
INTRODUCTION
Development of new chemical entities is costly and time-consuming. Drug repurposing (also known as drug repositioning), which refers to the discovery of new indications from existing drugs, has been a popular approach in recent years. After successful use of several non-anticancer drugs for cancer therapy, drug repurposing has attracted particular attention in both pre-clinical and clinical studies [1, 2].
Chk2 is a tumor susceptibility gene encoding for a serine/threonine protein kinase responsive to cellular DNA damage [3, 4]. Activated Chk2 protein kinase serves as a signal transducer, phosphorylating a wide variety of substrates including cell division cycle 25 (Cdc25) family members, promyelocytic leukemia protein (PML), E2F-1, BRCA1 (BReast CAncer gene 1) and p53, which have been highly associated with regulating DNA damage response and repair, cell cycle arrest as well as apoptosis [3–5]. During DNA damage response, Chk2 phosphorylates downstream substrates (e.g., BRCA1), leading to initiation of homologous recombination DNA repair signaling which may crossly interact non-homologous end joining repair pathway [6, 7]. When DNA damage cannot be repaired, damaged cells are able to introduce Chk2 kinase-mediated p53-dependent apoptotic programs [8]. Notably, Chk2 may also induce p53-independent apoptosis through phosphorylation at a Chk2 consensus phosphorylation site in E2F1, leading to protein stabilization, transcriptional activation and localization of phosphorylated E2F1 to discrete nuclear structures and induce apoptosis [9]. Based on the critical role during DNA damage response and cell apoptosis, Chk2 has been suggested as a potential target for anticancer therapy.
AMP-activated protein kinase (AMPK), existing as a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ subunits, acts as a metabolic master switch in regulating various intracellular processes including uptake of glucose, oxidation of fatty acids and biogenesis of mitochondria and glucose transporter 4 [10, 11]. Cellular stresses which result in depletion of cellular ATP and an increase in AMP:ATP ratio make AMPK a more susceptible substrate for phosphorylation in the activation loop of α subunit by the upstream AMPK kinase, LKB1. AMPK can also be phosphorylated and activated by calcium/calmodulin protein kinase kinase (CAMKK) 2 through changes in intracellular calcium levels [11–14]. Several lines of evidence reveal that AMPK and downstream mTOR pathways are potential therapeutic targets for the treatment of cancer [11, 13–15].
Nitroxoline, which functions by chelating Zn2+ and Fe2+ from biofilm matrix, is an antibiotic with effective activity at combating biofilm infections [16]. Nitroxoline has been suggested to induce intracellular oxidant stress [17]. Shim and the colleagues have performed a high throughput screening test for discovering the inhibitors of type 2 methionine amino peptidases (MetAP2) and have found that nitroxoline inhibited MetAP2 activity in human umbilical vein endothelial cells, leading to inhibition of endothelial tube formation and proliferation [18]. Furthermore, nitroxoline caused a reduction in tumor volume in breast cancer xenografts and in bladder cancer in an orthotopic mouse model [19]. Nitroxoline derivatives have been suggested to inhibit cathepsin B activity and to abrogate the invasion of Ras-transformed MCF-10A neoT cells [19]. However, the anticancer activity and mechanism of nitroxoline against human prostate cancer are yet to be identified. In this study, the role of AMPK/mTOR signaling pathway and the interplay with Chk2 in apoptosis have been identified in nitroxoline-mediated anticancer effect in both hormone-sensitive and hormone-refractory prostate cancer cells.
RESULTS
Nitroxoline induces anti-proliferative effect in human prostate cancer cells
The anti-proliferative effects of nitroxoline in both hormone-sensitive (LNCaP) and hormone-refractory prostate cancer cells (PC-3 and DU-145) were determined using sulforhodamine B (SRB) assay. Nitroxoline induced time-dependent inhibition of cell proliferation in these cell lines (Table 1). Flow cytometric analysis of carboxyfluorescein succinimidyl ester (CFSE) staining was applied to further confirm anti-proliferative activity. The data demonstrated that nitroxoline prevented the decrease of fluorescence intensity in PC-3 cells, indicating the inhibition of cell proliferation (Figure 1A). Similar effect was obtained in both DU-145 and LNCaP cells (fluorescence intensity at 72 h of control vs. nitroxoline: 144 ± 4 vs. 326 ± 9, P < 0.001 in DU145; 258 ± 24 vs. 498 ± 15, P < 0.001 in LNCaP). The long-term effect of nitroxoline was examined using colonogenic assay. After a 10-day exposure, nitroxoline resulted in a profound inhibition of colony formation with an IC50 of 3.2 ± 0.6 μM (Figure 1B).
Table 1: Effect of nitroxoline on cell proliferation in human prostate cancer cells
Cell line |
Time (hour) |
||
|---|---|---|---|
24 |
48 |
72 |
|
PC-3 |
8.3±0.5 |
5.5±0.1 |
4.6±0.4 |
DU-145 |
16.6±1.0 |
7.4±0.3 |
5.5±0.1 |
LNCaP |
6.6±0.3 |
5.0±0.3 |
4.2±0.1 |
Cells were incubated in the absence or presence of graded concentrations of nitroxoline for the indicated time. After the treatment, cells were fixed and stained for SRB assay. IC50 values (μM) are expressed as mean±SEM of three to four independent determinations.
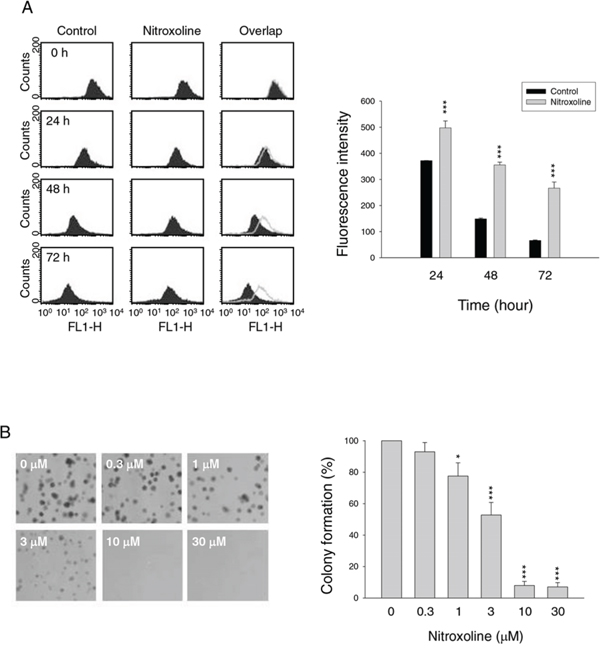
Figure 1: Effect of nitroxoline on anti-proliferation. PC-3 cells were incubated in the absence or presence of nitroxoline (A, 10 μM; B, 0.3 to 30 μM) for the indicated time (A, 24 to 72 hours; B, ten days). After treatment, the cells were harvested for flow cytometric analysis of CFSE staining A. or the clonogenic assay was applied B. Data are representative of three independent experiments. Quantitative data are expressed as mean ± SEM of three independent experiments. *P < 0.05 and ***P < 0.001 compared with the control.
Inhibition of cyclin D1-Rb-Cdc25A axis contributes to nitroxoline-induced G1 arrest
Flow cytometric analysis of propidium iodide (PI) staining in PC-3 cells revealed that nitroxoline induced G1 arrest of the cell cycle (Figure 2A and 2B) followed by a significant increase of apoptosis after a 48-hour treatment (25.3 ± 0.6% compared with control of 4.7 ± 1.1%, n = 3, P < 0.001). Cell cycle progression is controlled by periodic activation of several Cdk/cyclin complexes. Cyclin D1 and its catalytic partner Cdk4 are key players in G1 phase progression. Nitroxoline induced a time-related decrease of protein expression of cyclin D1 other than cyclin E, A and B1 in PC-3 cells (Figure 2C). Rb protein, a tumor suppressor responsible for G1 checkpoint, blocks the entry of S phase and cell cycle progression. Cyclin D1/Cdk4 complex inhibits Rb by partial phosphorylation, decreasing its association with E2F transcription factor and allowing E2F-regulated activation of downstream gene transcription [20, 21]. As a result, nitroxoline decreased cyclin D1 protein expression, leading to the inhibition of Rb phosphorylation (Figure 2C). Furthermore, nitroxoline decreased Cdc25A protein expression (Figure 2C). Cdc25A, a member of Cdc25 family of dual-specificity phosphatases, is required for S phase entry and is induced in G1 phase by E2F [22, 23]. These data suggest that nitroxoline induces G1 arrest through the inhibition of cyclin D1-Rb-Cdc25A axis in PC-3 cells. Similar effects were obtained in LNCaP cells (Supplementary Figure 1).
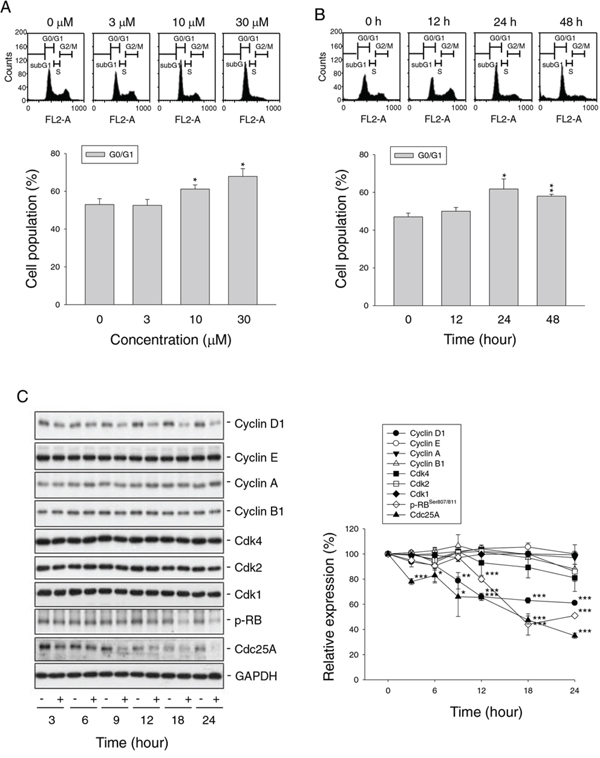
Figure 2: Effect of nitroxoline on cell cycle progression and related regulator proteins. PC-3 cells were incubated in the absence or presence of nitroxoline (A, 3 to 30 μM; B, 10 μM; C, 10 μM) for the indicated time (A, 24 hours; B, 12 to 48 hours; C, 3 to 24 hours). The cells were fixed and stained with propidium iodide to analyze DNA content by flow cytometric analysis A and B. or the cells were harvested and lysed for the detection of the indicated protein by Western blot analysis C. Data are representative of three to four independent experiments. Quantitative data are expressed as mean ± SEM of three to four independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with 100% control.
Nitroxoline regulates AMPK-mTOR pathway and induces autophagy
AMPK, a cellular energy sensor and signal transducer, is induced by a wide variety of metabolic stresses. mTOR, a downstream effector of AMPK, is a crucial player in cell growth and proliferation via regulating various cellular processes including transcription and translation [25]. Nitroxoline induced both concentration-dependent (Figure 3A) and time-dependent (Figure 3B) increases of AMPKα phosphorylation at Thr172 and decreases of mTOR phosphorylation at Ser-2448 in PC-3 cells. Nitroxoline also inhibited p70S6K phosphorylation at Thr389, a downstream substrate of mTOR (Figure 3A and 3B). The data indicate that nitroxoline induces the activation of AMPK but inhibition of mTOR signaling pathway in PC-3 cells. Similar effects were obtained in LNCaP cells (Supplementary Figure 1).
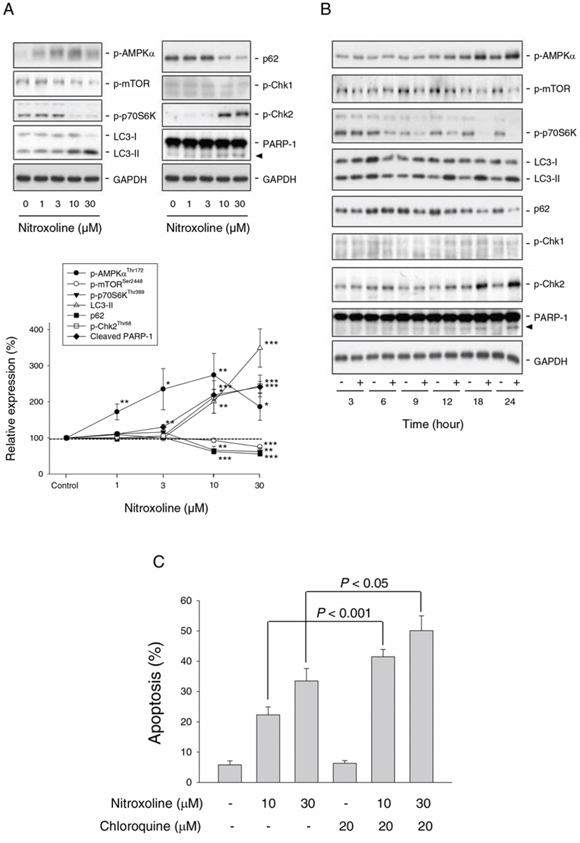
Figure 3: Effect of nitroxoline on the expression of several proteins and examination of autophagy-mediated cytoprotective effect. PC-3 cells were incubated in the absence or presence of nitroxoline (A, 1 to 30 μM; B, 10 μM) for the indicated time (A, 24 hours; B, 3 to 24 hours). Cells were harvested and lysed for the detection of the indicated protein expression by Western blot analysis. Protein expression was quantified using computerized image analysis system ImageQuant (Amersham Biosciences, NJ, USA). Data are expressed as mean ± SEM of three to four independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the control. C. PC-3 cells were incubated in the absence or presence of the indicated compound for 48 hours. The cells were fixed and stained with propidium iodide to analyze hypodiploid DNA content (apoptotic sub-G1 population) by flow cytometric analysis. Data are expressed as mean ± SEM of four independent experiments.
Autophagy is a catabolic process involving lysosomal degradation of dysfunctional or unnecessary cellular components. Autophagy is promoted by AMPK but inhibited by mTOR [26, 27]. Detection of LC3 is a reliable method for monitoring autophagy since lysosomal turnover of LC3-II reflects autophagic activity. Moreover, the level of p62 which is a ubiquitin- and LC3-binding protein is increased when autophagy is impaired [28]. As a result, nitroxoline induced a dramatic increase of LC3-II protein level but a decrease of p62 expression (Figure 3A and 3B), indicating an increase of autophagic activity. Increasing lines of evidence suggest that autophagy can serve as a survival pathway after cellular stresses by chemotherapy or radiation [27]. Chloroquine, an inhibitor of autophagy, was used to examine whether nitroxoline-induced autophagy represented a mechanism for cancer cells to survive or to promote programmed cell death. The data demonstrated that chloroquine, by itself, did not affect cell viability but significantly potentiated nitroxoline-induced cell apoptosis (Figure 3C and supplementary Figure 2), indicating a pro-survival role of autophagy.
siRNA-mediated knockdown of AMPKα was used to determine its functional regulation. AMPKα knockdown significantly inhibited nitroxoline-induced effects, including down-regulation of cyclin D1, inhibition of p70S6K phosphorylation, increase of LC3-II turnover and cleavage of PARP-1 (Figure 4). The data suggest that AMPK activation plays an upstream role to control cell cycle regulators and autophagic activity.
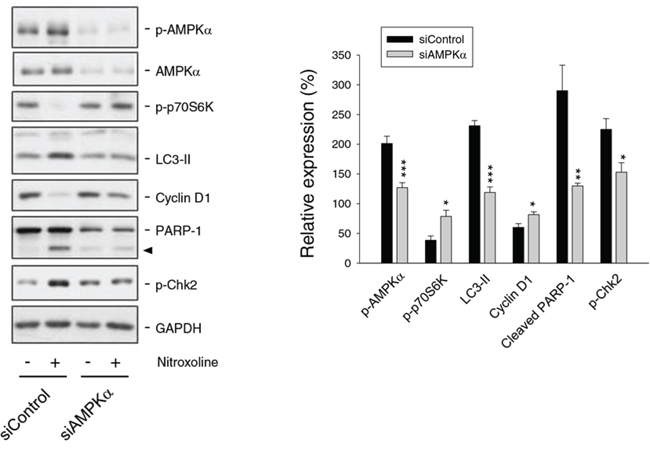
Figure 4: Examination of AMPK-regulated protein expression in PC-3 cells. Both the control and AMPKα knockdown cells were treated with or without nitroxoline (10 μM) for 24 hours. Cells were harvested and lysed for the detection of the protein expression using Western blot analysis. The protein expression was quantified using computerized image analysis system ImageQuant (Amersham Biosciences, NJ, USA). The data are quantified relative to nitroxoline-free group and are expressed as mean ± SEM of three to four independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the respective control.
Nitroxoline induces DNA damage and activates Chk2
Chk2 and Chk1 pathways that are activated by DNA double-strand breaks and single-stranded DNA, respectively, coordinate cellular responses to DNA damage [29]. Nitroxoline induced the phosphorylation and activation of Chk2 other than Chk1 in both LNCaP (Supplementary Figure 1) and PC-3 cells (Figure 3A and 3B), which were AMPK-dependent since the effect was significantly reduced by AMPKα knockdown (Figure 4). Recent work has revealed DNA damage-independent function of Chk2 involving in mitotic spindle assembly and chromosomal stability [30]. The data showed that nitroxoline resulted in a significant increase of DNA damage using comet assay (Figure 5A). The data suggest DNA damage-dependent function of Chk2. Activated Chk2 kinase may serve as a signal transducer in promoting cell survival or death. It phosphorylates numerous substrates including p53, E2F, Cdc25 phosphatases and BRCA1 which are related to halting cell cycle progression, introducing DNA repair, and inducing apoptosis following DNA damage response [30]. The application of both pharmacological Chk2 inhibitor and siRNA to knockdown Chk2 did not prevent nitroxoline-induced cyclin D1 down-regulation, p70S6K inhibition and Cdc25A down-regulation, but significantly blunted the cleavage of PARP-1 (Figure 5B and 5C), supporting a pro-apoptotic role of Chk2. The data were confirmed using SRB assay.
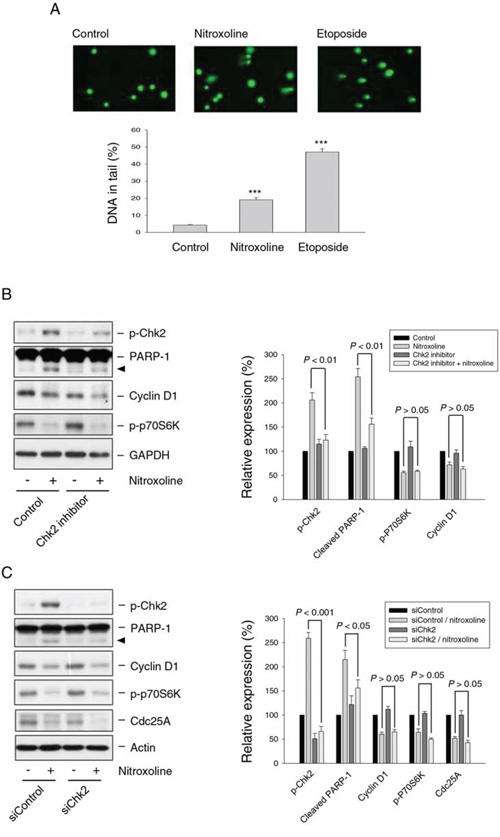
Figure 5: Effect of nitroxoline on DNA damage response and examination of Chk2-mediated protein expression. A. PC-3 cells were incubated in the absence or presence of nitroxoline (10 μM) or etoposide (50 μM) for 12 hours. Comet assay was employed to examine the integrity of chromosome DNA. Comet tail-positive cells indicate under DNA damage stress. Data are expressed as mean ± SEM of three independent determinations. ***P < 0.001 compared with the control. B and C. PC-3 cells were pre-treated with or without Chk2 inhibitor (20 μM) or transfected with siChk2. The cells were incubated in the absence or presence of nitroxoline (10 μM) for 24 hours. Cells were harvested and lysed for the detection of the protein expression using Western blot analysis. The protein expression was quantified using computerized image analysis system ImageQuant (Amersham Biosciences, NJ, USA). Data are expressed as mean ± SEM of three independent experiments.
H2AX, a variant of the H2A protein family, is phosphorylated by Ataxia telangiectasia mutated (ATM)- and Rad3-related protein (ATR) when DNA damage forms double-strand breaks. The newly phosphorylated H2AX, γ-H2AX, is the initial step in recruiting DNA repair proteins [29, 31, 32]. Both immunofluorescence microscopic examination and flow cytometric analysis showed that nitroxoline did not induce γ-H2AX formation but increased Chk2 phosphorylation. In contrast, camptothecin and etoposide which were DNA damage inducers through inhibition of topoisomerase I and II, respectively, caused increased levels of γ-H2AX and Chk2 phosphorylation (Figure 6A and 6B). The data suggest the absence of γ-H2AX-related DNA repair mechanism to nitroxoline action.
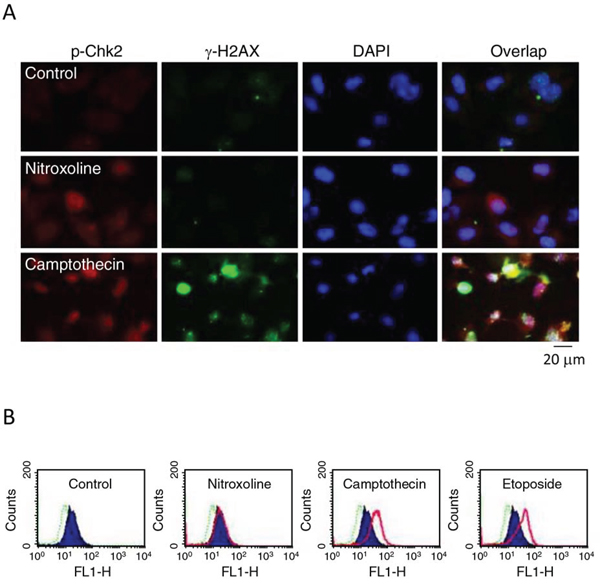
Figure 6: Effect of nitroxoline on protein expression of Chk2 and γ-H2AX. PC-3 cells were incubated in the absence or presence of nitroxoline (10 μM), camptothecin (10 μM) or etoposide (25 μM) for 24 hours. A. The cells were fixed for immunofluorescence microscopic examination by staining with anti-Chk2 (red fluorescence) and anti-γ-H2AX (green fluorescence) antibodies and DAPI (blue fluorescence, for nuclear detection). Scale bar, 20 μm. B. The cells were harvested for detection of γ-H2AX using flow cytometric analysis. Dashed line area, basal; blue area, vehicle control; pink area, the indicated drug. The data are representative of two independent experiments.
ZnCl2 supplement does not rescue nitroxoline-mediated anti-proliferative effect and cellular signaling
Nitroxoline is an antibiotic functioning by chelating Fe2+ and Zn2+. Zn2+ serves as structural ions in transcription factors and is a cofactor in more than 300 enzymes affecting a wide variety of cellular functions [33]. The re-introduction of Zn2+ was applied to determine whether the ions chelating effect contributed to nitroxoline-induced anticancer activity. As a result, ZnCl2 supplement did not significantly prevent Chk2 activation, cyclin D1 down-regulation, AMPK activation, p70S6K inhibition and PARP-1 cleavage (Figure 7). SRB assay also showed that ZnCl2 supplement did not modify nitroxoline-induced anti-proliferative effect (IC50 of 6.1 ± 0.6 μM vs. 5.5 ± 0.1 μM of nitroxoline alone, n = 4, P > 0.05).
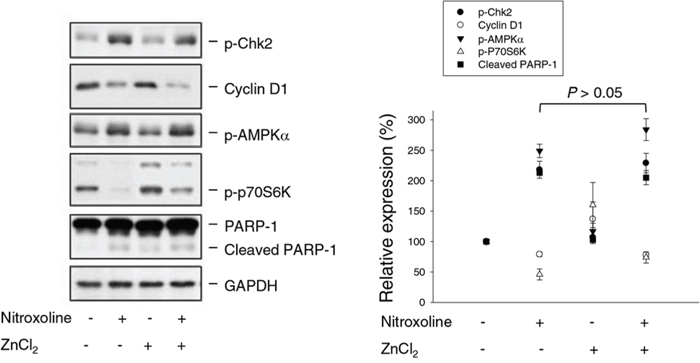
Figure 7: Effect of ZnCl2 supplementation on nitroxoline-induced effect. PC-3 cells were incubated in the absence or presence of ZnCl2 (100 μM) or nitroxoline (10 μM) for 24 hours. Cells were harvested and lysed for the detection of protein expression using Western blot analysis. Expression levels were quantified using computerized image analysis system ImageQuant (Amersham Biosciences, NJ, USA). Data are expressed as mean ± SEM of three independent experiments.
DISCUSSION
Nitroxoline, an FDA-approved antibiotic, has been identified with potential anticancer activity in both in vitro and in vivo studies [17–19]. In this study, nitroxoline induced anti-proliferative effects in both hormone-sensitive and hormone-refractory prostate cancers through induction of G1 arrest of the cell cycle. Cyclin D1 is a specific G1 phase protein forming a complex with and serving as a regulatory subunit of Cdk4 or Cdk6. Overexpression of cyclin D1 has been detected in numerous types of cancers, including cancers of the breast, lung and prostate [34–36] and has been considered as an oncogenic mechanism in hormone-refractory metastatic prostate cancer to the bone [36]. Rb is a critical tumor suppressor for G1 checkpoint, inhibiting the entry of cell cycle into S phase. Cyclin D1/Cdk4 complex can partially and appropriately phosphorylate Rb protein, leading to its dissociation from E2F1 transcription factor and allowing the activation of gene transcription [20, 21]. Many cancers have been shown to exhibit chromosomal abnormalities which directly result in the functional inactivation of Rb and promote cell proliferation [37]. In the present work, nitroxoline induced a dramatic down-regulation of cyclin D1 expression and inhibition of Rb phosphorylation that explained the G1 arrest of cell cycle and indicated the potential against prostate cancers.
mTOR, a serine/threonine kinase, plays a crucial role in cell proliferation, growth, survival and metabolism through the regulation of multiple interacting proteins. mTORC1 is a major rapamycin-sensitive mTOR complex, promoting protein synthesis in response to growth factors through the phosphorylation of p70S6K and 4EBP1 [25, 38]. p70S6K activation involves the phosphorylation of several serine and threonine sites; the critical event is Thr389 phosphorylation. Both phosphoinositide 3-dependent protein kinase 1 and mTOR are potent Thr389 kinases [39, 40]. Pharmacogenomic profiling of mTOR-p70S6K pathway shows that the protein synthetic machinery is overexpressed and activated during tumor progression [41]. Immunohistochemistry analysis also reveals a strong association between mTOR-p70S6K activation and prostate cancer progression [42, 43]. During G1 phase, cells grow in size and synthesize mRNA and proteins required for DNA synthesis at S phase. Nitroxoline displayed profound inhibition of mTOR-p70S6K pathway supporting G1 arrest activity. AMPK, a cellular energy sensor and signal transducer, is a key player to switch off mTOR pathway through phosphorylating and thereby activating tuberous sclerosis protein (TSC) 2, leading to association of TSC1/TSC2 complex and inhibition of mTOR pathway. AMPK is a potential target of drugs for regulating numerous human diseases ranging from metabolic disorders to cancers [25, 44]. AMPK knockdown significantly prevented the inhibition of mTOR-p70S6K pathway and rescued the cyclin D1 expression, indicating the central role of AMPK in nitroxoline-mediated effect.
Nitroxoline also induced autophagy through an AMPK-dependent pathway. Recent studies suggest that Atg1/UNC-51-like autophagy activating kinase 1 (Ulk 1) complex is an essential player in autophagy initiation [45]. Ulk1, a homologue of yeast Atg1, is activated by AMPK via direct phosphorylation of Ser317 and Ser777. In contrast, mTOR phosphorylates Ulk1 at Ser757 and interrupts the interaction between AMPK and Ulk1 [46]. Coordination between AMPK and mTOR is critical for Ulk1 in autophagy induction and may explain nitroxoline-induced activation of AMPK and autophagy. However, the increased autophagy decreased apoptosis to nitroxoline action. The cytoprotective autophagy has been studied under cellular stresses, such as hypoxia, nutrient deprivation, oxidative stress and cytotoxic drug exposure. Therefore, autophagy advances survival and growth of cancer cells and resistance to chemotherapy [27, 47]. Targeting autophagy in cancer will provide novel strategy for drug development [47, 48].
Chk2 encodes for a serine/threonine kinase which coordinates cellular responses to DNA damage [29]. It phosphorylates diverse substrates, including p53, E2F and Cdc25 phosphatases, associated with the arrest of cell cycle and introduction of DNA repair [3, 30]. More specifically, Chk2 is able to phosphorylate and down-regulate Cdc25A, resulting in G1 arrest of the cell cycle [49]. However, the data showed that Chk2 activation contributed to nitroxoline-induced cleavage of PARP-1 (a caspase-3 substrate) other than the regulation of cyclin D1, Cdc25A and p70S6K. The data indicate that Chk2 serves as a pro-apoptotic activator but not a coordinator in cell cycle arrest. Recent work suggests that γ-H2AX plays a key role in the enrollment and maintenance of DNA repair and checkpoint proteins [29, 31, 32]. It has been reported that H2AX-/- mouse embryonic fibroblasts are susceptible to radiation, showing deficits in repairing DNA damage compared to the wild-type counterparts [32]. The present study showed that nitroxoline, at effective concentrations in displaying anti-proliferative and apoptotic activities, did not induce γ-H2AX formation and suggested the absence of γ-H2AX-related DNA repair mechanism. However, higher concentrations of nitroxoline could induce γ-H2AX formation (data not shown) and, therefore, the related DNA repair mechanism might be activated. Notably, the activation of Chk2 was AMPK-dependent in prostate cancers upon nitroxoline exposure. Recent work with AMPKα knockout mouse embryonic fibroblasts has demonstrated a reduced phosphorylation and activity of Chk2 kinase in response to ionizing radiation [50]. Moreover, AMPK has been suggested to function in a point of convergence of metabolic and genomic stress signals, regulating downstream signaling upon growth factor stimulation and propagating DNA damage response through ATM/Chk2 pathway [51]. These studies support an upstream role of AMPK in regulating Chk2 activity. However, defect in DNA repair mechanism during nitroxoline treatment shifted the pro-survival nature of Chk2 into pro-apoptotic disposition. Similar pro-apoptotic function of Chk2 also has been suggested. Small molecules like Nutlin-3 and RITA (reactivation of p53 and induction of tumor cell apoptosis) have been reported to induce apoptosis in human uveal melanoma cells and colorectal carcinomas through p53-dependent Chk2 activation [52]. In contrast, Chk2 expression in p53-mutated cancer cells, in spite of the absence of DNA damaging stimuli, is sufficient to induce apoptosis [53]. These studies suggest that direct activation of Chk2 can be explored as a novel anticancer approach.
Taken together, the data suggest nitroxoline induces anti-proliferative and apoptotic signaling cascades against prostate cancer in a sequential manner (Figure 8). Nitroxoline induces the activation of AMPK, which in turn inhibits mTOR signaling pathway and cyclin D1-Rb-Cdc25A axis, leading to G1 arrest of cell cycle and apoptosis. Nitroxoline also induces AMPK-dependent activation of Chk2 that, at least partly, contributes to cell apoptosis. However, the activated autophagy may blunt apoptosis. The data suggest the potential of nitroxoline for therapeutic development against prostate cancer. Inhibitors of autophagy may potentiate apoptotic cell death and provide novel strategy for the development of nitroxoline.
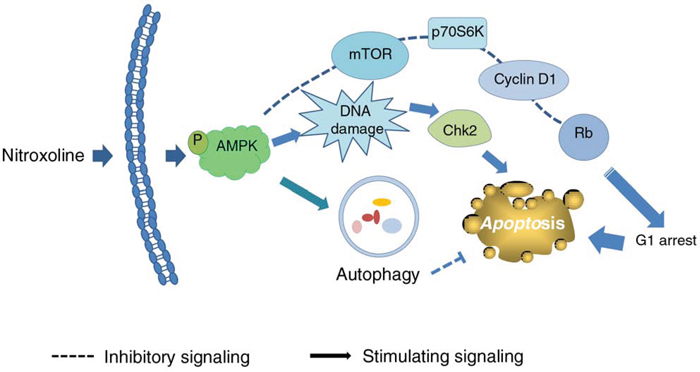
Figure 8: Schematic figure for nitroxoline-mediated signaling pathways on the inhibition of cell proliferation and apoptosis. Nitroxoline induces the activation of AMPK, which in turn inhibits mTOR signaling pathway and cyclin D1-Rb-Cdc25A axis, leading to G1 arrest of cell cycle and apoptosis. Nitroxoline also induces AMPK-dependent activation of Chk2 contributing to cell apoptosis. However, the activated autophagy may blunt the apoptosis.
MATERIALS AND METHODS
Materials
RPMI 1640 medium and fetal bovine serum (FBS) were obtained from GIBCO/BRL Life Technologies (Grand Island, NY). Antibodies to cyclin D1, cyclin E, cyclin A, cyclin B1, Cdk4, Cdk2, Cdk1, p-RbSer807/811, Cdc25A, PARP-1, AMPK siRNA and anti-mouse and anti-rabbit IgGs were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies to p-Chk1Ser345, p-Chk2Thr68, p-AMPKαThr172, p-mTORSer2448, p-p70S6KThr389 (p70 ribosomal S6 kinase), p62, p-H2AXSer139 and GAPDH were from Cell Signaling Technologies (Boston, MA). Antibody against LC3 was purchased from Novus Biologicals (Littleton, CO). The siRNA specifically targeting Chk2 mRNA was purchased from Dharmacon (Lafayette, CO). SRB, PI, CFSE, 4,6-diamidino-2-phenylindole dihydrochloride (DAPI), nitroxoline, paclitaxel, etoposide, Chk2 inhibitor and all other chemical compounds were obtained from Sigma-Aldrich (St. Louis, MO).
Cell lines and cell culture
PC-3, DU-145 (hormone-refractory) and LNCaP (hormone-sensitive) prostate cancer cell lines were from American Type Culture Collection (Rockville, MD). Cells were cultured in RPMI 1640 medium with 10% FBS (v/v), penicillin (100 units/ml) and streptomycin (100 μg/ml). Cultures were maintained in a 37°C incubator with 5% CO2.
SRB assay
Cells were seeded in 96-well plates in medium with 10% FBS. After 24 hours, cells were fixed with 10% trichloroacetic acid (TCA) to represent cell population at the time of agent addition (T0). After additional incubation of 0.1% dimethylsulfoxide (DMSO) or the agent for 48 hours, cells were fixed with 10% TCA and SRB at 0.4% (w/v) in 1% acetic acid was added to stain cells. Unbound SRB was washed out by 1% acetic acid. SRB bound cells were solubilized with 10 mM Trizma base. The absorbance was read at a wavelength of 515 nm. Using the following absorbance measurements, such as time zero (T0), control growth (C), and cell growth in the presence of the agent (Tx), the percentage growth was calculated at each of the agent concentrations levels. Percentage growth inhibition was calculated as: [1- (Tx-T0)/(C-T0)] × 100%. Growth inhibition of 50% (IC50) is determined at the agent concentration which results in 50% reduction of total protein increase in control cells during the agent incubation.
Cell proliferation assay with CFSE labeling
The cells were treated with CFSE at a final concentration of 10 μM. After incubation at 37°C for 10 minutes, RPMI medium with 10% FBS was added. Tubes were placed in ice for 5 minutes and then washed. After centrifugation, the cells were seeded in RPMI medium with 10% FBS for 24, 48 and 72 hours at 37°C under 5% CO2/95% air. After treatment, the fluorescence intensity was determined by flow cytometric analysis (Becton Dickinson, Mountain View, CA).
Colony formation assay
To assess anchorage-dependent colony formation effect, the cells (100 cells/well) were seeded in a 6-well plate. After a 10-day treatment with nitroxoline, the cell colonies were rinsed with phosphate-buffered saline (PBS), stained with 0.25% crystal violet/20% ethanol and photographed by a camera with Copy Stan (Nikon Model No. CS-920; Shimadzu, Japan).
Flow cytometric analysis of PI staining
Cells were harvested by trypsinization, fixed with 70% (v/v) alcohol at 4°C for 30 minutes and washed with PBS. After centrifugation, cells were incubated in 0.1 M phosphate-citric acid buffer (0.2 M NaHPO4, 0.1 M citric acid, pH7.8) for 30 minutes. Cells were centrifuged and resuspended with 0.5 ml PI solution containing Triton X-100 (0.1% v/v), RNase (100 μg/ml) and PI (80 μg/ml). DNA content was analyzed with FACScan and CellQuest software (Becton Dickinson, Mountain View, CA).
DNA fragmentation assay
The DNA fragmentation was determined using the Cell Death Detection ELISAplus kit (Roche, Mannheim, Germany). The assay was based on the quantitative in vitro determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) after induced cell death. After the treatment, the cells were lysed and centrifuged, and the supernatant was used for the detection of nucleosomal DNA according to the manufacturer's protocol.
Western blotting
After treatment, the cells were washed twice with ice-cold PBS and reaction was terminated by the addition of 100 μl ice-cold lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1% Triton X-100). For western blot analysis, the amount of proteins (40 μg) were separated by electrophoresis in a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. After an overnight incubation at 4°C in PBS/5% nonfat milk, the membrane was washed with PBS/0.1% Tween 20 for 1 hour and immuno-reacted with the indicated antibody for 2 hours at room temperature. After four washings with PBS/0.1% Tween 20, the anti-mouse or anti-rabbit IgG (dilute 1:2000) was applied to the membranes for 1 hour at room temperature. The membranes were washed with PBS/0.1% Tween 20 for 1 hour and the detection of signal was performed with an enhanced chemiluminescence detection kit (Amersham Biosciences, NJ, USA).
Small interfering RNA (siRNA) transfection
Cells were seeded into 60-mm tissue culture dishes with 30% confluence and grown for 24 hours to 50% confluence. Each dish was washed with serum-free Opti-MEM (Life Technologies, Ground Island, NY), and 2 ml of same medium was added. Aliquots containing siRNA in serum-free Opti-MEM were transfected into cells using Lipofectamine 2000 according to the instructions. After incubation for 24 to 48 hours, cells were washed with medium and incubated in 10% FBS-containing RPMI-1640 medium for 24 hours. The cells were treated with or without nitroxoline and the protein expression was detected using Western bot analysis.
Comet assay
After treatment, the cells were pelleted and resuspended in ice-cold PBS. The resuspended cells were mixed with 1.5% low melting point agarose. This mixture was loaded onto a fully frosted slide that had been pre-coated with 0.7% agarose and a coverslip was then applied to the slide. The slides were submerged in pre-chilled lysis solution (1% Triton X-100, 2.5 M NaCl, and 10 mM EDTA, pH 10.5) for 1 hour at 4°C. After soaking with pre-chilled unwinding and electrophoresis buffer (0.3 N NaOH and 1 mM EDTA) for 20 minutes, the slides were subjected to electrophoresis for 15 minutes at 0.5 V/cm (20 mA). After electrophoresis, slides were stained with 1X Sybr Gold (Molecular Probes) and nuclei images were visualized and captured at 400X magnifications with an Axioplan 2 fluorescence microscope (Zeiss, Germany) equipped with a CCD camera (Optronics, Goleta, CA). Over hundreds of cells were scored to calculate the overall percentage of comet tail-positive cells.
Immunofluorescence microscopic examination
After treatment, the cells were fixed with 100% methanol at −20°C for 20 minutes and incubated in 1% bovine serum albumin containing 0.1% Triton X-100 at 37°C for 30 minutes. For double staining, cells were first processed the single staining procedure. The cells were washed twice with PBS for 5 minutes and stained with the p-chk2 antibody at room temperature for 1 hour and then, the Cyt3-conjugated secondary antibody at room temperature for 1 hour. After washed with PBS, this process was repeated for staining γ-H2AX with FITC-conjugated secondary antibody. Nuclear staining was performed by 1 μg/ml DAPI. The cells were analyzed by an Axio Imager A1 microscope (Carl Zeiss).
Flow cytometric assay of γ-H2AX
After treatment, the cells were harvested by trypsinization, fixed with 70% (v/v) ethanol at −20°C for 30 minutes and washed with PBS. After centrifugation, cells were permeabilized with 0.1% Triton X-100/PBS and then incubated with anti-γ-H2AX antibody for 1 hour at room temperature. The cells were washed and incubated with FITC-labeled anti-rabbit secondary antibody for 1 hour at room temperature. Cells were washed and re-suspended in PBS for the analysis with FACScan and CellQuest software (Becton Dickinson, Mountain View, CA).
Data analysis
Data are presented as the mean ± SEM for the indicated number of independent experiment. Statistical analysis of data for multiple groups is performed with one-way analysis of variance (ANOVA). Student's t-test is applied for comparison of two groups. P-values less than 0.05 are statistically considered significant.
ACKNOWLEDGEMENTS AND FUNDING
We acknowledge the support provided by the Ministry of Science and Technology in Taiwan (MOST 103-2320-B-002-009-MY3 and MOST 104-2320-B-002-005-MY3). The support by the Center for Innovative Therapeutics Discovery at National Taiwan University is also acknowledged.
CONFLICTS OF INTEREST
All authors declared no conflicts of interest.
REFERENCES
1. Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014; 10:654–663.
2. Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, Uren A, Brown ML, Byers SW. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012; 55:6832–6848.
3. Stolz A, Ertych N, Bastians H. Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability. Clin Cancer Res. 2011; 17:401–405.
4. Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer. 2003; 7:925–936.
5. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003; 3:421–429.
6. Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004; 24:708–718.
7. Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, Hallahan DE, Powell SN, Xia F. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006; 66:1401–1408.
8. Jack MT, Woo RA, Hirao A, Cheung A, Mak TW, Lee PW. Chk2 is dispensable for p53-mediated G1 arrest but is required for a latent p53-mediated apoptotic response. Proc Natl Acad Sci USA. 2002; 99:9825–9829.
9. Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol. 2003; 5:401–409.
10. Ojuka EO. Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle. Proc Nutr Soc. 2004; 63:275–278.
11. Russo GL, Russo M, Ungaro P. AMP-activated protein kinase: a target for old drugs against diabetes and cancer. Biochem Pharmacol. 2013; 86:339–350.
12. Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2014; 33C:1–7.
13. Hardie DG, Alessi DR. LKB1and AMPK and the cancer-metabolism link - ten years after. BMC Biol. 2013; 11:36.
14. Lee CR, Chun JN, Kim SY, Park S, Kim SH, Park EJ, Kim IS, Cho NH, Kim IG, So I, Kim TW, Jeon JH. Cyclosporin A suppresses prostate cancer cell growth through CaMKKβ/AMPK-mediated inhibition of mTORC1 signaling. Biochem Pharmacol. 2012; 84:425–431.
15. Colamonici M, Blyth G, Saleiro D, Szilard A, Bliss-Moreau M, Giles FJ, Altman JK, Beauchamp EM, Platanias LC. Dual targeting of acute myeloid leukemia progenitors by catalytic mTOR inhibition and blockade of the p110α subunit of PI3 kinase. Oncotarget. 2015; 6:8062–8070.
16. Pelletier C, Prognon P, Bourlioux P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob Agents Chemother. 1995; 39:707–713.
17. Jiang H, Taggart JE, Zhang X, Benbrook DM, Lind SE, Ding WQ. Nitroxoline (8-hydroxy-5-nitroquinoline) is more a potent anti-cancer agent than clioquinol (5-chloro-7-iodo-8-quinoline). Cancer Lett. 2011; 312:11–17.
18. Shim JS, Matsui Y, Bhat S, Nacev BA, Xu J, Bhang HE, Dhara S, Han KC, Chong CR, Pomper MG, So A, Liu JO. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J Natl Cancer Inst. 2010; 102:1855–1873.
19. Sosič I, Mirković B, Arenz K, Stefane B, Kos J, Gobec S. Development of new cathepsin B inhibitors: combining bioisosteric replacements and structure-based design to explore the structure-activity relationships of nitroxoline derivatives. J Med Chem. 2013; 56:521–533.
20. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006; 25:5220–5227.
21. Sellers WR, Kaelin WG Jr. Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol. 1997; 15:3301–3312.
22. Fernandez-Vidal A, Mazars A, Manenti S. CDC25A: a rebel within the CDC25 phosphatases family?. Anticancer Agents Med Chem. 2008; 8:825–8231.
23. Vigo E, Müller H, Prosperini E, Hateboer G, Cartwright P, Moroni M C Helin K. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999; 19:6379–6395.
24. Inoki K, Kim J, Guan KL. AMPK, and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012; 52:381–400.
25. Cuyàs E, Corominas-Faja B, Joven J, Menendez JA. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Methods Mol Biol. 2014; 1170:113–144.
26. Hsueh YS, Chang HH, Chiang NJ, Yen CC, Li CF, Chen LT. MTOR inhibition enhances NVP-AUY922-induced autophagy-mediated KIT degradation and cytotoxicity in imatinib-resistant gastrointestinal stromal tumors. Oncotarget. 2014; 5:11723–11736.
27. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013; 4:e838.
28. Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol. 2010; 648:193–214.
29. Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010; 108:73–112.
30. Stolz A, Ertych N, Bastians H. Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability. Clin Cancer Res. 2011; 17:401–405.
31. Kuo LJ, Yang LX. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. in vivo. 2008; 22:305–309.
32. Kao J, Milano MT, Javaheri A, Garofalo MC, Chmura SJ, Weichselbaum RR, Kron SJ. gamma-H2AX as a therapeutic target for improving the efficacy of radiation therapy. Curr Cancer Drug Targets. 2006; 6:197–205.
33. Haase H, Rink L. Zinc signals and immune function. Biofactors. 2014; 40:27–40.
34. He Y, Liu Z, Qiao C, Xu M, Yu J, Li G. Expression and significance of Wnt signaling components and their target genes in breast carcinoma. Mol Med Rep. 2014; 9:137–143.
35. Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer. 2007; 55:1–14.
36. Drobnjak M, Osman I, Scher HI, Fazzari M, Cordon-Cardo C. Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin Cancer Res. 2000; 6:1891–1895.
37. Palmero I, Peters G. Perturbation of cell cycle regulators in human cancer. Cancer Surv. 1996; 27:351–367.
38. Bracho-Valdés I, Moreno-Alvarez P, Valencia-Martínez I, Robles-Molina E, Chávez-Vargas L, Vázquez-Prado J. mTORC1- and mTORC2-interacting proteins keep their multifunctional partners focused. IUBMB Life. 2011; 63:896–914.
39. Weng QP, Kozlowski M, Belham C, Zhang A, Comb MJ, Avruch J. Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J Biol Chem. 1998; 273:16621–16629.
40. Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002; 16:1472–1487.
41. Xu G, Zhang W, Bertram P, Zheng XF, McLeod H. Pharmacogenomic profiling of the PI3K/PTEN-AKT-mTOR pathway in common human tumors. Int J Oncol. 2004; 24:893–900.
42. Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008; 118:3051–3064.
43. Kremer CL, Klein RR, Mendelson J, Browne W, Samadzedeh LK, Vanpatten K, Highstrom L, Pestano GA, Nagle RB. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate. 2006; 66:1203–1212.
44. van Veelen W, Korsse SE, van de Laar L, Peppelenbosch MP. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene. 2011; 30:2289–2303.
45. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010; 22:132–139.
46. Kim J, Kundu M, Viollet B, Guan KL. AMPK, and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13:132–141.
47. Viry E, Paggetti J, Baginska J, Mgrditchian T, Berchem G, Moussay E, Janji B. Autophagy: An adaptive metabolic response to stress shaping the antitumor immunity. Biochem Pharmacol. 2014; 92:31–42.
48. Zhang JW, Zhang SS, Song JR, Sun K, Zong C, Zhao QD, Liu WT, Li R, Wu MC, Wei LX. Autophagy inhibition switches low-dose camptothecin-induced premature senescence to apoptosis in human colorectal cancer cells. Biochem Pharmacol. 2014; 90:265–275.
49. Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001; 410:842–847.
50. Sanli T, Storozhuk Y, Linher-Melville K, Bristow RG, Laderout K, Viollet B, Wright J, Singh G, Tsakiridis T. Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: modulation of cellular signals regulating cell cycle and survival. Radiother Oncol. 2012; 102:459–465.
51. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014; 15:156–169.
52. de Lange J, Verlaan-de Vries M, Teunisse AF, Jochemsen AG. Chk2 mediates RITA-induced apoptosis. Cell Death Differ. 2012; 19:980–989.
53. Chen CR, Wang W, Rogoff HA, Li X, Mang W, Li CJ. Dual induction of apoptosis and senescence in cancer cells by Chk2 activation: checkpoint activation as a strategy against cancer. Cancer Res. 2005; 65:6017–6021.