
INTRODUCTION
Colorectal cancer (CRC) is thought to arise through the accumulation of multiple genetic and epigenetic alterations, leading to the activation of oncogenes and loss of function of tumor suppressor genes [1]. Cumulative evidence suggests that numerous changes in DNA methylation act as premalignant steps toward malignancy during colorectal tumorigenesis, and that aberrant CpG island methylation is a major mechanism driving these epigenetic alterations [2, 3]. A subset of CRCs is known to exhibit concurrent hypermethylation of multiple CpG islands, which is referred to as the CpG island methylator phenotype (CIMP) [4]. In addition, a number of studies have shown that aberrant DNA methylation is involved in determining tumor invasiveness or its tendency toward metastasis in CRC [5–8].
Most CRCs are thought to develop through the adenoma-carcinoma sequence, though a subset of CRCs develop from nonpolypoid lesions. Laterally spreading tumors (LSTs) are generally defined as lesions that extend laterally along the luminal wall and are greater than 10 mm in diameter with a low vertical axis [9]. These flat lesions are thought to be less invasive because they are more likely to be found at the adenoma stage or an early CRC stage [9–11]. LSTs are usually categorized into two types: tumors with granular morphology (LST-G) and those with flat or nongranular morphology (LST-NG) [12]. Although the molecular mechanism underlying LST development is not fully understood, there have also been multiple reports of frequent KRAS mutation in LSTs [13–15]. And there is reportedly a high prevalence of CIMP-high in this type of tumor [14]. In addition, one recent study reported that LST-G is characterized by an intermediate-methylation epigenotype and KRAS mutation, while LST-NG is associated with a low-methylation epigenotype and frequent CTNNB1 mutation [16]. An inverse association between APC methylation and submucosal invasion by LSTs has also been reported [17].
The above-summarized findings indicate epigenetic alterations may be an important determinant of the lateral or vertical growth pattern and invasiveness of colorectal neoplasms. To identify these molecular alterations, we carried out high-throughput CpG island methylation profiling in sets of tumors with lateral or vertical growth. We identified a number of CpG islands differentially methylated between the two groups, including that of NTSR1, which was preferentially methylated in laterally growing and noninvasive colorectal tumors. We show that methylation-associated silencing of NTSR1 is inversely associated with the invasiveness of colorectal tumors, and that its methylation could be a predictive biomarker of a better prognosis in CRC patients.
RESULTS
Identification of NTSR1 methylation in large noninvasive colorectal tumors
Our first aim was to identify changes in DNA methylation that could be causally related to the growth pattern and invasiveness of colorectal tumors. To accomplish this, we categorized endoscopically or surgically resected colorectal tumors based on the presence or absence of submucosal invasion – i.e., whether they were invasive or noninvasive tumors (ITs or NTs). The tumors were also categorized according to their size, large being ≥ 20 mm in diameter along the colonic surface and small being < 20 mm in diameter along the colonic surface. Then using methylated CpG island amplification-microarray (MCAM) analysis, we assessed the global methylation pattern in large NTs (n = 3), small ITs (n = 3) and normal colonic mucosa adjacent to the tumors (n = 3) (Figure 1A), and screened differentially methylated CpG islands. Unsupervised hierarchical clustering analysis of the MCAM data revealed a substantial number of differentially methylated genes (n = 575), the majority of which were methylated at higher levels in large NTs than in small ITs (Figure 1B, Supplementary Table S1). Among these, we focused on neurotensin receptor 1 (NTSR1) because although neurotensin (NTS) signaling has been strongly implicated in tumorigenesis, methylation of NTSR1 has not yet been reported in colorectal tumors (Figure 1B) [18].
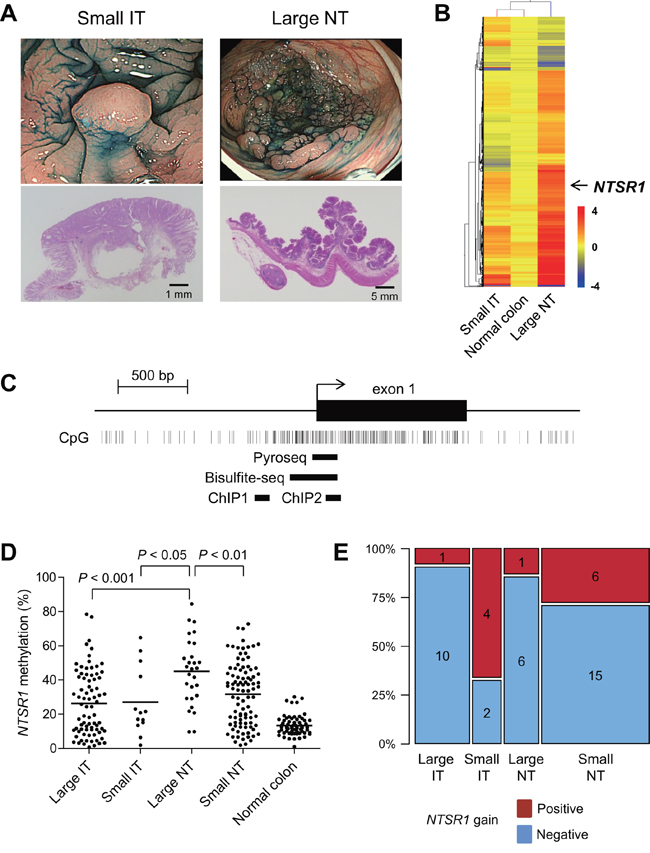
Figure 1: Identification of NTSR1 methylation in large noninvasive tumors. A. Representative examples of a small invasive tumor (IT) and a large noninvasive tumors (NT). Endoscopic views are shown on the top and histological views are blow. B. Summary of MCAM results from small ITs, large NTs and normal colon. Genes differentially methylated between small ITs and large NTs were selected, after which unsupervised hierarchical clustering was carried out. Each row represents a single probe, and each column represents an average of each category. C. Diagram of the NTSR1 CpG island. The transcription start site and exon 1 are shown on the top, and the regions analyzed using bisulfite pyrosequencing, bisulfite sequencing and ChIP-PCR are shown below. D. Summarized results of the bisulfite pyrosequencing analysis of NTSR1 in specimens from the indicated colorectal tumor types and adjacent normal colonic tissue. E. Frequencies of NTSR1 copy number gain in the indicated types of colorectal tumors. The number of samples in each portion is also shown.
We validated the MCAM results by performing quantitative bisulfite pyrosequencing with a set of clinical samples (large IT, n = 78; small IT, n = 13; large NT, n = 28; small NT, n = 96; normal colon, n= 66; CRCs in Dukes’ stages C and D were not included) (Figure 1C, 1D). Methylation in the promoter CpG island of NTSR1 was relatively limited in normal colon but was elevated to differing degrees in primary tumors (Figure 1D). Notably, large NTs showed significantly greater methylation than other tumor types, which is consistent with the MCAM results (Figure 1D). Because expression of NTSR1 is reportedly upregulated in various malignancies [18], we also determined whether any chromosomal aberrations were associated with NTSR1 in colorectal tumors. Among the tumor specimens analyzed, array-based comparative genomic hybridization (array CGH) results were available for 45 samples from an earlier study (large IT, n = 11; large NT, n = 7; small IT, n = 6; small NT, n = 21) [2]. Those data showed that NTSR1 copy number gains were substantially more frequent in small ITs than other types of tumors (Figure 1E). These results suggest that elevated levels of NTSR1 methylation are associated with laterally spreading and noninvasive tumor growth, while lower levels of NTSR1 methylation are related to smaller tumors with more invasive growth patterns. Our data also suggest that NTSR1 copy number gain may also be related to the invasive tumor growth.
Association between NTSR1 methylation and clinicopathological features
We next assessed the association between NTSR1 methylation and other molecular and clinicopathological features of colorectal tumors. Survival data were available for 91 patients with invasive tumors, and Cox regression analysis revealed the lowest hazard ratio for the high-methylation group (HR = 0.186; 95% CI, 0.051–0.676), when we employed a cutoff value of 29%. Kaplan-Meier analysis showed better overall survival among patients with high NTSR1 methylation (Figure 2A). We thus categorizing the tumors into high- (≥29%) or low-methylation groups (<29%) based on the level of NTSR1 methylation, but we detected no statistically significant differences with respect to age, gender or tumor location between the two groups (Tables 1, 2). NTSR1 methylation also did not correlate with BRAF or TP53 mutation or CIMP status, though KRAS mutation was more prevalent in the high-methylation group (Table 1, 2). Among invasive tumors, distant metastasis showed a weak correlation with NTSR1 methylation, while other clinicopathological characteristics had no significant association (Table 1). Among noninvasive tumors, including precursor lesions and carcinoma in situ, levels of NTSR1 methylation were significantly higher in LST-G lesions than other morphological types (Figure 2B), but histological type did not correlate with NTSR1 methylation (Table 2). We further examined the association between NTSR1 expression and prognosis using a publicly available data set from 62 CRC patients (GSE12945) [19]. When we categorized CRC patients according to their NTSR1 expression levels, we found shorter overall survival among those with tumors that strongly expressed NTSR1 (Supplementary Figure S1). These results suggest that higher methylation and weaker expression of NTSR1 is associated with a better prognosis in CRC patients.
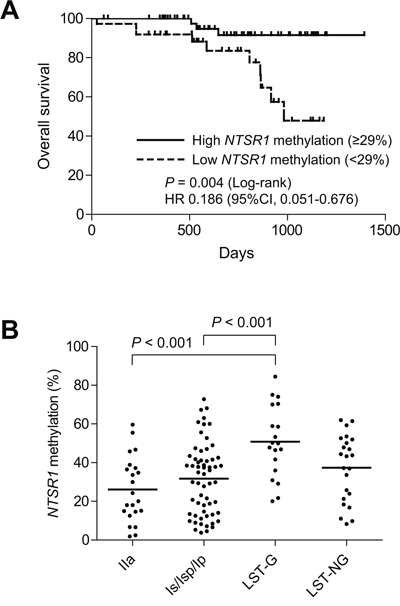
Figure 2: Analysis of NTSR1 methylation in colorectal tumors. A. Kaplan-Meier curves showing the effect of NTSR1 methylation (high, ≥ 29%; low, < 29%) on overall survival of patients with invasive colorectal tumors (n = 91). B. Summarized results of bisulfite pyrosequencing of NTSR1 in the indicated morphological types of noninvasive tumors.
Table 1: Correlation between NTSR1 methylation and the clinicopathological features of invasive colorectal tumors
NTSR1 methylation |
P |
||
<29% (n = 64) |
≥29% (n = 67) |
||
Age (y, mean ± SD) |
66.33 ± 12.92 |
68.81 ± 10.21 |
NS |
Gender |
|||
Female |
21 |
30 |
NS |
Male |
43 |
37 |
|
Location |
|||
Right |
29 |
28 |
NS |
Left |
11 |
23 |
|
Rectum |
24 |
16 |
|
BRAF |
|||
Mut |
1 |
5 |
NS |
Wt |
63 |
62 |
|
KRAS |
|||
Mut |
16 |
21 |
NS |
Wt |
48 |
46 |
|
TP53 |
|||
Mut |
24 |
26 |
NS |
Wt |
40 |
41 |
|
CIMP |
|||
Positive |
4 |
10 |
NS |
Negative |
60 |
57 |
|
pT category |
|||
pT1 |
12 |
8 |
NS |
pT2 |
8 |
13 |
|
pT3 |
34 |
41 |
|
pT4 |
6 |
5 |
|
NA |
4 |
0 |
|
pN category |
|||
pN0 |
40 |
51 |
NS |
pN1 |
20 |
15 |
|
NA |
4 |
1 |
|
pM category |
|||
pM0 |
56 |
66 |
0.018 |
pM1 |
5 |
0 |
|
NA |
3 |
1 |
|
Dukes’ stage |
|||
A |
18 |
18 |
NS |
B |
22 |
33 |
|
C |
16 |
15 |
|
D |
5 |
0 |
|
NA |
3 |
1 |
|
Lymphatic invasion |
|||
Negative |
5 |
2 |
NS |
Positive |
55 |
64 |
|
NA |
4 |
1 |
|
Vascular invasion |
|||
Negative |
8 |
6 |
NS |
Positive |
51 |
60 |
|
NA |
5 |
1 |
|
NA, not available; NS, not significant
Table 2: Correlation between NTSR1 methylation and the clinicopathological features of noninvasive colorectal tumors
NTSR1 methylation |
P |
||
<29% (n = 47) |
≥29% (n = 77) |
||
Age (y, mean ±SD) |
69.21 ± 9.78 |
68.91 ± 10.03 |
NS |
Gender |
|||
Female |
14 |
27 |
NS |
Male |
33 |
50 |
|
Location |
|||
Right |
24 |
37 |
NS |
Left |
13 |
16 |
|
Rectum |
10 |
24 |
|
BRAF |
|||
Mut |
9 |
8 |
NS |
Wt |
38 |
69 |
|
KRAS |
|||
Mut |
12 |
32 |
NS |
Wt |
35 |
45 |
|
TP53 |
|||
Mut |
3 |
7 |
NS |
Wt |
44 |
70 |
|
CIMP |
|||
Positive |
10 |
15 |
NS |
Negative |
37 |
62 |
|
Pathology |
|||
HP |
2 |
5 |
NS |
TSA |
3 |
9 |
|
SSA |
6 |
5 |
|
T-ad |
17 |
20 |
|
TV-ad |
10 |
22 |
|
Cancer (Tis) |
9 |
16 |
|
NA, not available; NS, not significant
Methylation of NTSR1 is associated with gene silencing in CRC cells
To determine whether NTSR1 methylation is associated with the gene's silencing, we analyzed the methylation and expression statuses of NTSR1 in a series of CRC cell lines (Figure 3A). Various levels of NTSR1 methylation were detected in 6 of the 9 CRC cell lines tested, with DLD1 and LoVo cells exhibiting the highest levels. Bisulfite sequencing was then used to obtain a more detailed picture of the methylation status in selected cell lines. We found that the CpG island of NTSR1 is densely methylated in these cells, but is nearly completely unmethylated in HCT116 cells (Figure 3B, Supplementary Figure S2). Reverse transcription-polymerase chain reaction (RT-PCR) analysis revealed a loss of NTSR1 expression in DLD1 and LoVo cells, which was restored by treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza-dC), suggesting DNA methylation is responsible for the transcriptional silencing (Figure 3A, 3C, Supplementary Figure S3). By contrast, in 5 of 9 CRC cell lines NTSR1 expression was higher than in normal colon, irrespective of their methylation levels (Figure 2A). We also analyzed in detail the methylation status of NTSR1 in SW48 cells, because these cells exhibited a moderate level of DNA methylation and elevated NTSR1 expression (Figure 3A). Bisulfite sequencing revealed a mixture of methylated and unmethylated alleles in SW48 cells, suggesting the gene is expressed from the unmethylated alleles (Supplementary Figure S2).
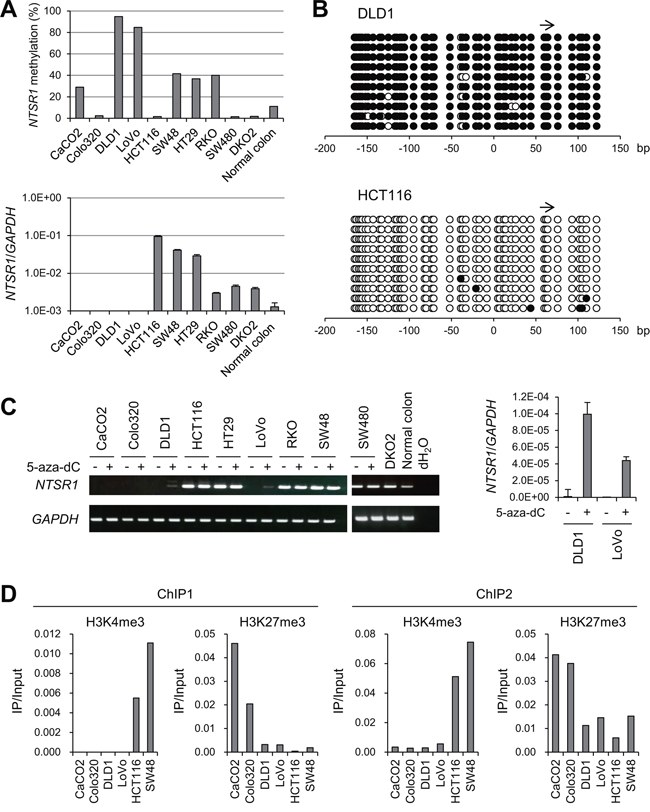
Figure 3: Analysis of NTSR1 methylation and expression in CRC cell lines. A. Bisulfite pyrosequencing (top) and quantitative RT-PCR (bottom) analyses of NTSR1 in the indicated CRC cell lines and normal colonic tissue. B. Bisulfite sequencing analysis of the NTSR1 CpG island in the indicated CRC cell lines. Open and filled circles represent unmethylated and methylated CpG sites, respectively. The arrow indicates the region analyzed with bisulfite pyrosequencing. C. RT-PCR results (left) and quantitative RT-PCR (right) analysis of NTSR1 in the indicated CRC cell lines, with or without 5-aza-dC treatment. Cells were treated with 2 μM 5-aza-dC for 72 h. D. ChIP-PCR analyses of NTSR1 in CRC cells. Levels of H3K4me3 and H3K27me3 in two regions around the NTSR1 transcription start site (see Figure 1C) are shown.
NTSR1 was also not expressed in CaCO2 or Colo320 cells, but we detected low levels of DNA methylation and a lack of re-expression after 5-aza-dC treatment, indicating that a different mechanism may be responsible for the gene silencing (Figure 3A, 3C). We therefore analyzed the histone modification status in selected CRC cell lines. Using chromatin immunoprecipitation (ChIP)-PCR, we assessed trimethylated histone H3 lysine 4 (H3K4me3), an active histone mark, and H3K27me3, a repressive histone mark, in two regions around the NTSR1 transcription start site (Figure 1C). We found that levels of H3K4me3 were markedly higher in cells in which NTSR1 was expressed (HCT116 and SW48) than in cells in which NTSR1 was silenced (Figure 3D). By contrast, H3K27me3 levels were significantly higher in CaCO2 and LoVo cells than other cells, suggesting that a repressive histone modification is involved in the silencing of NTSR1 in these cells.
To confirm whether DNA methylation is associated with NTSR1 silencing in primary CRCs, we analyzed data sets from The Cancer Genome Atlas (TCGA). We observed inverse relationships between the methylation levels on multiple probe sets of an Infinium BeadChip and levels of NTSR1 expression in CRC tissues (Supplementary Figure S4). Taken together, these results suggest that CpG island methylation and histone modifications are associated with the silencing of NTSR1.
Functional analysis of NTSR1 in CRC cells
To clarify the function of NTSR1 in CRC cells, we next assessed the expression of NTS in a series of CRC cell lines, most of which exhibited detectable expression in RT-PCR analyses (Figure 4A). We then transfected two cell lines in which NTSR1 was silenced (DLD1 and LoVo) with a NTSR1 expression vector and performed a flow cytometric analysis. We found that ectopic NTSR1 expression did not induce cell cycle arrest or apoptosis in CRC cells (Figure 4B, Supplementary Figure S5). Colony formation assays showed that cells expressing ectopic NTSR1 formed much larger colonies than those transfected with an empty vector (Figure 4C), and Matrigel invasion assays showed that NTSR1 promoted CRC cell invasion (Figure 4D). Conversely, NTSR1 knockdown in CRC cells otherwise exhibiting strong NTSR1 expression (HCT116) suppressed cell proliferation (Figure 4E). Following NTSR1 depletion, HCT116 cells also showed a tendency toward reduced invasiveness, though the effect was not statistically significant (Figure 4E).
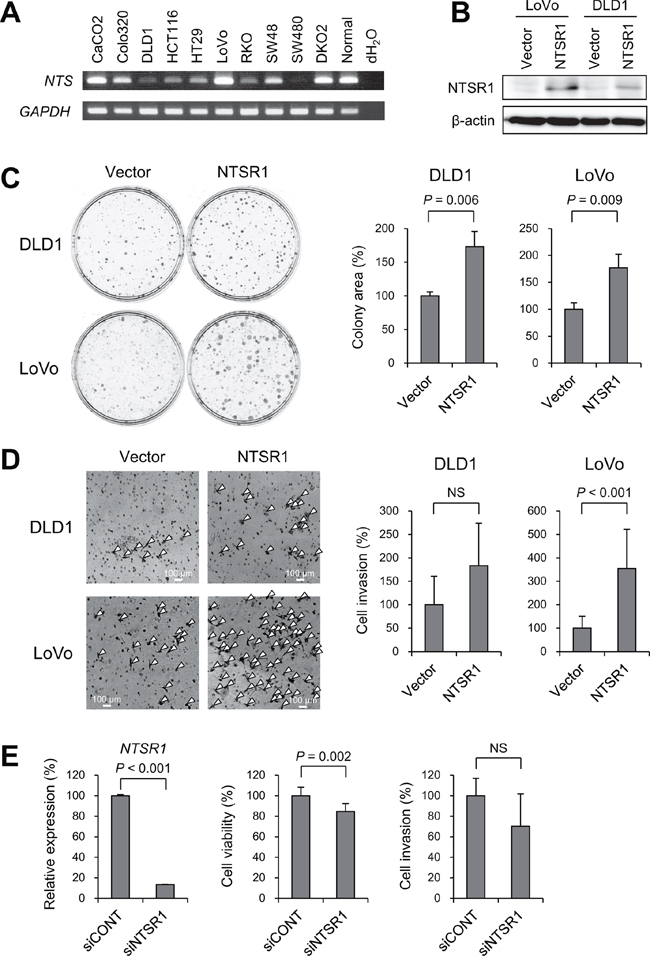
Figure 4: Functional analysis of NTSR1 in CRC cells. A. RT-PCR analysis of NTS in CRC cell lines and normal colonic tissue. B. Western blot analysis of NTSR1 in the indicated CRC cells transfected with a NTSR1 expression vector or a control vector (Vector). C. Colony formation assays using the indicated CRC cell lines transfected with a NTSR1 expression vector or a control vector. Representative results are on the left, and relative colony formation efficiencies are on the right. Shown are means of 3 replications; error bars represent SDs. D. Matrigel invasion assays using the indicated CRC cell lines transfected with a NTSR1 expression vector or a control vector. Invading cells are indicated by arrowheads. Shown on the right are the means of 5 random microscopic fields per membrane; error bars represent SDs. E. Results of quantitative RT-PCR analysis of NTSR1 expression (left), cell viability assays (middle) and Matrigel invasion assays (right) using HCT116 cells transfected with siRNA targeting NTSR1 (siNTSR1) or a control siRNA (siCONT). Values were normalized to cells transfected with the control siRNA. Cell viability assay results are means of 8 replications. Matrigel invasion assay results are means of 5 random microscopic fields per membrane. Error bars represent SDs. NS, not significant.
Alteration of NTSR1 methylation during the progression of colorectal tumorigenesis
The results summarized above suggest NTSR1 is a target of epigenetic silencing in colorectal tumors, despite it functions as an oncogene. This prompted us to examine the genetic and epigenetic alterations occurring to NTSR1 during tumor progression by analyzing a mixed colorectal lesion consisting of adenomatous and malignant portions (Figure 5A). Using biopsy specimens from adenomatous and cancerous regions, we analyzed NTSR1 methylation and copy number alterations. Bisulfite pyrosequencing revealed high levels of NTSR1 methylation in the adenomatous regions and significantly lower levels in the cancerous regions (Figure 5B). This difference in methylation status was further confirmed by bisulfite sequencing (Figure 5C). In addition, array CGH analysis revealed a NTSR1 copy number gain in only the cancerous component (Figure 5D). Correspondingly, immunohistochemical analysis of the same tumor specimens showed low and elevated levels of NTSR1 protein in the adenomatous and cancerous regions, respectively (Figure 5E, Supplementary Figure S6). Further analysis of NTSR1 methylation in additional mixed colorectal lesions containing adenomatous and malignant portions (n = 22) showed that methylation levels were significantly higher in the adenomas than adjacent normal colonic tissue, but they were generally lower in the malignant portions (Figure 5F). These results suggest that NTSR1 is a methylation-prone gene in early colorectal lesions, but reduced methylation and may activate NTSR1 during the malignant progression of colorectal tumorigenesis.
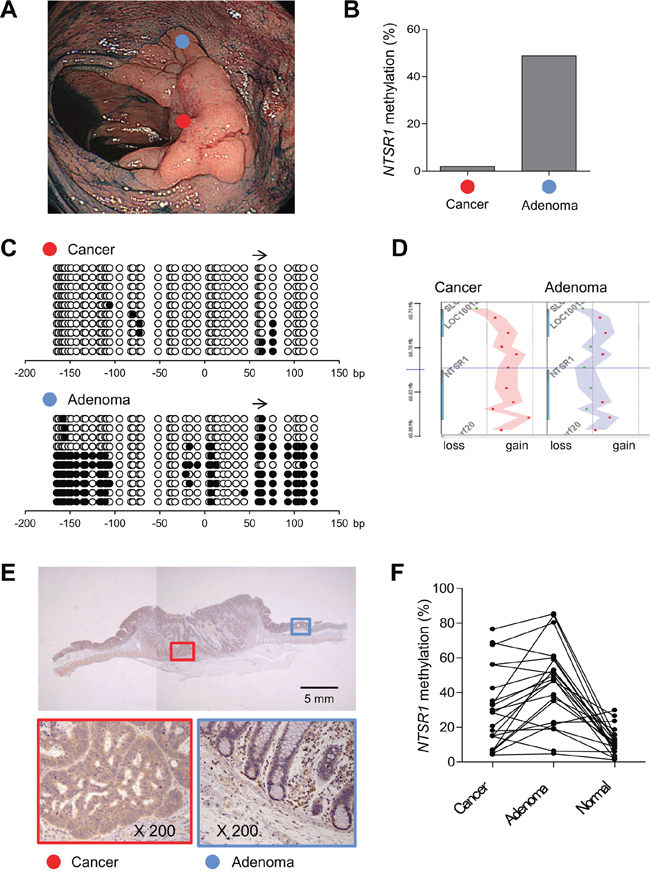
Figure 5: Alteration of NTSR1 methylation during the progression of colorectal tumorigenesis. A. Endoscopic view of a colorectal tumor consisting of an adenomatous portion (blue dot) and an invasive cancerous portion (red dot). B. Levels of NTSR1 methylation in biopsy specimens from the indicated portions analyzed by bisulfite pyrosequencing. C. Bisulfite sequencing analysis of NTSR1 in the indicated portions. The arrow indicates the region analyzed with bisulfite pyrosequencing. D. Array CGH results of the NTSR1 locus in the indicated portions. Each dot represents a single probe and genomic gains are indicated by dots on the right; losses are on the left. E. Immunohistochemical analysis of NTSR1. Magnified views of the cancerous and adenomatous portions (indicated by red and blue boxes, respectively) are shown below. F. Summarized results of bisulfite pyrosequencing of NTSR1 in colorectal tumors containing adenomatous and cancerous portions (n = 22). Methylation levels in adjacent normal colonic tissue from the same patients are also shown.
DISCUSSION
In the present study, we searched for DNA methylation changes associated with the growth pattern of colorectal tumors. We found that the CpG island of NTSR1 is highly methylated in laterally spreading and noninvasive tumors, that methylation of NTSR1 is associated with transcriptional silencing of the gene, and that higher levels of the methylation in primary tumors are associated with better patient survival. Functional analysis suggested that NTSR1 expression promotes tumor growth and invasion, which is consistent with the better prognosis of patients with NTSR1 methylation.
NTS is a 13-amino acid neuropeptide initially isolated from bovine hypothalamus [20]. Since then it has been determined that NTS is localized mainly in the central nervous system and distal small bowel, and is released from the gut upon ingestion of fats [21, 22]. The physiological functions of NTS include modulation of GI tract motility [23–25], stimulation of intestinal secretion [26, 27] and stimulation of the growth and regeneration of the intestinal epithelial cells [28, 29]. The NTS signal is transmitted via two G protein-coupled receptors (GPCRs), NTSR1 and NTSR2, which respectively exhibit higher and lower affinity for NTS [30].
An abundance of data implicate NTS and NTSR1 in the progression of several cancers, including pancreatic, breast, lung and colorectal cancers [18], and NTS is expressed at higher levels in cultured and primary CRC cells than in normal colon [31]. Epithelial crypts isolated from normal human adult colon lack NTSR1 expression, whereas it is overexpressed in many CRC cell lines [32]. NTSR1 is a target of the Tcf/β-catenin complex in the Wnt signaling pathway, and activation of NTSR1 expression is an early event in colorectal tumorigenesis [33]. Another study reported that NTSR1 expression progressively increases during the process of colorectal tumorigenesis [34]. Overexpression of NTSR1 is also observed during the progression of inflammatory bowel disease-related oncogenesis [35, 36]. Through NTSR1, NTS signaling mediates cancer cell proliferation, survival, migration and invasiveness by activating a variety of signaling molecules, including protein kinase C (PKC), extracellular signal-regulated kinase 1 and 2 (ERK1/2) and interleukin (IL)-8 [18, 37, 38]. Administration of NTS significantly increases the growth of CRC cell xenografts in mice, and the effect is blocked by SR 48692, a specific NTSR1 antagonist [39]. Moreover, a recent study using Ntsr1-deficient mice demonstrated that targeted disruption of Ntsr1 reduces susceptibility to azoxymethane (AOM)-induced colon tumorigenesis [40].
Given the apparent oncogenic function of NTSR1, it seems paradoxical that NTSR1 would be a target of aberrant DNA methylation in colorectal tumors. Histone modifications also play important roles in epigenetic regulation, and H3K27me3, which is mediated by the polycomb group proteins, is well known to be a repressive mark. Recent studies showed that genes marked by the polycomb complex in embryonic stem (ES) cells have a predisposition toward DNA hypermethylation in cancer [41, 42]. Moreover, many of the genes hypermethylated in adult cancers are held in a transcription-ready state in ES cells by a bivalent chromatin domain containing both active (H3K4me3) and repressive (H3K27me3) histone marks [43]. NTSR1 is one such gene, as its promoter contains both H3K4me3 and H3K27me3 marks in both mouse and human ES cells [44]. In addition, we observed that H3K27me3 is also involved in the epigenetic silencing of NTSR1 in CRC cells. Methylation of NTSR1 has been reported in pancreatic and lung cancers, suggesting NTSR1 is a methylation-prone gene in tumors of multiple organs [45, 46].
In colorectal lesions that contain both adenoma and carcinoma components, we often observed NTSR1 hypermethylation in the adenomatous portion and generally lower methylation levels in the cancerous portions. The mechanism underlying the changes of NTSR1 methylation remains unclear, but what is known suggests several possibilities: (1) precursor cells without NTSR1 methylation may selectively survive during the malignant progression, (2) methylated CpG island of NTSR1 in precursor cells may be actively demethylated during the progression from adenoma to carcinoma, and/or (3) the unmethylated NTSR1 allele may be amplified in cancer cells. Our array CGH results indicate that amplification of the NTSR1 locus may be one of the mechanisms by which NTSR1 methylation is reduced in cancer cells. In addition, as mentioned above, NTSR1 is a target of the Wnt signaling pathway and is transcriptionally activated by the Tcf/β-catenin complex [33]. The progression from early adenoma to invasive carcinoma is associated with an increase of nuclear β-catenin levels [47], which could promote transcriptional activity at NTSR1 that prevents the NTSR1 promoter from being methylated in advanced tumors.
In conclusion, we found that NTSR1 is frequently methylated in colorectal tumors and that elevated levels of NTSR1 methylation are associated with laterally spreading and noninvasive growth patterns. We also found that levels of NTSR1 methylation are likely downregulated during the development of CRC, leading to NTSR1 activation. Because NTSR1 functions as an oncogene and NTS-NTSR1 signaling is a potential target of anti-cancer therapy, methylation of NTSR1 could be a predictive biomarker of the responsiveness of CRC patients to such treatment.
MATERIALS AND METHODS
Study population and cell lines
Colorectal tumor tissues were collected from Japanese patients who underwent endoscopic or surgical resection at Akita Red Cross Hospital. A total of 309 specimens from 99 precursor lesions, 156 CRCs and 54 samples of adjacent normal tissue were obtained through surgical resection or endoscopic biopsy. Informed consent was obtained from all patients before collection of the specimens. Approval of this study was obtained from Institutional Review Board of Akita Red Cross Hospital and Sapporo Medical University. CRC cell lines (DLD1, LoVo, HCT116, SW48, CaCO2, Colo320, HT29, RKO and SW480) were obtained and cultured as described previously [48]. HCT116 cells harboring genetic disruptions within the DNMT1 and DNMT3B loci (DKO2) are as described [48]. To restore epigenetically silenced genes, cells were treated with 2 μM 5-aza-dC (SIGMA, St Louis, MO, USA) for 72 h, replacing the drug and medium every 24 h. Genomic DNA was extracted using the standard phenol-chloroform procedure. Total RNA was extracted using TRIZOL reagent (Life Technologies, Carlsbad, CA) and then treated with a DNA-free kit (Life Technologies).
Endoscopic and histological analysis
High-resolution magnifying endoscopes (CF260AZI; Olympus, Tokyo, Japan) were used for all colonoscopic analyses. All detected colorectal tumors were observed at high magnification after staining with indigo carmine dye and 0.05% crystal violet. Surface microstructures were classified according to Kudo's pit pattern classification system [49]. Most often one biopsy specimen was collected from each lesion for the purpose of extracting genomic DNA. However, when at least two components with different surface microstructures were found in a single lesion, biopsy specimens were obtained for each portion, as described previously [2]. The lesions were then treated through endoscopic mucosal resection (EMR), endoscopic submucosal dissection (ESD) or surgical resection, after which histological analyses were carried out. The morphology of noninvasive tumors was determined according to the Paris classification [50]. LSTs were defined as lesions ≥ 10 mm in diameter with a low vertical axis extending laterally along the interior luminal wall [51].
MCAM analysis
MCAM was performed as described previously [2]. A BioPrime Plus Array CGH Genomic Labeling System (Life Technologies) was used to label MCA amplicons from tumor samples with Alexa Fluor 647, and those from a pooled mixture of normal colonic tissue with Alexa Fluor 555. Labeled MCA amplicons were then hybridized to a custom human CpG island microarray (G4497A; Agilent Technologies, Santa Clara, CA, USA), which included 15,134 probes covering 6,157 unique genes. After washing, the array was scanned using an Agilent DNA Microarray Scanner (Agilent technologies), and the data were processed using Feature Extraction ver. 10.7 (Agilent Technologies) and analyzed using GeneSpring GX ver. 11 (Agilent technologies).
Methylation analysis using bisulfite-pyrosequencing and bisulfite sequencing
Genomic DNA (1 μg) was modified with sodium bisulfite using an EpiTect Bisulfite Kit (Qiagen, Hilden, Germany), after which bisulfite sequencing and pyrosequencing were carried out as described previously [2]. For bisulfite pyrosequencing, the biotinylated PCR product was purified, made single-stranded and used as a template in a pyrosequencing reaction run according to the manufacturer's instructions. The pyrosequencing reaction was carried out using a PSQ96 system with a PyroGold reagent Kit (Qiagen), and the results were analyzed using Q-CpG software (Qiagen). Tumors were defined as CIMP-positive when methylation was detected in the loci of three or more of five markers: MINT1, MINT2, MINT12, MINT31 and p16 [2]. For bisulfite sequencing, amplified PCR products were cloned into pCR2.1-TOPO vector (Life Technologies), and 10 clones from each sample were sequenced using an ABI3130x automated sequencer (Life Technologies). Primer sequences and PCR product sizes are listed in Supplementary Table S2.
Mutation analysis
Mutations within codon 600 of BRAF and codons 12 and 13 of KRAS were examined by pyrosequencing using BRAF and KRAS pyro kits (Qiagen) according to the manufacturer's instructions [2]. Mutation of TP53 was initially determined by PCR-SSCP followed by direct sequencing, as described [2].
RT-PCR
Single-stranded cDNA was prepared using SuperScript III reverse transcriptase (Life Technologies), after which the integrity of the cDNA was confirmed by amplifying glyceraldehydes-3-phosphate dehydrogenase (GAPDH). Primer sequences and PCR product sizes are listed in Supplementary Table S2. Quantitative RT-PCR was carried out using TaqMan Gene Expression Assays (NTSR1, Hs00901549_m1; GAPDH, Hs02758991_g1; Life Technologies) and a 7500 Fast Real-Time PCR System (Life Technologies). SDS ver. 1.4 (Life Technologies) was used for comparative delta Ct analysis.
ChIP-PCR
ChIP was carried out as described previously [52]. Briefly, 5 × 105 cells were cross-linked with formaldehyde (final concentration is 0.5%) for 10 min at room temperature. After washing, the cells were resuspended in 110 μl of SDS lysis buffer and sonicated. The resultant sheared chromatin was immunoprecipitated overnight at 4°C using 0.2 μg of anti-H3K4me3 (#04–745, Millipore, Billerica, Massachusetts, USA) or 2 μg of anti-H3K27me3 (#9733, Cell Signaling Technology, Danvers, MA, USA) antibody. Before adding the antibodies, 10 μl of the sheared chromatin were saved as input DNA. Samples were incubated for 4 h at 65°C to reverse crosslinking, treated with RNase A and proteinase K, and purified. Input DNA and the immunoprecipitates were then subjected to quantitative PCR analysis using Fast SYBR Green PCR Master Mix (Life Technologies) according to the manufacturer's instructions. Primer sequences and PCR product sizes are listed in Supplementary Table S2.
Expression vector and siRNA
An expression construct containing full-length NTSR1 (pReceiver-M11-NTSR1) was purchased from Gene Copoeia (Rockville, MD, USA). CRC cells (5 × 105 cells/well in 6-well plates) were transfected with 2.5 μg of the expression vector or empty pcDNA3.1 using Lipofectamine 3000 (Life Technologies) according to the manufacturer's instructions. For RNA interference (RNAi)-mediated knockdown of NTSR1, 5 × 105 cells were transfected with 25 pmol of Stealth RNAi siRNA (Life Technologies) or a negative control (Life Technologies) using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instructions.
Western blot analysis
Samples of total cell lysate (20 μg of protein) were separated using SDS-PAGE (12.5% acrylamide) and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were then blocked with Block Ace (Dainippon Pharmaceutical, Tokyo, Japan) and incubated with goat anti-NTSR1 polyclonal Ab (1:1000 dilution, C-20; Santa Cruz Biotechnology) and mouse anti-β-actin mAb (1:10000 dilution, clone AC-15; Sigma-Aldrich, St. Louis, MO, USA). The immunoreactive bands were visualized using horseradish peroxidase-conjugated anti-mouse or anti-goat IgG (1:10000 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and ECL (GE Healthcare Bio-Sciences KK, Tokyo, Japan). Images were acquired using a chemiluminescent CCD imager (Image Quant LAS 4000; GE Healthcare Bio-Sciences KK, Tokyo, Japan).
Colony formation assay
Cells were transfected with a NTSR1 expression vector or a control vector as described above. After incubation for 24 h, the transfectants were plated on 60-mm culture dishes and selected for 10 to 14 days in 1.0 mg/ml G418, after which colonies were stained with Giemsa.
Cell viability assay
Cells were transfected with siRNA as described above. After incubation for 24 h, the transfectants were seeded into 96-well plates to a density of 5 × 103 cells per well and incubated for an additional 72 h. Cell viability assays were then carried out using a Cell Counting kit-8 (Dojindo, Tokyo, Japan) according to the manufacturer's instructions.
Matrigel invasion assay
Cells were transfected with the expression construct or siRNA as described above. After incubation for 24 h, 1 × 105 transfectants suspended in 500 μl of serum-free culture medium were added to the tops of BD BioCoat Matrigel Invasion Chambers (BD Biosciences, Franklin Lakes, NJ, USA) prehydrated with phosphate buffered saline (PBS), and 700 μl of medium supplemented with 10% fetal bovine serum (FBS) were added to the lower wells of the chambers. After incubation for 24 h, the invading cells were stained with 1% toluidine and counted in 5 randomly selected microscopic fields per membrane.
Flow cytometry
Cells were transfected with the expression construct as described above. After incubation for 72 h, apoptosis was measured using an ApoScreen Annexin V Apoptosis Kit (Southern Biotech, Birmingham, AL, USA). Briefly, 1 × 106 cells were washed twice in cold PBS and then resuspended in cold binding buffer, after which 10 μL of annexin V-FITC was added to 100 μL of the cell suspension. The mixture was then incubated for 15 min on ice in the dark before addition of 380 μL of cold binding buffer and 10 μL of propidium iodide. For cell cycle analysis, cells were fixed in ethanol, and DNA was stained with propidium iodide. For each sample, data were acquired from a minimum of 1 × 105 cells using a BD FACSCant II (BD Biosciences) with BD FACSDiva software (BD Biosciences), and were analyzed using FlowJo ver. 10 (FlowJo, LLC, Ashland, OR, USA).
Immunohistochemistry
Specimens were fixed in buffered formalin and embedded in paraffin using routine procedures. Sections (4-μm thick) were prepared, dried, deparaffinized and rehydrated before microwave treatment (H2500, Microwave Processor, Bio Rad) in citrate buffer (pH 6.0) for 5 min. A goat anti-NTSR1 polyclonal Ab (1:100 dilution, C-20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) [53] and an automatic staining machine (Envision(00000FE) system; DAKO, Glostrup, Denmark) were used for immunohistochemical labeling. The slides were counterstained with hematoxylin, then dehydrated and mounted.
Statistical analysis
To compare differences in continuous variables between groups, t tests or ANOVA with post hoc Tukey's tests were performed. Fisher's exact test or chi-squared test was used for analysis of categorical data. Values of P < 0.05 (two-sided) were considered statistically significant. To assess the association between survival and methylation levels, Cox regression analyses were performed. Kaplan-Meier curves were plotted to compare survival in two groups stratified based on gene methylation or expression status. The minimum P-value method was used to determine the best cutoff value of the NTSR1 methylation level for survival analysis. After excluding data within the highest and the lowest 10 percentiles, all cutoff values were tested using the log-rank test, and a cutoff value with the lowest P-value was determined. Statistical analyses were carried out using JMP ver. 10 (SAS Institute Inc., Cary, NC, USA) and GraphPad Prism ver. 5.0.2 (GraphPad Software, La Jolla, CA, USA).
ACKNOWLEDGMENTS
The authors dedicate this manuscript to the memory of Dr. Minoru Toyota. The authors also thank Dr. William F. Goldman for editing the manuscript.
FUNDING
This study was supported in part by a Grant-in-Aid for Scientific Research (B) from the Japan Society for Promotion of Science (H. Suzuki), a Grant-in-Aid for Young Scientist (B) from the Japan Society for Promotion of Science (E. Yamamoto), a Grant-in-Aid for the Third-term Comprehensive 10-year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare, Japan (H. Suzuki), and the Suhara Memorial Foundation (H. Suzuki).
CONFLICTS OF INTEREST
The authors disclose no conflict of interests.
REFERENCES
1. Suzuki H, Tokino T, Shinomura Y, Imai K, Toyota M. DNA methylation and cancer pathways in gastrointestinal tumors. Pharmacogenomics. 2008; 9:1917–1928.
2. Yamamoto E, Suzuki H, Yamano HO, Maruyama R, Nojima M, Kamimae S, Sawada T, Ashida M, Yoshikawa K, Kimura T, Takagi R, Harada T, Suzuki R, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol. 2012; 181:1847–1861.
3. Yagi K, Takahashi H, Akagi K, Matsusaka K, Seto Y, Aburatani H, Nakajima A, Kaneda A. Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am J Pathol. 2012; 180:616–625.
4. Suzuki H, Yamamoto E, Maruyama R, Niinuma T, Kai M. Biological significance of the CpG island methylator phenotype. Biochem Biophys Res Commun. 2014; 455:35–42.
5. Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB. Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene. 2006; 25:4986–4997.
6. Cho CY, Wang JH, Chang HC, Chang CK, Hung WC. Epigenetic inactivation of the metastasis suppressor RECK enhances invasion of human colon cancer cells. J Cell Physiol. 2007; 213:65–69.
7. Harder J, Engelstaedter V, Usadel H, Lassmann S, Werner M, Baier P, Otto F, Varbanova M, Schaeffner E, Olschewski M, Blum HE, Opitz OG. CpG-island methylation of the ER promoter in colorectal cancer: analysis of micrometastases in lymph nodes from UICC stage I and II patients. Br J Cancer. 2009; 100:360–365.
8. Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008; 105:13556–13561.
9. Kudo S. Endoscopic mucosal resection of flat and depressed types of early colorectal cancer. Endoscopy. 1993; 25:455–461.
10. Yoshikane H, Hidano H, Sakakibara A, Mori S, Takahashi Y, Niwa Y, Goto H. Endoscopic resection of laterally spreading tumours of the large intestine using a distal attachment. Endoscopy. 1999; 31:426–430.
11. Tanimoto T, Tanaka S, Haruma K, Yoshihara M, Sumii K, Kajiyama G, Shimamoto F. Growth patterns in various macroscopic types of noninvasive intramucosal colorectal carcinoma with special reference to apoptosis and cell proliferation. Dis Colon Rectum. 1998; 41:1376–1384.
12. Hurlstone DP, Cross SS, Adam I, Shorthouse AJ, Brown S, Sanders DS, Lobo AJ. Efficacy of high magnification chromoscopic colonoscopy for the diagnosis of neoplasia in flat and depressed lesions of the colorectum: a prospective analysis. Gut. 2004; 53:284–290.
13. Noro A, Sugai T, Habano W, Nakamura S. Analysis of Ki-ras and p53 gene mutations in laterally spreading tumors of the colorectum. Pathol Int. 2003; 53:828–836.
14. Hiraoka S, Kato J, Tatsukawa M, Harada K, Fujita H, Morikawa T, Shiraha H, Shiratori Y. Laterally spreading type of colorectal adenoma exhibits a unique methylation phenotype and K-ras mutations. Gastroenterology. 2006; 131:379–389.
15. Kaji E, Kato J, Suzuki H, Akita M, Horii J, Saito S, Higashi R, Ishikawa S, Kuriyama M, Hiraoka S, Uraoka T, Yamamoto K. Analysis of K-ras, BRAF, and PIK3CA mutations in laterally-spreading tumors of the colorectum. J Gastroenterol Hepatol. 2011; 26:599–607.
16. Sakai E, Ohata K, Chiba H, Matsuhashi N, Doi N, Fukushima J, Endo H, Takahashi H, Tsuji S, Yagi K, Matsusaka K, Aburatani H, Nakajima A, et al. Methylation epigenotypes and genetic features in colorectal laterally spreading tumors. Int J Cancer. 2014;
17. Hashimoto K, Shimizu Y, Suehiro Y, Okayama N, Hashimoto S, Okada T, Hiura M, Ueno K, Hazama S, Higaki S, Hamanaka Y, Oka M, Sakaida I, et al. Hypermethylation status of APC inversely correlates with the presence of submucosal invasion in laterally spreading colorectal tumors. Mol Carcinog. 2008; 47:1–8.
18. Wu Z, Martinez-Fong D, Tredaniel J, Forgez P. Neurotensin and its high affinity receptor 1 as a potential pharmacological target in cancer therapy. Front Endocrinol (Lausanne). 2012; 3:184.
19. Staub E, Groene J, Heinze M, Mennerich D, Roepcke S, Klaman I, Hinzmann B, Castanos-Velez E, Pilarsky C, Mann B, Brummendorf T, Weber B, Buhr HJ, et al. An expression module of WIPF1-coexpressed genes identifies patients with favorable prognosis in three tumor types. J Mol Med (Berl). 2009; 87:633–644.
20. Carraway R, Leeman SE. The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J Biol Chem. 1973; 248:6854–6861.
21. Evers BM, Izukura M, Townsend CM Jr, Uchida T, Thompson JC. Differential effects of gut hormones on pancreatic and intestinal growth during administration of an elemental diet. Ann Surg. 1990; 211:630–636. discussion 636–638.
22. Evers BM, Izukura M, Townsend CM Jr, Uchida T, Thompson JC. Neurotensin prevents intestinal mucosal hypoplasia in rats fed an elemental diet. Dig Dis Sci. 1992; 37:426–431.
23. Bueno L, Fioramonti J, Fargeas MJ, Primi MP. Neurotensin: a central neuromodulator of gastrointestinal motility in the dog. Am J Physiol. 1985; 248:G15–19.
24. Wiklund B, Liljeqvist L, Rokaeus A. Neurotensin increases net fluid secretion and transit rate in the small intestine of man. Regul Pept. 1984; 8:33–39.
25. Thor K, Rosell S. Neurotensin increases colonic motility. Gastroenterology. 1986; 90:27–31.
26. Reasbeck PG, Barbezat GO, Shulkes A, Leader J. Secretion of neurotensin and its effects on the jejunum in the dog. Gastroenterology. 1984; 86:1552–1556.
27. Riegler M, Castagliuolo I, Wang C, Wlk M, Sogukoglu T, Wenzl E, Matthews JB, Pothoulakis C. Neurotensin stimulates Cl(-) secretion in human colonic mucosa In vitro: role of adenosine. Gastroenterology. 2000; 119:348–357.
28. Wood JG, Hoang HD, Bussjaeger LJ, Solomon TE. Neurotensin stimulates growth of small intestine in rats. Am J Physiol. 1988; 255:G813–817.
29. Izukura M, Evers BM, Parekh D, Yoshinaga K, Uchida T, Townsend CM Jr, Thompson JC. Neurotensin augments intestinal regeneration after small bowel resection in rats. Ann Surg. 1992; 215:520–526. discussion 526–527.
30. Vincent JP, Mazella J, Kitabgi P. Neurotensin and neurotensin receptors. Trends Pharmacol Sci. 1999; 20:302–309.
31. Evers BM, Ishizuka J, Chung DH, Townsend CM Jr, Thompson JC. Neurotensin expression and release in human colon cancers. Ann Surg. 1992; 216:423–430. discussion 430–421.
32. Maoret JJ, Pospai D, Rouyer-Fessard C, Couvineau A, Laboisse C, Voisin T, Laburthe M. Neurotensin receptor and its mRNA are expressed in many human colon cancer cell lines but not in normal colonic epithelium: binding studies and RT-PCR experiments. Biochem Biophys Res Commun. 1994; 203:465–471.
33. Souaze F, Viardot-Foucault V, Roullet N, Toy-Miou-Leong M, Gompel A, Bruyneel E, Comperat E, Faux MC, Mareel M, Rostene W, Flejou JF, Gespach C, Forgez P. Neurotensin receptor 1 gene activation by the Tcf/beta-catenin pathway is an early event in human colonic adenomas. Carcinogenesis. 2006; 27:708–716.
34. Gui X, Guzman G, Dobner PR, Kadkol SS. Increased neurotensin receptor-1 expression during progression of colonic adenocarcinoma. Peptides. 2008; 29:1609–1615.
35. Bossard C, Souaze F, Jarry A, Bezieau S, Mosnier JF, Forgez P, Laboisse CL. Over-expression of neurotensin high-affinity receptor 1 (NTS1) in relation with its ligand neurotensin (NT) and nuclear beta-catenin in inflammatory bowel disease-related oncogenesis. Peptides. 2007; 28:2030–2035.
36. Gui X, Liu S, Yan Y, Gao Z. Neurotensin receptor 1 overexpression in inflammatory bowel diseases and colitis-associated neoplasia. World J Gastroenterol. 2013; 19:4504–4510.
37. Muller KM, Tveteraas IH, Aasrum M, Odegard J, Dawood M, Dajani O, Christoffersen T, Sandnes DL. Role of protein kinase C and epidermal growth factor receptor signalling in growth stimulation by neurotensin in colon carcinoma cells. BMC Cancer. 2011; 11:421.
38. Massa F, Tormo A, Beraud-Dufour S, Coppola T, Mazella J. Neurotensin-induced Erk1/2 phosphorylation and growth of human colonic cancer cells are independent from growth factors receptors activation. Biochem Biophys Res Commun. 2011; 414:118–122.
39. Maoret JJ, Anini Y, Rouyer-Fessard C, Gully D, Laburthe M. Neurotensin and a non-peptide neurotensin receptor antagonist control human colon cancer cell growth in cell culture and in cells xenografted into nude mice. Int J Cancer. 1999; 80:448–454.
40. Bugni JM, Rabadi LA, Jubbal K, Karagiannides I, Lawson G, Pothoulakis C. The neurotensin receptor-1 promotes tumor development in a sporadic but not an inflammation-associated mouse model of colon cancer. Int J Cancer. 2012; 130:1798–1805.
41. Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007; 39:157–158.
42. Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I, Cedar H. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007; 39:232–236.
43. Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007; 39:237–242.
44. Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008; 4:e1000242.
45. Hagihara A, Miyamoto K, Furuta J, Hiraoka N, Wakazono K, Seki S, Fukushima S, Tsao MS, Sugimura T, Ushijima T. Identification of 27 5′ CpG islands aberrantly methylated and 13 genes silenced in human pancreatic cancers. Oncogene. 2004; 23:8705–8710.
46. Guo S, Yan F, Xu J, Bao Y, Zhu J, Wang X, Wu J, Li Y, Pu W, Liu Y, Jiang Z, Ma Y, Chen X, et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin Epigenetics. 2015; 7:3.
47. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001; 1:55–67.
48. Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008; 68:4123–4132.
49. Kudo S, Tamura S, Nakajima T, Yamano H, Kusaka H, Watanabe H. Diagnosis of colorectal tumorous lesions by magnifying endoscopy. Gastrointest Endosc. 1996; 44:8–14.
50. Update on the paris classification of superficial neoplastic lesions in the digestive tract: Endoscopy. 2005; 37:570–578.
51. Kudo S, Lambert R, Allen JI, Fujii H, Fujii T, Kashida H, Matsuda T, Mori M, Saito H, Shimoda T, Tanaka S, Watanabe H, Sung JJ, et al. Nonpolypoid neoplastic lesions of the colorectal mucosa. Gastrointest Endosc. 2008; 68:S3–47.
52. Maruyama R, Choudhury S, Kowalczyk A, Bessarabova M, Beresford-Smith B, Conway T, Kaspi A, Wu Z, Nikolskaya T, Merino VF, Lo PK, Liu XS, Nikolsky Y, et al. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet. 2011; 7:e1001369.
53. Alifano M, Souaze F, Dupouy S, Camilleri-Broet S, Younes M, Ahmed-Zaid SM, Takahashi T, Cancellieri A, Damiani S, Boaron M, Broet P, Miller LD, Gespach C, et al. Neurotensin receptor 1 determines the outcome of non-small cell lung cancer. Clin Cancer Res. 2010; 16:4401–4410.