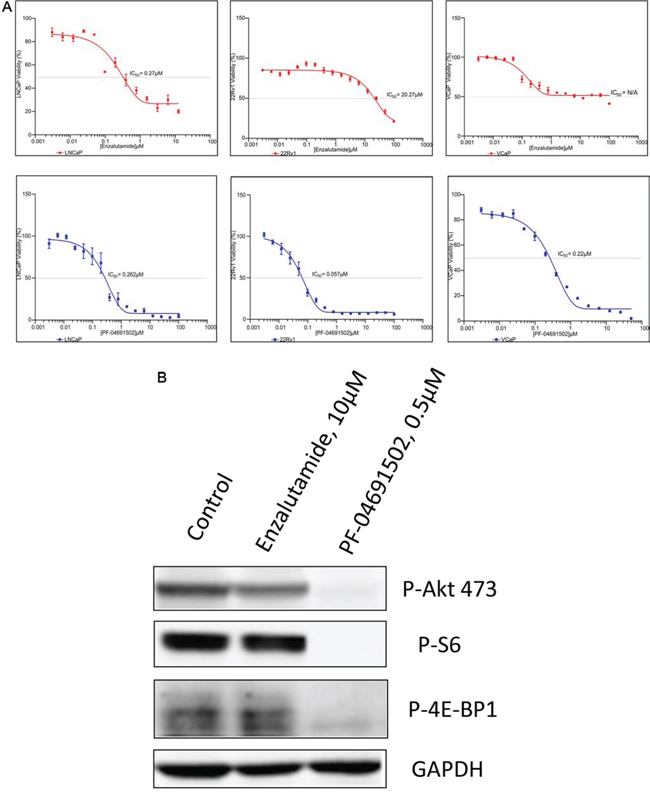
Figure 3: Antiproliferative activity of PI3K/mTOR inbibitor and AR inhibitor in prostate cancer cell lines. A. LNCaP, 22Rv1 and VCaP cells were exposed to varying concentrations of Enzalutamide or PF-04691502 for 4 days. Cell viability was assessed by MTS analysis. Points are the means of triplicate determinations ± SD. Inset: The IC50s of Enzalutamide and PF-04691502 were calculated in these cell lines and shown. B. LNCaP cells were untreated or treated with 10 μM of Enzalutamide and 0.5 μM of PF-04691502 for 18 h. Cells were collected for protein isolation. 50 μg of total protein from each lysate was resolved by SDS-PAGE and immunoblotted with antibodies specific for phosphorylated Akt (Ser473), phosphorylated S6 and phosphorylated 4E-BP1. GAPDH was used as a loading control.
Table 1: IC50s (μM) of PI3K/mTOR and AR signaling inhibitors in castration-sensitive LNCaP and castration-resistant 22Rv1 and VCaP cell lines
Inhibitors |
GDC-0980 |
GDC-0941 |
LY294002 |
PF-04691502 |
Enzalutamide |
Abiraterone |
|---|---|---|---|---|---|---|
LNCaP |
0.32 |
0.40 |
23.63 |
0.26 |
0.27 |
10.49 |
22Rv1 |
0.13 |
0.43 |
16.91 |
0.06 |
20.27 |
25.47 |
VCaP |
0.21 |
0.39 |
19.30 |
0.22 |
N/A |
30.32 |
Dual inhibition of PI3K/mTOR and AR pathways led to synergistic suppression of proliferation and increased apoptosis in LNCaP and castration-resistant 22Rv1 and VCaP cell lines
Since inhibition of the AR pathway results in activation of the PI3K/Akt/mTOR pathway by reciprocal feedback activation, we hypothesized that dual inhibition of PI3K/mTOR and AR has synergistic inhibition of cell proliferation and may overcome drug resistance. As expected, a variety of combinations of PI3K/mTOR and AR inhibitors resulted in synergism or strong synergism of inhibition of cell viability in not only LNCaP, but also in CRPC cell lines 22Rv1 and VCaP (Fig. 4 and Table 2). It is known that apoptosis is induced when the PI3K pathway is suppressed. To examine apoptosis, LNCaP cells were treated with 10 μM of Enzalutamide or 0.2 μM of PF-04691502 or 10 μM of Enzalutamide combined with 0.2 μM of PF-04691502 for 72 hours, stained with Annexin V and PI and evaluated by flow cytometry. PF-04691502 but not Enzalutamide induced apoptosis in LNCaP cells (Fig. 5A). However, apoptosis was more prominent with combination treatment (Fig. 5A). These results were confirmed by demonstrating an increased level of cleaved PARP in both LNCaP and 22Rv1 cells (Fig. 5B). Enzalutamide did not induce PARP cleavage, however, PF-04691502 alone led to PARP cleavage and combination of Enzalutamide and PF-04691502 resulted in additional PARP cleavage. In summary, dual targeting of PI3K/mTOR and AR resulted in synergistic inhibition of cell proliferation and significant induction of apoptosis in castration-sensitive LNCaP and castration-resistant 22Rv1 and VCaP cell lines.
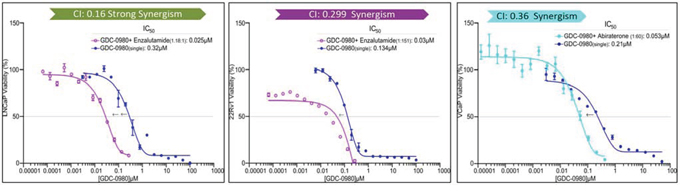
Figure 4: PI3K/mTOR inhibitor synergizes with AR inhibitor to decrease cell viability. LNCaP, 22Rv1 and VCaP cells were treated with PI3K/mTOR inhibitor, GDC-0980 plus AR signaling inhibitor, Enzalutamide for 4 days. The dose ratio was calculated for each combination based on the IC50 of single agents. MTS assays were carried out and the combination-index (CI) was calculated. Representative graphs were shown for the synergistic effect of each combination treatment.
Table 2: Combination indices derived from the median-effect principle of Chou and Talalay for different combination treatments in prostate cancer cell lines
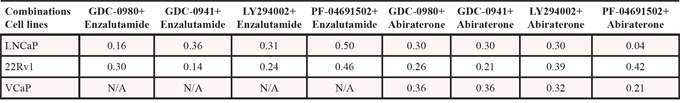
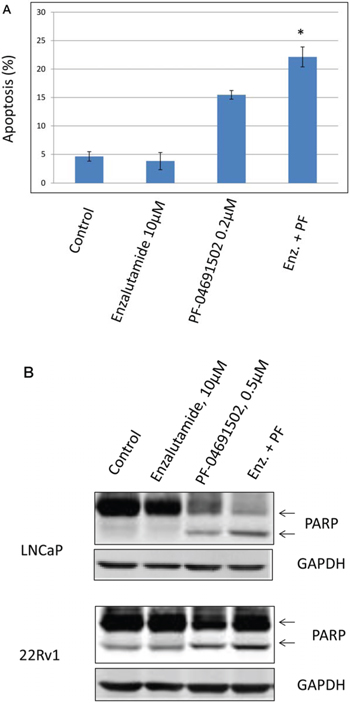
Figure 5: The combination treatment of Enzalutamide and PF-04691502 increases apoptosis in LNCaP and 22Rv1 cells. A. LNCaP cells were treated with 10 μM of Enzalutamide (Enz.) or 0.2 μM of PF-04691502 (PF) or 10 μM of Enzalutamide combined with 0.2 μM of PF-04691502 for 72 h. Apoptosis was analyzed by flow cytometry after annexin V and PI staining. Columns show means of triplicate analysis ± S.D. *, P < 0.05 differ from control and single agents by Student t test. B. LNCaP and 22Rv1 cells were treated with 10 μM of Enzalutamide or 0.5 μM of PF-04691502 or 10 μM of Enzalutamide combined with 0.5 μM of PF-04691502 for 72 h. PARP-cleavage levels were analyzed by Western blotting. GAPDH was used as a loading control.
AR inhibitor and PI3K/mTOR inhibitor induced G1 and G2 cell cycle arrest in prostate cancer cells
Activation of the AR or PI3K pathway regulates cell cycle progression. To examine cell cycle progression affected by the AR or PI3K/mTOR inhibitor in prostate cancer cells, LNCaP and 22Rv1 cells were treated with 10 μM of Enzalutamide or 0.2 μM of PF-04691502 or 10 μM of Enzalutamide combined with 0.2 μM of PF-04691502 for 48 hours and DNA content was evaluated using flow cytometry (Fig. 6). Treatment with Enzalutamide dramatically increased G0/G1 and decreased S phase populations in both cell lines. However, treatment with PF-04691502 induced G2/M arrest and also reduced S populations. Similarly, combination treatment induced G2/M arrest in LNCaP cells and continued to reduce S phase in both of cell lines. These effects may contribute to inhibition of cell proliferation and induction of apoptosis by AR and PI3K/mTOR inhibitors in these cells.
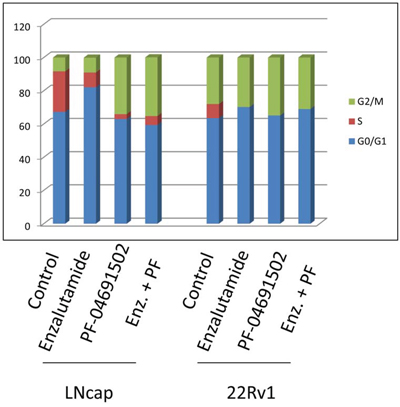
Figure 6: AR and PI3K/mTOR inhibitor induced cell cycle arrest in prostate cancer cells. LNCaP and 22Rv1 cells were treated with 10 μM of Enzalutamide or 0.2 μM of PF-04691502 or combination of 10 μM of Enzalutamide and 0.2 μM of PF-04691502 for 48 h. Samples were harvested and stained with propidium iodide. DNA contents were evaluated by flow cytometry. Percentages of G0/G1, S and G2/M phase cell populations were analyzed by ModFit software and graphed.
DISCUSSION
Patients with castrate-resistant prostate cancer (CRPC) have poor survival rates due to limited effective therapies. Gain-of-function of the androgen receptor (AR) and activation of PI3K/Akt/mTOR signaling pathway correlate with progression to castration-resistant prostate cancer (CRPC). However, as single agents, AR or PI3K/mTOR inhibitors result in a reciprocal feedback activation loop. Therefore, combination of a PI3K/mTOR inhibitor with an AR inhibitor might result in a more profound suppression of CRPC. In this study, we demonstrated that acute depletion of androgen increased AR expression and Akt activity in LNCaP cells. Further, chronic inhibition of AR by Enzalutamide (MDV3100) resulted in a decrease of AR protein level and loss of AR activity implicated by a markedly reduced PSA expression, increased Akt phosphorylation, and resistance to AR and PI3K/mTOR inhibitors. On the other hand, chronic inhibition of PI3K/mTOR pathway by PF-04691502, a potent, selective pan-PI3K/mTOR inhibitor resulted in suppression of Akt activation, increased AR and PSA expression, and robust resistance to Enzalutamide. Inhibition of PI3K/mTOR but not AR alone suppressed cell proliferation in CRPC cell lines 22Rv1 and VCaP. However, combination of PI3K/mTOR and AR led to synergistic suppression of proliferation and induction of further apoptosis and cell cycle arrest.
It has been reported that inhibition of the PI3K pathway increased AR protein levels and its target gene activity in PTEN-negative prostate cancers. Conversely, blockade of AR resulted in activation of PI3K. Combined PI3K and AR pathway inhibition gave profound tumor regressions in preclinical models of prostate cancer [19]. It is also reported that the AKT inhibitor AZD5363 potently inhibits proliferation and induces apoptosis in prostate cancer cell lines and has anticancer activity in vivo in androgen-sensitive and castration-resistant phases of the LNCaP xenograft model. However, the effect of castration-resistant tumor growth inhibition and prostate-specific antigen (PSA) stabilization is transient and resistance occurs with increasing PSA after approximately 30 days of treatment. The combination of AZD5363 with the antiandrogen bicalutamide results in synergistic inhibition of cell proliferation and induction of apoptosis in vitro, and prolongation of tumor growth inhibition and PSA stabilization in CRPC in vivo [20]. Consistent with these reports, our results support the combined inhibition of PI3K/mTOR and AR to represent a novel therapeutic strategy that warrants clinical trial evaluation in patients with CRPC. Currently there are two clinical trials investigating PI3Kβ inhibitor GSK2636771 (NCT02215096) and pan-PI3K/mTOR inhibitor LY3023414 (NCT02407054) in combination with enzalutamide in metastatic castration-resistant prostate cancer patients.
The PI3K/Akt/mTOR pathway is frequently activated and plays a central role in tumorigenesis across a variety of malignancies [21, 22]. It is upregulated and has been implicated in the survival and metastasis of prostate cancer cells, especially in high Gleason score and in CRPC [23, 24]. Thus, selective targeting of this pathway may provide opportunities to affect prostate cancer growth. Currently, several small-molecule inhibitors targeting different proteins of the PI3K/AKT/mTOR pathway, especially a class of dual PI3K/mTOR inhibitors, which bind to and inactivate both PI3K and mTOR, have shown potent anticancer activity in prostate cancer in pre-clinical and clinic trials [25, 26] (Table 3). PF-04691502 and GDC-0980 are novel, potent, selective class I PI3K and mTOR inhibitors [27, 28]. GDC-0941 is a potent inhibitor of PI3Kα/δ [29]. All three drugs have entered phase I or II clinical trials [30, 31]. Our data here indicated that GDC-0980, GDC-0941 or PF-04691502 alone efficiently inhibited cell proliferation (Fig. 3A) with inactivation of the PI3K pathway, including Akt, S6 and 4E-BP1 (Fig. 3B), and induced apoptosis in both castration-sensitive LNCaP cells and castration-resistant 22Rv1 and VCaP cells. In contrast, AR-resistant LNCaP cells (LNCaP ‘MDV-R’) are insensitive to PI3K/mTOR inhibition (Fig. 2A). Similar to the AKT inhibitor, anti-tumor effects are likely transient and resistance occurs after chronic exposure. However, combinations of LY294002, GDC-0980, GDC-0941 or PF-04691502 with AR signaling inhibitor, Enzalutamide or Cyp450 17A1 inhibitor Abiraterone significantly delays CRPC growth associated with induction of enhanced apoptosis compared to single agent therapy. Hence, a combination is likely to be more effective than monotherapy and also may reduce drug dosage to minimize side effect to normal tissue in elderly patients with CRPC.
Table 3: Summary of drugs targeting the PI3K and AR pathways
Inhibitor |
Target |
Reference |
|---|---|---|
GDC-0980 |
PI3Kα/β/δ/γ |
|
GDC-0941 |
PI3Kα/δ |
|
LY294002 |
PI3Kα/δ/β |
|
PF-04691502 |
PI3K(α/β/δ/γ)/mTOR |
|
Enzalutamide |
Androgen-Receptor (AR) |
|
Abiraterone |
Cyt P450-17A1 |
Studies of cell cycle progression in prostate cancer cells have shown that androgen is a critical regulator of the G1-S transition. Mechanistic investigation has revealed that AR mediates cdk4/6 activation and subsequent phosphorylation and inactivation of the retinoblastoma tumor suppressor (RB) to promote G1-S phase progression and thereby govern androgen-dependent proliferation [32, 33]. Knudsen et al. and Xu et al. reported that prostate cancer cells deprived of androgen arrest in early G1 phase [34, 35]. Similarly, our data in this study show inhibition of AR by Enzalutamide dramatically blocked prostate cancer cells entering S phase from G1 (Fig. 6), especially in androgen-dependent LNCaP cells. Interestingly, the PI3K/Akt/mTOR signaling has been implicated to regulate both G1/S and G2/M transition [36]. Inhibition of PI3K by LY294002 or other PI3K inhibitors and mTOR by rapamycin have been demonstrated to induce G1 cell cycle arrest in the prostate cancer cells [37] and other human malignances [38, 39]. However, recently LY294002 has been shown to block G2/M transition in retinal cells [40]. The novel PI3K/mTOR inhibitors, GDC-0980, GDC-0941 and PF-04691502 have been reported to induce G1 cell-cycle arrest in breast, lung, glioblastoma and hepatocellular carcinoma cells [27, 41–43]. In contrast, here exposure to PF-04691502 markedly resulted in G2/M arrest in both castration-sensitive and resistant cell lines (Fig. 6). The detailed molecular mechanisms need further investigation.
In conclusion, our findings indicate that disruption of AR signaling by androgen depletion or AR inhibitor upregulate the PI3K/Akt/mTOR pathway and subsequently acquire resistance to PI3K/mTOR inhibitor. Conversely, chronic inhibition of the PI3K/Akt/mTOR pathway acquires resistance to AR inhibitor therapy. Suppression of PI3K/mTOR alone results in inactivation of Akt, S6 and 4EBP1, reduction of cell proliferation induction of apoptosis and G2/M cell cycle arrest. Most important, we show that dual inhibition of AR and PI3K/mTOR signaling pathways has synergistic inhibitory effect of cell growth associated with increase of apoptosis in both castration-sensitive and resistant prostate cancer cells. These results suggest that PI3K/mTOR axis should be inhibited in combination with AR inhibition in hormone sensitive and CRPC in early therapeutic trials.
MATERIALS AND METHODS
Cells and reagents
LNCaP, Vcap and 22Rv1 cell lines used in this study were from ATCC (Rockville, MD) and maintained in RPMI 1640 medium (Mediatech, VA) supplemented with 10% fetal bovine serum, 2 mM sodium pyruvate and 100 units/ml penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. For the androgen-depletion experiments, LNCaP cells were grown in androgen-depleted medium, phenol red-free RPMI 1640 supplemented with 10% charcoal/dextran-treated FBS (HyClone, Logan, UT). For resistance cell lines development, the LNCaP cells were exposed intermittently and incrementally to various concentrations (5–10 μM) of Enzalutamide or 0.1–0.3 μM of PF-04691502. A total of 12 months later, LNCaP cells were grown stably in Enzalutamide (10 μM) or PF-04691502 (0.3 μM)-containing medium, and these resistant cells were evaluated in an MTS assay for cell viability and labeled LNCaP ‘MDV-R’ and LNCaP ‘PF-R’. AR inhibitor Enzalutamide was provided by Medivation, Inc. and Astellas Pharma Inc. Abiraterone and PI3K/mTOR inhibitors: GDC-0980, GDC-0941, LY294002 and PF-04691502 were purchased from http://Selleckchem.com (Houston, TX). The compounds were dissolved at 50 mM in DMSO as a stock solution, and then further diluted to desired concentrations for in vitro experiments. Anti-AR, Anti-PSA and anti-PARP (H-250) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-Akt (Ser473 and Thr308), anti-phospho-S6, anti-phospho-4E-BP1 and anti-GAPDH (14C10) antibodies were from Cell Signaling Technology (Danvers, MA), and anti-β-actin antibody was purchased from Sigma (St Louis, MO).
Cell proliferation assay (MTS assay)
Cells were seeded at 10,000 per well in 96-well culture plates and allowed to grow for 24 hr followed by the desired treatment with increasing concentrations of the indicated agents (Enzalutamide, Abiraterone, GDC-0980, GDC-0941, LY294002 and PF-04691502) for 4 days. Viable cell densities were determined using a CellTiter 96 Cell Proliferation Assay kit (Promega). Absorbance readings at 490 nm were analyzed against the control group for each drug treatment to determine cell viability. The studies were performed in triplicates x 4 and IC50 values were estimated by Calcusyn software (Biosoft, UK). For combination studies of AR inhibition plus PI3K/mTOR inhibition, an equipotent ratio was calculated to determine a combined graded combination treatment. The equipotent ratio is the ratio of the median effects resulting from the single dose treatments of AR inhibitors and PI3K/mTOR inhibitors. A control group was established for each drug treatment in six replicates. The effects of the combined treatments were determined by the combination-index (CI) and isobologram methods derived from the median-effect principle of Chou and Talalay.
Apoptosis assay
Cells were treated with 10 μM of Enzalutamide or 0.2 μM of PF-04691502 or combination of 10 μM of Enzalutamide and 0.2 μM of PF-04691502 for 72 h. Using Annexin V staining to detect apoptosis, treated cells were harvested and rinsed with cold PBS once. After centrifugation for 5 min, cells were resuspended in 500 μl of 1x Annexin V binding buffer (BioVision, Annexin V-FITC Reagent Kit, Cat.#1001–1000) and then added 5 μl of Annexin V-FITC and 5 μl of Propidium Iodide (BioVision, Annexin V-FITC Reagent Kit). After incubation for 5 min at room temperature in the dark, the samples were analyzed by flow cytometry.
Cell Cycle analysis
LNCaP and 22Rv1 cells were treated with 10 μM of Enzalutamide or 0.2 μM of PF-04691502 or combination of 10 μM of Enzalutamide and 0.2 μM of PF-04691502 for 48 h. Treated cells were centrifuged at 1,500 g for 5 min at 4oC and resuspended in PBS, fixed by drop-wise addition of ice-cold ethanol (100%) to a final concentration of 70%, and incubated for 30 min on ice. Fixed cells were pelleted and treated with 100 μl of RNase A (0.2 mg/ml in PBS) for 5 min at room temperature, then suspended in 0.5 ml ddH2O. After staining with 4 μg/ml propidium iodide, the DNA content was determined using a Becton Dickson flow cytometer and the cell cycle profile was analyzed by ModFit software. Cell aggregates were gated out of the analysis, based on the width of the propidium iodide fluorescence signal. Each profile was compiled from 10,000 gated events.
Immunoblotting
The cells were lysed in NP-40 lysis buffer containing 50 mM Tris.Cl (pH 7.4), 0.15 M NaCl, 0.5% NP-40, 1 mM DTT, 50 mM Sodium Fluoride, and 2 ml/ml Protease inhibitor cocktail (Sigma, St. Louis, MO). Protein concentrations were determined using the BioRad protein assay kit (Hercules, CA) and 50 μg of protein was resolved by electrophoresis on a 10% SDS-PAGE gel. The proteins were then transferred onto a nitrocellulose membrane and non-specific binding was blocked by incubating with 5% nonfat milk in TBST buffer (0.01 M Tris-Cl, 0.15 M NaCl, 0.5% Tween-20, pH 8.0) at room temperature for 1 hr. The membrane was subjected to the indicated antibodies and the proteins were detected by a LI-COR Odyssey Infrared Imaging System.
Statistical analysis
All in vitro experiments were performed in triplicate. The results were expressed as mean± S.D. The difference between two mean values were measured by the Student’s t-test (Excel) and considered to be statistically significant when p ≤ 0.05.
ACKNOWLEDGMENTS AND FUNDING
We wish to thank Medivation, Inc. and Astellas Pharma Inc. for providing enzalutamide. Also we wish to thank UTHSC for providing seed funding for this project.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin. 2014; 64:9–29.
2. Bonkhoff H, Berges R. From pathogenesis to prevention of castration resistant prostate cancer. Prostate. 2010; 70:100–112.
3. Rini BI, Small EJ. Hormone-refractory Prostate Cancer. Curr Treat Options Oncol. 2002; 3:437–446.
4. Singh P, Yam M, Russell PJ, Khatri A. Molecular and traditional chemotherapy: a united front against prostate cancer. Cancer Lett. 2010; 293:1–14.
5. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011; 364:1995–2005.
6. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014; 371:424–433.
7. Han G, Buchanan G, Ittmann M, Harris JM, Yu X, Demayo FJ, Tilley W, Greenberg NM. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci U S A. 2005; 102:1151–1156.
8. Culig Z, Comuzzi B, Steiner H, Bartsch G, Hobisch A. Expression and function of androgen receptor coactivators in prostate cancer. J Steroid Biochem Mol Biol. 2004; 92:265–271.
9. Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001; 61:4315–4319.
10. Shi XB, Xue L, Zou JX, Gandour-Edwards R, Chen H, deVere White RW. Prolonged androgen receptor loading onto chromatin and the efficient recruitment of p160 coactivators contribute to androgen-independent growth of prostate cancer cells. Prostate. 2008; 68:1816–1826.
11. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008; 68:4447–4454.
12. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001; 61:3550–3555.
13. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10:33–39.
14. Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Janne OA, Visakorpi T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009; 69:8141–8149.
15. Makkonen H, Kauhanen M, Jaaskelainen T, Palvimo JJ. Androgen receptor amplification is reflected in the transcriptional responses of Vertebral-Cancer of the Prostate cells. Mol Cell Endocrinol. 2011; 331:57–65.
16. El Sheikh SS, Romanska HM, Abel P, Domin J. Predictive value of PTEN and AR coexpression of sustained responsiveness to hormonal therapy in prostate cancer—a pilot study. Neoplasia. 2008; 10:949–953.
17. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22.
18. Reid AH, Attard G, Ambroisine L, Fisher G, Kovacs G, Brewer D, Clark J, Flohr P, Edwards S, Berney DM, et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br J Cancer. 2010; 102:678–684.
19. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011; 19:575–586.
20. Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, Fazli L, Kim S, Thaper D, Gleave ME, Zoubeidi A. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2013; 12:2342–2355.
21. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010; 28:1075–1083.
22. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8:627–644.
23. Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R, Kreisberg JI. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin Cancer Res. 2002; 8:1168–1171.
24. McCall P, Gemmell LK, Mukherjee R, Bartlett JM, Edwards J. Phosphorylation of the androgen receptor is associated with reduced survival in hormone-refractory prostate cancer patients. Br J Cancer. 2008; 98:1094–1101.
25. Tang KD, Ling MT. Targeting drug-resistant prostate cancer with dual PI3K/mTOR inhibition. Curr Med Chem. 2014; 21:3048–3056.
26. Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009; 9:237–249.
27. Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011; 10:2189–2199.
28. Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J, Folks A, Friedman L, Goldsmith R, Gunzner J, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011; 54:7579–7587.
29. Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008; 51:5522–5532.
30. Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, Clarke PA, Raynaud FI, Levy G, Ware JA, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015; 21:77–86.
31. Britten CD, Adjei AA, Millham R, Houk BE, Borzillo G, Pierce K, Wainberg ZA, LoRusso PM. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs. 2014; 32:510–517.
32. Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008; 6:e001.
33. Schiewer MJ, Augello MA, Knudsen KE. The AR dependent cell cycle: mechanisms and cancer relevance. Mol Cell Endocrinol. 2012; 352:34–45.
34. Knudsen KE, Arden KC, Cavenee WK. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998; 273:20213–20222.
35. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006; 66:7783–7792.
36. Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003; 2:339–345.
37. Gao N, Zhang Z, Jiang BH, Shi X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem Biophys Res Commun. 2003; 310:1124–1132.
38. Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Rasp KH, Illing B, Dohner H, Dohner K, Schittenhelm MM. Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol Cancer. 2013; 12:46.
39. Hill R, Kalathur R, Callejas S, Colaco L, Brandao R, Serelde B, Cebria A, Blanco-Aparicio C, Pastor J, Futschik M, et al. A novel Phosphatidylinositol 3-Kinase (PI3K) inhibitor directs a potent FOXO-dependent, p53-independent cell cycle arrest phenotype characterized by the differential induction of a subset of FOXO-regulated genes. Breast Cancer Res. 2014; 16:482.
40. Ornelas IM, Silva TM, Fragel-Madeira L, Ventura AL. Inhibition of PI3K/Akt pathway impairs G2/M transition of cell cycle in late developing progenitors of the avian embryo retina. PLoS One. 2013; 8:e53517.
41. Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011; 10:2426–2436.
42. Zou ZQ, Zhang LN, Wang F, Bellenger J, Shen YZ, Zhang XH. The novel dual PI3K/mTOR inhibitor GDC-0941 synergizes with the MEK inhibitor U0126 in non-small cell lung cancer cells. Mol Med Rep. 2012; 5:503–508.
43. Wang FZ, Peng J, Yang NN, Chuang Y, Zhao YL, Liu QQ, Fei HR, Zhang JG. PF-04691502 triggers cell cycle arrest, apoptosis and inhibits the angiogenesis in hepatocellular carcinoma cells. Toxicol Lett. 2013; 220:150–156.