INTRODUCTION
Inactivation of the retinoblastoma (RB1) tumor suppressor occurs with such a high frequency in cancer — either directly, through mutations, or indirectly, through the altered expression of RB1 regulators — to be proposed as a fundamental event for tumor development [1]. Therefore, many efforts have been dedicated to investigate RB1 mechanisms of action in order to shed light on key events in cancer development.
Early studies identified RB1 and other cell cycle regulatory proteins as intracellular targets of transforming viruses [2–5]. So, initial studies on RB1 function focused on its role as a cell cycle regulator and correlated its oncosuppressive activity mainly with its capability to negatively regulate cell cycle through the interaction with members of the E2F family of transcription factors [6]. Indeed, the canonical model for the tumor suppressive action of RB1, which emerged from these early studies, is based on its ability to inhibit the G1-S transition through the repression of E2F target genes involved in DNA synthesis and cell cycle progression. This function of RB1 is regulated through changes in its phosphorylation status, which are mediated by cyclin-cyclin-dependent kinase (CDK) complexes, CDK inhibitors and phosphatase activity (for a review see [7]). When RB1 is in its active hypophosphorylated state, it represses E2F-mediated transcription by binding and blocking the E2F transactivation domain and forming complexes with E2Fs, together with their heterodimerization partners DPs, at cell cycle gene promoters. Conversely, RB1 phosphorylation, which is initiated by cyclin D-CDK4/6 in response to mitogenic signals, inactivates the RB1 repressive function by dissociating the RB1-E2F-DP complexes. The transcriptional repressive activity of RB1 also relies on its ability to recruit chromatin remodelling enzymes to E2F-regulated promoters. However, the action of these enzymes seems to be more related to chromatin changes associated with a stable gene silencing during cell cycle exit rather than to the negative control of the G1-S transition in cycling cells [8].
RB1 belongs to a family of three proteins, including also retinoblastoma-like 1 (RBL1/p107) and retinoblastoma-like 2 (RBL2/p130) [7]. RBL2/p130 altered expression and delocalization was found in various cancers, in which it functions as a valuable prognostic marker [9–21]. Both RBL1/p107 and RBL2/p130 are involved in the negative regulation of cell cycle by interacting with E2F family members although in different combinations with respect to RB1 [7]. Recently, it has been shown that RBL2/p130, together with E2F4-DP and the multi-vulval class B (MuvB) core component, form the DREAM complex (DP, RB-like, E2F and MuvB), which mediates cell cycle gene repression during quiescence and coordinates periodic gene expression during the G1-S and G2-M transitions by releasing RBL2/p130 and recruiting different transcription factors [22].
However, although classically the tumor suppressive function of the RB proteins has been mainly attributed to their ability to arrest cell cycle by repressing E2F target genes, these proteins can also control cell cycle progression through E2F-independent mechanisms. Indeed, both RB1 and RB-like proteins are able to negatively regulate cell cycle by inhibiting CDK activity through either indirect or direct mechanisms, respectively [8, 23]. Moreover, beyond their function as G1 checkpoint regulators, RB proteins are involved in many other cellular processes, such as preservation of chromosomal stability, induction and maintenance of senescence, regulation of cellular differentiation, angiogenesis, and apoptosis, which could all contribute to the RB oncosuppressive activity [24, 25]. At the molecular level, RB1 is now viewed as a platform for multiple protein interactions through which it regulates different cellular pathways [8].
Given the central role of RB1 in regulating cellular processes that are crucial for both defense against tumor progression and response to cancer treatments, a careful analysis of RB1 status could be critical to guide therapeutic decisions. Furthermore, unleashing RB1 oncosuppressive potential is an appealing strategy for cancer therapy. Considering that the main goal of most anticancer therapeutic interventions is to induce apoptotic cell death or inhibit proliferation, a fundamental requirement to exploit RB1 for therapeutic purposes is to fully understand its role in these processes. However, whereas the role of RB proteins as negative regulators of cell proliferation is well-understood, their role in apoptosis is still very controversial. Here, we provide an overview of the main findings on the function of RB1 and the other family members in the apoptotic process and in the coordinated control of cell death and proliferation in both normal cells and preclinical cancer models. Moreover we describe clinical studies exploring the possible utility of RB1 as a predictive marker of response to both cytotoxic and cytostatic treatments and studies investigating the possible use of RB proteins as ultimate therapeutic targets.
RB family and apoptosis regulation
The role of RB1 in apoptosis control is still very controversial and limited data are available for as concerns the role of the other RB family members in this process. The first observations indicated an antiapoptotic role of RB1. However, emerging data now show a proapoptotic role of RB1 in different cellular contexts and recent advances in the understanding of RB1 mechanisms of action in apoptosis suggest multifunctional roles for this protein, which can be modulated by several post-translational modifications [8].
Early findings revealing the antiapoptotic role of RB1
The ability to evade apoptosis is a hallmark of cancer [26]. Consistently, alterations that inhibit apoptosis can favor tumor progression and cause resistance to treatments [27]. So, it was quite surprising when findings from mouse studies first revealed an antiapoptotic role for RB1, considering its well-established oncosuppressive function. Indeed, studies with Rb1-null mice showed that RB1 loss induced not only deregulated proliferation, as expected, but also massive apoptosis in the nervous system, lens and skeletal muscles (see [28] for a thorough description of these studies).
Genetic crosses demonstrated that the RB1-binding partners E2Fs have a crucial role in triggering apoptosis in Rb1-null mice [28]. Indeed, as a consequence of RB1 loss, the release and de-repression of the E2F1 transcription factor, which among the E2F family members has a unique role in apoptosis induction [29], can trigger cell death by the activation of E2F1 target genes encoding proapoptotic proteins, such as p73, caspases, apoptotic peptidase activating factor-1 (APAF-1), and BCL-2 homology 3 (BH3)-only family members [30].
However, later it became clear that the apoptotic phenotypes of Rb1-null mice are not entirely caused by E2F deregulation but are also largely due to the defective development of extra-embryonic tissues rather than only to the lack of Rb1 cell autonomous function [31–33]. Moreover, RB1 is able to bind and inhibit proapoptotic factors other than E2F1 [28].
The analysis of tissue-specific Rb1 mutant mouse models showed that RB1 loss in some tissues induced unscheduled proliferation without having effects on apoptosis, whereas in other tissues (lens and myoblasts) induced apoptosis, specifically in differentiating cells [34]. It has been suggested that RB1 loss can induce either apoptosis or uncontrolled proliferation depending on different cellular contexts: in cells committed to a specific differentiation program RB1 deficiency triggers apoptosis, whereas in cycling cells RB1 loss leads to uncontrolled proliferation [35]. A possible explanation on how cells lacking RB1 can proliferate rather than undergo apoptosis is that mitogenic stimulation activates prosurvival factors that counteract the proapoptotic gene induction resulting from RB1 loss [28].
Role of RB1 in the coordinated control of proliferation and apoptosis
RB1 dual role as inhibitor of both cell division and apoptosis raises the question of how normal cells can inactivate RB1 to enable cell division without inducing apoptosis. A possible mechanistic explanation is that the RB1 reversible inhibition occurring during cell cycle through phosphorylation is functionally different from the RB1 complete loss that induces apoptosis in Rb1-null mice [36]. Support for this hypothesis comes from the observation that Cdkn2a-null animals, in which RB1 is expected to be in a hyperphosphorylated state, do not show the massive apoptosis observed in Rb1-null mice [36]. So, the “inactive” hyperphosphorylated form of RB1 seems to maintain the antiapoptotic activity. Indeed, although the commonly accepted model for RB1 function would predict that phosphorylated RB1 releases its E2F partners during G1-S cell cycle transition, it has been observed that RB1-E2F1 complexes persist beyond the S phase, regardless of RB1 hyperphosphorylation [37]. Moreover, in proliferating cells RB1 and E2F1 occupy apoptotic gene promoters, whereas are dissociated from cell cycle genes [38]. The ability of RB1-E2F1 complexes to persist when RB1 is phosphorylated during cell cycle progression seems to rely on an E2F1-specific binding domain in the RB1 C-terminal region [8]. Therefore, a simplified model that emerges from these observations is that phosphorylation of RB1 by CDKs during G1-S transition causes the release of most E2Fs to induce transcription of cell cycle genes, but at least a fraction of RB1-E2F1 complexes remains stable thanks to the unique interaction of these proteins and persist at the promoters of apoptotic genes, thus repressing their expression [8] (Fig. 1A).
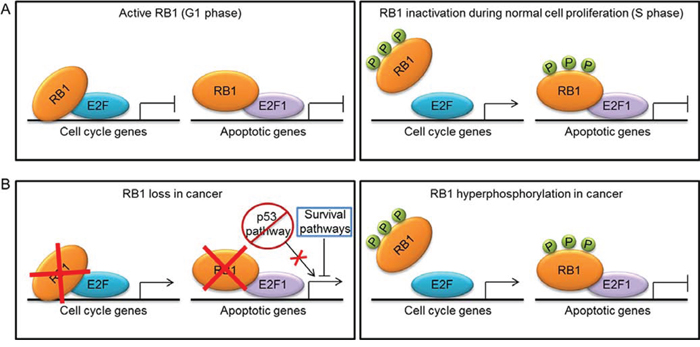
Figure 1: Mechanisms of RB1 inactivation during normal cell proliferation and tumorigenesis and their effect on transcription of cell cycle and apoptotic genes. A. In G1 phase, the active hypophosphorylated form of RB1 binds to E2F transcription factors to repress the expression of cell cycle and apoptotic genes. In S phase, RB1 is inactivated by phosphorylation (P) and releases most E2Fs to induce transcription of cell cycle genes. A fraction of RB1-E2F1 complexes persist at the promoters of apoptotic genes, thus repressing their expression. B. In cancer, RB1 can be inactivated by either mutations or hyperphosphorylation. RB1 loss leads to de-repression of both cell cycle and apoptotic genes, whereas hyperphosphorylation causes de-repression only of cell cycle genes. Thus, in cells lacking RB1, tumorigenesis can occur only if survival pathways protect cells from RB1-loss induced apoptosis by limiting E2F1 proapoptotic activity or if a second alteration, such as the abrogation of the p53 proapoptotic pathway, occurs.
RB1 antiapoptotic role and tumorigenesis
RB1 role in inhibiting apoptosis has important implications for tumorigenesis. RB1 ability to prevent apoptosis might seem inconsistent with its oncosuppressive role, since apoptosis inhibition is more likely to favor tumor growth. However, the induction of apoptosis, which occurs as a consequence of RB1 loss, can represent a safeguard mechanism to hinder the expansion of potentially malignant cells [39]. Indeed, RB1 inhibition confers a proliferative advantage to cells, which could favor tumor development, but the massive apoptosis resulting from RB1 loss could overcome the increased proliferative potential and hinder cancer growth. This can explain why most sporadic cancers have RB1 inactivated because of defects in the pathways that regulate its phosphorylation rather than by mutations [28]. In this way, tumors gain a proliferative advantage and, at the same time, can be protected from apoptosis by the hyperphosphorylated form of RB1, which could preserve its antiapoptotic activity through the same mechanism described above (Fig. 1B). Another proposed mechanism whereby phosphorylated RB1 can protect cells from apoptosis is by directly binding and inhibiting the proapoptotic factor ANP32A (acidic nuclear phosphoprotein 32A) [40]. Therefore, RB1 phosphorylation can provide cancer cells with both proliferative and survival advantages.
Conversely, following RB1 loss, cancer can develop only if cells are intrinsically resistant to RB1-deficiency-induced apoptosis, or if cell death is counteracted by parallel activation of survival pathways, or, finally, if a second alteration, such as the abrogation of the p53 proapoptotic pathway (Fig. 1B), occurs during tumor development to allow cell expansion and cancer growth [28]. A recently described mechanism of apoptosis inhibition whereby RB1-deficient cells can undergo malignant transformation involves the de-repression of the S-phase kinase-associated protein 2 (SKP2), which suppresses apoptosis by limiting E2F1 activity [41]. A further mechanism of resistance to apoptosis that could promote tumorigenesis following RB1 loss could rely on the increased levels of the antiapoptotic protein survivin. Indeed, it has been demonstrated that RB1 is able to inhibit transcription of the gene encoding survivin [42–44] and homozygous deletion of Rb1 in mouse embryonic fibroblasts (MEFs) led to survivin induction [42]. Consistently, high levels of survivin were found in the RB1-null cancer retinoblastoma [45].
RB1 antiapoptotic role in response to cell death inducers
Several observations from RB1 knockdown and overexpression studies confirmed the antiapoptotic role of RB1 also in response to different apoptotic stimuli. In particular, RB1 knockdown has been shown to enhance the sensitivity to cell death induced by different anticancer agents, such as DNA-damaging and microtubule interfering agents, in cells from several cancer types, including lymphoma, breast, lung, and prostate cancer, and glioblastoma [46–50]. Similarly, RB1 ablation in mouse embryonic and adult fibroblasts increased the sensitivity to chemotherapy-induced cell death [51–53]. Analogously, restoration of the wild-type RB1 protein in RB1-deficient cells from several cancer types (osteosarcoma and different carcinomas) inhibited apoptosis upon various apoptotic stimuli, such as ionizing radiation, p53 overexpression, ceramide, and interferon (IFN)-γ [54–57]. Therefore, all these data point to a protective role of RB1 against different cell death inducers in several cell types. Some studies suggested that this protective action could be a secondary consequence of RB1 ability to arrest cell cycle in response to stress signals [52, 58, 59]. However, the ectopic expression of a mutated form of RB1, which was unable to induce growth arrest, protected RB1 deficient osteosarcoma and breast cancer cells from DNA damage-induced apoptosis [60]. Thus, RB1 can exert an antiapoptotic activity independent of growth suppression, probably mainly through the direct inhibition of apoptotic genes.
Role of RB1 dephosphorylation and caspase cleavage during apoptosis
Apoptosis is often accompanied by a shift from the hyperphosphorylated to the hypophosphorylated form of RB1 [61–67]. Consistently, phosphatase activity directed toward RB1 seems to be required for apoptosis induction in cells from different cancer types [61, 65, 67, 68] and the antiapoptotic protein BCL2 can prevent RB1 dephosphorylation and apoptosis [63, 64]. Moreover, RB1 hyperphosphorylation seems to be correlated with resistance to apoptotic treatments [69, 70]. All these studies suggest that RB1 dephosphorylation is required for apoptosis to occur, and, in particular, it has been recently reported that dephosphorylation at threonine-821 has a key role in this process [71].
Studies conducted on promyelocytic leukemia and breast cancer cell lines suggested that dephosphorylation of RB1 during apoptosis could be necessary for its cleavage by caspases and consequent degradation, which would eliminate its antiapoptotic action and allow cells to undergo death in response to apoptotic stimuli, such as DNA damage [65, 67, 72, 73]. Indeed, RB1 dephosphorylation seems to be a prerequisite for caspase cleavage, since the hyperphosphorylated form of RB1 is a poor caspase substrate [65], and different studies support the notion that caspase cleavage is a crucial step for eliminating RB1 during apoptosis in various cell types [74–78].
So, it was tempting to speculate that, unlike the promoters of cell cycle genes where RB1 activity is mostly modulated through phosphorylation/dephosphorylation processes, at the promoters of apoptotic genes RB1 degradation could play an important part [28] (Fig. 2). However, although caspase-resistant forms of RB1 hindered apoptosis induced by tumor-necrosis factor-alpha (TNF-α) in mouse fibroblasts [79, 80] and by potassium deprivation in mouse cerebellar neurons [81], the cleavage-resistant RB1 did not decrease cell death triggered by DNA damage in mouse fibroblasts [80] and did not protect human T lymphocytes from apoptosis induced by the ligation of FAS death receptor [79]. Thus, RB1 cleavage by caspases is not an absolute prerequisite for apoptosis, but it can be a crucial step for certain types of apoptotic events, depending on both the cell type and the nature of the death inducers.
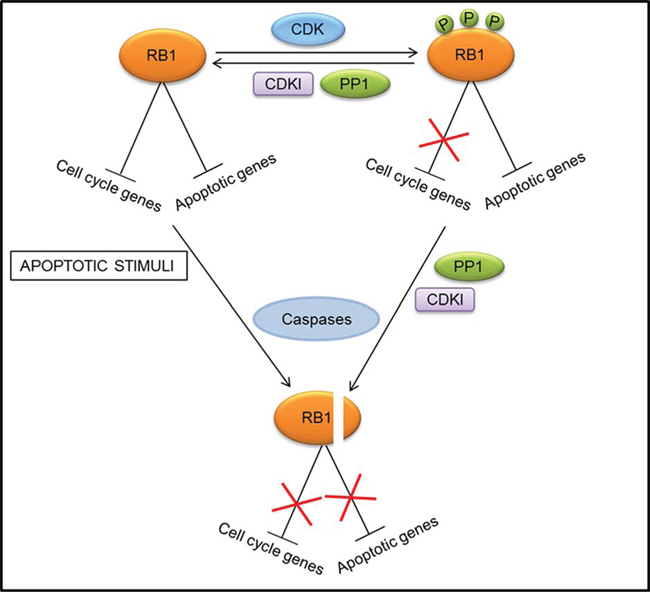
Figure 2: Different mechanisms of regulation of RB1 activity at the promoters of cell cycle and apoptotic genes. At the promoters of cell cycle genes, RB1 activity is modulated through phosphorylation (P)/dephosphorylation processes mediated by cyclin dependent kinases (CDKs), CDK inhibitors (CDKIs), and protein phosphatase 1 (PP1). Conversely, phosphorylation by CDKs does not affect the RB1 repressive action at promoters of apoptotic genes. A mechanism to prevent this inhibitory activity and induce apoptotic gene expression consists in RB1 dephosphorylation and subsequent cleavage by caspases.
Emerging data revealing a proapoptotic role for RB1
The RB1 dephosphorylation observed during apoptosis is not always linked to a subsequent RB1 degradation, which, as described above, could serve to eliminate its antiapoptotic action. Conversely, the active hypophosphorylated form of RB1 seems to have a crucial role in apoptosis induction in some circumstances. Indeed, accumulation of the active hypophosphorylated form of RB1 seemed to be required for apoptosis induced by matrix contact deprivation in prostate cancer cells [64]. Consistently, the induced expression of RB1 forms that cannot be phosphorylated and/or degraded by caspases stimulated the apoptotic response to TNF in Rat-16 cells [59]. Overall these data point to a proapoptotic role of RB1 in some contexts, although the phosphorylation resistant form of RB1 causes contrasting effects in response to different apoptotic stimuli [59].
Support for the hypothesis of a proapoptotic role of RB1 in some circumstances also comes from a study reporting that RB1 knockdown reduced the toxicity of histone deacetylase (HDAC) inhibitors in colon cancer cells [82]. Moreover, the restoration of wild-type RB1 in RB1-deficient prostate cancer cells promoted apoptosis after radiation or ceramide treatment [83, 84]. This is, however, in sharp contrast with the aforementioned studies showing apoptosis inhibition following the restoration of wild-type RB1 in several RB1-deficient cell types upon different treatments.
A molecular mechanism whereby RB1 can exert a proapoptotic function in some cellular contexts in response to apoptotic stimuli, such as DNA damage, has been recently proposed [8, 34]. In response to DNA damage, both RB1 and E2F1 undergo extensive post-translational modifications: RB1 is dephosphorylated at CDK target sites but is phosphorylated by checkpoint kinase 1/2 (CHEK1/2), acetylated, and methylated; E2F1 is phosphorylated by ataxia-telangiectasia mutated (ATM) and CHEK2, as well as acetylated and demethylated [8, 85]. Phosphorylated RB1 remains bound to E2F1 and the recruitment of the histone acetyltransferase p300/CBP-associated factor (P/CAF; also known as KAT2B) leads to the formation of a transcriptionally active complex, which promotes the expression of pro-apoptotic genes (Fig. 3). Consistently, a transcriptionally active complex containing RB1, E2F1 and P/CAF has also been reported to have a key role in the apoptotic response following hepatocarcinoma cell treatment with transforming growth factor-β1 (TGF-β1) [86].
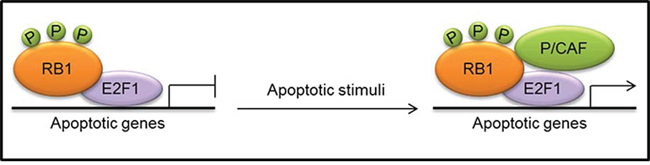
Figure 3: Formation of a transcriptionally active complex, consisting of phosphorylated (P) RB1, E2F1 and the histone acetyltransferase p300/CBP-associated factor (P/CAF), at the promoters of apoptotic genes in response to apoptotic stimuli.
Most of the findings on the role of RB1 in apoptosis derived from studies on its activity as transcriptional regulator. Conversely, in a recent study RB1 was shown able to promote apoptosis in response to apoptotic signals at mithocondria, by directly interacting with BAX and conformationally activating it to induce mitochondrial outer membrane permeabilization and cytochrome c release [30, 87]. Moreover, hyperphosphorylated RB1 is still competent to induce mitochondrial apoptosis. However, another study showed that RB1 dephosphorylation caused RB1-BAX complex dissociation and apoptosis induction [88]. These incongruous results could be explained by the different cell types used in the two studies (MEF and human breast cancer cell, respectively) or by the different methods used for manipulating phosphorylation, which might result in differences in site-specific phosphorylation of RB1 [88].
In conclusion, all these observations indicate that RB1 is a multifunctional protein, whose role in apoptosis depends on both the cell type and the nature of the death inducers and whose function can be modulated by different combinations of phosphorylated sites and other post-translational modifications.
Role of the other RB family members in apoptosis
Although data on the function of RBL1/p107 and RBL2/p130 in apoptosis are more limited than those available for RB1, the picture that emerges is similarly complex.
Studies with mutant mice suggested an antiapoptotic role for RBL1/p107 and RBL2/p130. Indeed, although Rbl1-null mice did not show increased cell death during embryogenesis, mouse embryos lacking both Rb1 and Rbl1 showed accelerated apoptosis in liver and central nervous system with respect to embryos lacking only Rb1 [89]. Therefore, RBL1/p107 seems to cooperate with RB1 in protecting some cell types from apoptosis. Balb/cJ mouse embryos lacking Rbl2 showed extensive apoptosis in the neural tube, brain and dermomyotome, thus suggesting an antiapoptotic role for RBL2/p130 at least in this mouse strain [90]. Moreover, although the individual loss of each Rb family member did not significantly affect proliferation and survival of mouse pancreatic β cells in vivo, the combined loss of Rb1 and Rbl2 resulted in increased proliferation and cell death [91].
Consistent with a possible antiapoptotic role of RBL2/p130, it has been observed that its overexpression inhibits apoptosis in response to apoptotic stimuli in neurons [92] and ovarian cancer cells [93].
At the molecular level, the RBL2/p130-E2F4 repressor complex has been shown to inhibit directly the transcription of apoptotic genes in postmitotic cardiomyocytes [94] and neurons [92]. Apoptotic stimuli can induce RBL2/p130 hyperphosphorylation, dissociation of the RBL2/p130-E2F4 complex and de-repression of apoptotic genes in neurons [92]. So, unlike the RB1 repressive activity at the apoptotic genes, which seems to be maintained regardless of RB1 phosphorylation (Fig. 1), the RBL2/p130 inhibitory action at these genes can be inactivated through RBL2/p130 phosphorylation. Consistently, the RB family members are inactivated through different mechanisms during camptothecin-induced apoptosis in B lymphocytes: RB1 is degraded, in keeping with the above described role of RB1 caspase cleavage during apoptosis in various cell types, whereas RBL1/p107 and RBL2/p130 are phosphorylated, leading to the release of E2F proteins [95]. Consistent with this different regulation of RB protein activity during apoptosis, the C-terminal caspase cleavage site of RB1 is not conserved in RBL1/p107 and RBL2/p130 [74].
Although these observations indicate an antiapoptotic role for RBL1/p107 and RBL2/p130, the latter can also exert a proapoptotic activity in different contexts. In particular, in marked contrast to what observed in mouse pancreatic β cells lacking both Rb1 and Rbl2 [91], the combined ablation of these two genes in the mouse lung epithelium resulted in reduced apoptosis with respect to that observed in lung epithelium lacking only Rb1, thus suggesting a proapoptotic activity of RBL2/p130 [96]. Consistently, the expression of RBL2/p130 was correlated with high apoptotic indexes in retinoblastoma samples [97]. Moreover, RBL2/p130 overexpression induced apoptosis in Saos-2 osteosarcoma cells [97] and marrow stromal cells committed toward a neuron-like phenotype [98] and caused an increase in γ-radiation-induced apoptosis in hamster glioblastoma cells [99].
Interestingly, RBL2/p130 can be part of different protein complexes with opposite functions at apoptotic gene promoters. Indeed, it has been suggested that the difference in apoptotic response to chemotherapeutic treatment observed in drug-resistant osteosarcoma cells and their parental cell line could be due to the presence of distinct E2Fs–RBL2/p130 complexes on the p73 promoter: in the parental cell line, p73 transcription is activated by an E2F1–RBL2/p130–p300 complex, whereas, in the drug-resistant cells, it is repressed by an E2F4–RBL2/p130–HDAC1 complex [100]. So, the involvement in functionally different complexes provides a possible molecular explanation on how RBL2/p130 can exert opposite functions in apoptosis regulation in different circumstances. However, we are still far from understanding the mechanisms whereby RBL2/p130 can contribute to apoptosis control and further research in this field is ongoing.
RB1 as a predictive marker of response to therapy
The key dual role of RB1 in arresting cell cycle upon anti-proliferative signals and in apoptotic response to various anticancer treatments, observed in the preclinical studies described above, suggests that RB1 could be a crucial determinant of clinical response to therapies. Therefore, a careful evaluation of its functional status and context-dependent role in response to specific treatments in different tumors might be fundamental to guide therapeutic decisions. Indeed, several studies on patients with different cancer types suggest that RB1 status affects tumor sensitivity to treatments and clinical outcome (Table 1). Therefore, RB1 might represent a predictive marker of response to therapy for various tumor types.
Table 1: Studies on the role of RB1 as a predictive marker of response to therapies
Cancer type |
Treatments |
RB1 status and assessment method |
No of patients |
Association between RB1 status and clinical response |
References |
ALL |
Glucocorticoids |
Hypophosphorylation (HRE and WB) |
32 |
RB1 hypophosphorylation correlates with good responsiveness to glucocorticoid therapy. |
[115] |
Bladder cancer |
Radiation |
Loss of expression (IHC) |
98 |
Loss of RB1 expression is associated with improved response to radiation. |
[110] |
Bladder cancer |
Radiation |
Loss of expression (IHC) |
106 |
Loss of RB1 expression is associated with improved response to radiation and relapse-free survival. |
[111] |
Bladder cancer |
Transurethral resection and intravesical BCG +IFN-α |
Low expression (IHC) |
93 |
RB1 underexpression is associated with nonresponse to BCG +IFN-α treatment and tumor recurrence. |
[113] |
Bladder cancer |
Transurethral resection and intravesical BCG |
Loss of expression or overexpression (IHC) |
27 |
RB1 altered expression predicts recurrence and progression following BCG treatment. |
[114] |
Breast cancer |
Cyclophosphamide, methotrexate, 5-FU |
Loss of expression (IHC) |
518 |
Loss of RB1expression predicts a good clinical outcome for patients treated with adjuvant chemotherapy. |
[101] |
Breast cancer |
Paclitaxel, 5-FU, doxorubicin, cyclophosphamide |
LOH (analysis of polymorphic markers at RB1 locus) |
133 |
RB1-LOH signature is associated with a good response to neoadjuvant chemotherapy. |
[102] |
Breast cancer |
Cyclophosphamide, methotrexate, 5-FU |
Loss of expression (IHC) |
518 |
RB1 loss is higher in TNBCs than in other subtypes. In patients with TNBCs treated with adjuvant chemotherapy, RB1 loss is associated with a good prognosis. |
[103] |
Breast cancer |
Three different neoadjuvant chemotherapy regimens |
Pathway disruption (analysis of an RB1-loss gene expression signature) |
98 |
RB1 pathway disruption is associated with improved response to multiple chemotherapeutic regimens in both ER+ and ER-breast cancers. |
[104] |
Breast cancer |
Tamoxifen |
Pathway disruption (analysis of an RB1-loss gene expression signature) |
60 |
RB1 pathway deregulation is associated with early recurrence following tamoxifen monotherapy in ER+ tumors. |
[47] |
Breast cancer |
Tamoxifen |
Loss of function (discrepancy between RB1 phosphorylation and cell proliferation) |
500 |
Loss of RB1 function is associated with tamoxifen treatment resistance in ER+ tumors. |
[105] |
Ovarian carcinoma |
Platinum and paxlitaxel chemotherapy combinations after radical surgery |
High expression (IHC) |
300 |
High expression of RB1 is associated with a poor prognosis in patients who underwent radical surgery and postoperative chemotherapy. |
[116] |
Prostate cancer |
Combined androgen blockade |
Low mRNA levels (RT-PCR) |
81 tumors |
A higher frequency of RB1 mRNA downregulation was observed in cancers from treated patients with respect to those from untreated patients. |
[107] |
Prostate cancer |
Hormone deprivation |
Loss (RT-qPCR, IHC, gene expression signature, locus copy number) |
44 |
RB1 loss is associated with the transition to the incurable castration-resistant status and poor clinical outcome. |
[108] |
ALL: acute lymphoblastic leukemia; BCG: bacillus Calmette-Guerin; ER: estrogen receptor; 5-FU: 5-fluorouracil; HRE: high resolution electrophoresis; IFN-α: interferon-α; IHC: immunohistochemistry; LOH: loss of heterozygosity; RT-qPCR: reverse transcription-quntitative PCR; TNBC: triple-negative breast cancer; WB: western blotting
In particular, consistent with the above reported observation that RB1 knockdown enhances the sensitivity to cell death induced by DNA-damaging agents in breast cancer cells [47], the disruption of RB1 function was found to be associated with improved response to adjuvant/neoadjuvant chemotherapy and good clinical outcome for patients with breast cancer [101–104]. Conversely, RB1 pathway deregulation in breast cancers proved to be associated with resistance to hormone therapy with the antiestrogen tamoxifen and tumor recurrence [47, 105]. These observations are consistent with preclinical data showing that RB1 knockdown or functional inactivation in breast cancer cells prevents antiestrogen-induced cell cycle arrest [47, 106], thus resulting in continued proliferation and tumor xenograft growth [47]. Overall, these studies indicate that the evaluation of RB1 status is crucial for directing breast cancer therapy, suggesting, in particular, that patients with RB1-negative breast cancers might benefit from chemotherapeutic treatments, whereas disruption of RB1 function is associated with failure of hormone therapy.
Similar to breast cancer, also in prostate cancer RB1 deficiency seems to be associated with a poor response to hormone therapy (including surgical or chemical castration to suppress androgen production and/or administration of antiandrogens). Indeed, a higher frequency of RB1 mRNA downregulation was observed in recurrent prostate cancers from patients who had undergone combined androgen blockade with respect to cancers from untreated patients, suggesting that RB1 inactivation might be associated with hormone treatment resistance [107]. Also, RB1 loss in prostate cancer samples was found to be associated with the transition to the incurable castration-resistant status and poor clinical outcome [108]. These results are consistent with preclinical data showing that RB1 knockdown or functional inactivation in prostate cancer cells inhibits the androgen deprivation-induced proliferative stall [49, 109] and is sufficient to induce castration-resistant tumor xenograft growth [108]. Conversely, as also reported above, RB1 knockdown enhanced the sensitivity of prostate cancer cell lines to cell death triggered by antimicrotubule and DNA-damaging agents [49]. Therefore, RB1 status seems to have a key role in cellular response to therapeutic interventions in prostate cancer cells and could represent a marker for directing therapy also in this tumor type.
Consistent with the putative antiapoptotic role of RB1 in bladder cancer cells, as observed following ceramide treatment in the aforementioned study by McConkey [56], and in keeping with the above described protective role of RB1 against DNA-damaging agents in several cancer types, loss of RB1 expression was found to be associated with improved response to radiation and relapse-free survival in patients with bladder cancer [110, 111]. Therefore, the analysis of RB1 status might be useful to select patients with bladder cancer who would benefit from a treatment schedule including radiotherapy, which has been shown to induce ceramide [112]. Conversely, RB1 altered expression seems to be predictive of nonresponse to intravesical bacille Calmette-Guerin (BCG) +/− IFN-α treatment and tumor recurrence in bladder cancer patients [113, 114].
Interestingly, RB1 hypophosphorylation occurring upon treatment with glucocorticoids, the main pharmacological agents used against childhood acute lymphoblastic leukemia (ALL), correlates with a good responsiveness [115], suggesting that RB1 might represent a possible surrogate endpoint of therapeutic response for childhood ALL.
Finally, high RB1 expression was correlated with a poor prognosis in ovarian carcinoma patients who had undergone radical surgery and postoperative chemotherapy [116] and also to be associated with vascular invasion and recurrence in hepatocellular carcinoma patients after surgical treatments [117]. These studies suggest that high RB1 expression holds the potential to be a good prognosticator of postoperative disease for these tumor types, and could be used to stratify patients for postoperative treatment in certain instances.
In addition to affect cellular sensitivity to cytotoxic and hormonal agents routinely used in the clinic, RB1 status also impacts cellular response to a number of kinase inhibitors, which, although not yet in clinical use, represent the basis for the development of promising targeted therapies. In particular, similar to what described for hormone therapy, RB1 deficiency seems to limit the effectiveness of these agents that function through cytostatic mechanisms [118].
Although the aforementioned studies point to an important role of RB1 as a predictor of therapeutic response, the evaluation of its status is not yet used to guide therapeutic decisions in clinical practice. Indeed, the major hurdle associated with the development of RB1 as a predictive marker of response to treatments is the lack of standardized methods to assess its functional status in tumor specimens [118] and several different approaches have been used to detect the different mechanisms whereby RB1 is functionally inactivated in cancer (Table 1).
Targeting RB1 for cancer therapy
Owing to RB1 involvement in many cellular processes, all contributing to its oncosuppressive activity, and considering that its pathway is altered in most human cancers, RB1 represents an appealing target for cancer therapy. Two main strategies have been conceived to directly target RB1 for cancer treatment. The first aims to exploit RB1 loss for therapeutic purposes, whereas the second is intended to reactivate RB1 tumor suppressor function [119].
Exploiting RB1 deficiency to therapeutic purposes
Different approaches exploiting RB1 loss to kill cancer cells have been designed (Fig. 4A). For instance, oncolytic viruses carrying deletions in the E1A viral protein, which render viral replication dependent on RB1 inactivation, were developed to selectively kill RB1-defective tumor cells [120] (Fig. 4A). Moreover, other oncolytic viruses were designed to exploit the deregulated E2F transcriptional activity resulting from RB1 loss for their replication and tumor cell killing [120]. Similarly, another approach took advantage of deregulated E2F transcriptional activity to activate a prodrug selectively in cancer cells with inactive RB1 [121]. Furthermore, chemical/genetics screen have been performed to identify compounds or “synthetic lethal” genes to specifically kill RB1-defective cells [122, 123].
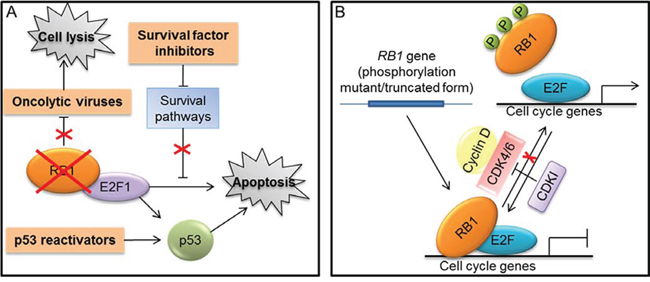
Figure 4: Strategies targeting RB1 for cancer treatment. A. Approaches that exploit RB1 loss to therapeutic purposes (indicated in orange boxes): use of oncolytic viruses that depend on RB1 inactivation for their replication and tumor cell killing; methods that exploit the loss of RB1 and the consequent E2F1 activation to kill tumor cells through the use of agents, such as inhibitors of survival factors and p53 reactivators, that enhance the E2F-1 mediated apoptosis. B. Approaches aiming to reactivate RB1 tumor suppressor function: gene-transfer strategies to express truncated or mutated forms of RB1 that remain in the hypophosphorylated status; RB1 reactivation through the use of cyclin dependent kinases inhibitors (CDKIs) to maintain an efficient transcriptional repression and induce cell cycle arrest.
Another strategy to selectively kill RB1-deficient cells is based on agents able to release the latent E2F1 proapoptotic activity in these cells [118, 119]. Indeed, as described above (Fig. 1B), in cancer cells lacking RB1, the E2F1-mediated apoptosis resulting from RB1 deficiency is suppressed through resistance mechanisms [28]. Different cellular pathways that protect cells from RB1-loss-induced apoptosis by limiting E2F1 proapoptotic activity have been identified [41, 118, 119]. Therefore, inhibitors of these survival pathways could be synthetically lethal with RB1 deficiency and could represent the basis to develop a therapeutic approach targeting RB1-defective cells [118, 119] (Fig. 4A).
The E2F1 proapoptotic activity is interconnected with that of p53 (see [124] for a thorough description of this molecular crosstalk) and, as illustrated above (Fig. 1B), the abrogation of p53 proapoptotic pathway could be required to limit apoptosis downstream of RB1 loss in order to enable tumorigenesis [28]. For instance, inhibition of p53 pathway is a crucial determinant in the genesis of the RB1-null cancer retinoblastoma [125]. Thus, it has been shown that compounds reactivating p53 could be useful in the treatment of retinoblastoma and other tumors lacking RB1, through the increase of E2F1-mediated apoptosis [118, 119, 126] (Fig. 4A).
Restoring RB1 tumor suppressor function
Compared to other tumor suppressors, such as p53, fewer studies aiming to restore RB1 function have been reported. Restoration of wild-type RB1 protein through gene-transfer approaches produced variable results on tumor cell growth and showed deleterious effects on p53-mediated apoptosis. Therefore, RB1 phosphorylation mutants and truncated variants that remain in the hypophosphorylated status have been used in preclinical models to enhance RB1 tumor suppressor function [120, 127] (Fig. 4B).
Another strategy to reactivate RB1 function is based on the use of CDK inhibitors that can prevent RB1 phosphorylation (Fig. 4B). However, whether these compounds exert their effects mainly acting on RB1 pathway remains an unanswered question, especially considering that most of the available molecules target multiple CDKs, including also transcription-related CDKs, can have also other targets, and seem to act also through mechanisms independent from cell cycle [128–132]. Nevertheless, a potent and highly selective inhibitor of CDK4 and CDK6, called Palbociclib (PD-0332991), recently proved to be dependent on the presence of RB1 for its antiproliferative effect in several cancer cell lines and xenograft models [133] and a Phase I trial of PD-0332991 utilized RB1-deficiency as an exclusion criterion [119]. In a Phase II trial, PD-0332991 treatment was associated with a favorable progression-free rate in patients with CDK4-amplified and RB-expressing liposarcomas [134]. Moreover, in patients with mantle cell lymphoma, the antiproliferative effect of PD-0332991 was significantly correlated with a marked decrease in the percentage of phospho-RB1 positive cells in lymph node biopsies [135]. The outcome of preclinical and clinical trials based on the use of PD-0332991 have been recently reviewed by Knudsen and colleagues [136].
Consistent with the above reported observations that RB1 pathway disruption is correlated with resistance to therapies that function through cytostatic mechanisms, such as hormone therapy, reactivation of the antiproliferative activity of RB1 through CDK4/6 inhibitors in combination with endocrine therapy have shown promise in breast cancer clinical trials [137]. However, considering that highly selective CDK4/6 inhibitors, such as PD-0332991, act only as cytostatic agents without inducing apoptosis [138–141], an important issue is to establish whether these compounds favor or antagonize apoptotic induction by the commonly used cytotoxic agents. It has been reported that G1 cell cycle arrest induced by pretreatment or concurrent treatment with highly selective CDK4/6 inhibitors prevented apoptotic triggering by different chemotherapeutics in RB1-positive breast cancer cells, melanoma cells and immortalized fibroblasts, thus suggesting that CDK4/6 inhibitors can antagonize chemotherapy [137, 139, 141]. Conversely, contrasting effects have been observed following combination treatments with CDK4/6 inhibitors and ionizing radiation. Indeed, pretreatment with highly selective CDK4/6 inhibitors (PD-0332991 or 2BrIC) increased radioresistance of RB1-positive melanoma cells and immortalized fibroblasts [139], whereas combination treatments with radiation administered either concurrently or sequentially with PD-0332991 resulted in increased anticancer activity in mice with RB1-positive glioblastoma intracranial xenografts [140]. Moreover, PD-0332991 sensitized myeloma cells to cell death induced by different cytotoxic agents, such as the glucocorticoid dexamethasone, the proteasome inhibitor bortezomib and the immunomodulatory agent lenalidomide, as indicated by both preclinical studies and clinical trials [138, 142–144]. Therefore, a thorough understanding of the effects of CDK4/6 inhibition in combination with different cytotoxic agents is required to define the best treatment schedule in a clinical setting. For instance, previous studies have shown that the “cell cycle-mediated resistance” to chemotherapy can be overcome by an appropriately scheduled administration [136, 145].
Data on the possible use of the other RB family members as targets in cancer therapy are very limited, although retrovirus-mediated transfer of RBL2/p130 proved to be able to inhibit lung carcinoma cell growth both in vitro and in a mouse model [11]. Moreover, we developed a 39-residue peptide, called Spa310, derived from the spacer region of RBL2/p130, which was able to inhibit CDK2 activity and lung cancer proliferation in xenotrasplanted nude mice, thus suggesting that this peptide could represent the basis for the development of new targeted therapeutic strategies [146–148].
CONCLUSIONS
Since its discovery as the first tumor suppressor gene almost 30 years ago, RB1 has represented an appealing target for cancer therapy. Indeed, decades of research have been dedicated to investigate the mechanisms of action of RB1 protein in order to exploit it therapeutically. In particular, considering that the main goal of most anticancer therapies is to inhibit proliferation and induce apoptosis, a major focus of this research has been to fully understand RB1 role in these processes both in physiological conditions and in response to treatments.
Unlike the well-established function of RB1 as a negative regulator of cell cycle, its role in apoptosis is still very controversial. Early studies with Rb1-null mice revealed an antiapoptotic role for RB1. Thus, reconciling the seemingly incongruous functions of RB1 as a negative regulator of both proliferation and apoptosis has represented a particularly tough challenge for many years. However, recent studies have identified molecular mechanisms, mainly based on the unique interaction between phosphorylated RB1 and the proapoptotic factor E2F1, able to explain RB1 function in the coordinated control of proliferation and apoptosis both in normal cells and during tumorigenesis.
RB1 has an antiapoptotic action also in response to different death stimuli and, at least in some apoptotic events, this action can be eliminated through RB1 dephosphorylation and subsequent caspase cleavage. However, to further complicate matters, emerging data have revealed that RB1 can exert a proapoptotic function in some cell types upon different treatments. Thus, RB1 is now viewed as a multifunctional protein, whose role in apoptosis depends on both the cell type and the nature of the death inducers and whose function can be modulated by several post-translational modifications. Therefore, a careful evaluation of its functional status and context-dependent role in response to specific treatments in different tumors might be fundamental to guide therapeutic decisions.
Several clinical studies suggest, indeed, that RB1 might represent a predictive marker of response to therapy for various tumor types. Overall, RB1 status seems to have opposite effects on responses to cytotoxic and cytostatic agents, respectively. In particular, RB1 deficiency seems to be associated with improved cytotoxic response to DNA damaging agents, at least for some cancer types. Conversely, the loss of RB1 function limits the effectiveness of anti-proliferative signals and hormone therapies, which function through cytostatic mechanisms.
Taken together, all these observations identify RB1 as crucial target of tailored anticancer strategies. Two main approaches have been conceived to exploit RB1 therapeutically. The first aims to take advantage of RB1 loss mainly through the research of synthetic lethal interactions and the use of oncolytic viruses, which depend on RB1 inactivation for their replication and tumor cell killing. Conversely, the second is intended to reactivate RB1 tumor suppressor function, principally through the use of CDK4/6 inhibitors that have shown promise in clinical trials.
Despite these promising results, to date a greater understanding of RB1 functions is required to successfully translate RB1-related research to the clinic. Indeed, the importance of RB1 functional status in therapeutic response has not yet been elucidated for many cancer types. Moreover, the development of more rigorous, standardized methods to assess RB1 status in tumor specimens is required before RB1 can be used as a predictive marker to guide therapeutic decisions in clinical practice. Furthermore, a deep understanding of the specific functions of different combinations of phosphorylations and other post-translational modifications is necessary to fully clarify the RB1 biological activity in different contexts and to define the best therapeutic strategy.
ACKNOWLEDGMENTS
We are thankful to the Sbarro Health Research Organization (http://www.shro.org), the Human Health Foundation (http://www.hhfonlus.org), AIRC-Associazione Italiana per la Ricerca sul Cancro, IG 2014-15690 and the Commonwealth of Pennsylvania for their support Prof Giordano is the director of research line 3 at CROM, Istituto Nazionale Tumori ‘Fondazione G. Pascale’-IRCCS, Naples, Italy.
Abbreviations
ALL, acute lymphoblastic leukemia; ANP32A, acidic nuclear phosphoprotein 32A; PAF-1, apoptotic peptidase activating factor-1; ATM, ataxia-telangiectasia mutated; BCG, bacillus Calmette-Guerin; BH3, BCL-2 homology 3; CDK, cyclin-dependent kinase; CDKI, CDK inhibitor; CHEK1/2, checkpoint kinase 1/2; DREAM, DP, RB like, E2F and MuvB; ER, estrogen receptor; 5-FU, 5-fluorouracil; HDAC, histone deacetylase; HRE, high resolution electrophoresis; IFN-α, interferon-α; IHC, immunohistochemistry; LOH: loss of heterozygosity; MEFs, mouse embryonic fibroblasts; MuvB, multi-vulval class B; P/CAF, p300/CBP-associated factor; PP1: protein phosphatase 1; RB, retinoblastoma; RBL, retinoblastoma-like; RT-qPCR, reverse transcriptionquntitative PCR; SKP2, S-phase kinase-associated protein 2; TGF-β1, transforming growth factor-β1; TNBC, triple-negative breast cancer; TNF-α, tumor-necrosis factor-alpha; WB, western blotting.
REFERENCES
1. Sherr CJ. Cancer cell cycles. Science. 1996; 274:1672–1677.
2. Giordano A, Whyte P, Harlow E, Franza BR Jr, Beach D, Draetta G. A 60 kd cdc2-associated polypeptide complexes with the E1A proteins in adenovirus-infected cells. Cell. 1989; 58:981–990.
3. Giordano A, Lee JH, Scheppler JA, Herrmann C, Harlow E, Deuschle U, Beach D, Franza BR Jr. Cell cycle regulation of histone H1 kinase activity associated with the adenoviral protein E1A. Science. 1991; 253:1271–1275.
4. Giordano A, McCall C, Whyte P, Franza BR Jr. Human cyclin A and the retinoblastoma protein interact with similar but distinguishable sequences in the adenovirus E1A gene product. Oncogene. 1991; 6:481–485.
5. Mayol X, Grana X, Baldi A, Sang N, Hu Q, Giordano A. Cloning of a new member of the retinoblastoma gene family (pRb2) which binds to the E1A transforming domain. Oncogene. 1993; 8:2561–2566.
6. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995; 81:323–330.
7. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006; 25:5220–5227.
8. Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013; 14:297–306.
9. Susini T, Baldi F, Howard CM, Baldi A, Taddei G, Massi D, Rapi S, Savino L, Massi G, Giordano A. Expression of the retinoblastoma-related gene Rb2/p130 correlates with clinical outcome in endometrial cancer. J Clin Oncol. 1998; 16:1085–1093.
10. Massaro-Giordano M, Baldi G, De Luca A, Baldi A, Giordano A. Differential expression of the retinoblastoma gene family members in choroidal melanoma: prognostic significance. Clin Cancer Res. 1999; 5:1455–1458.
11. Claudio PP, Howard CM, Pacilio C, Cinti C, Romano G, Minimo C, Maraldi NM, Minna JD, Gelbert L, Leoncini L, Tosi GM, Hicheli P, Caputi M, Giordano GG, Giordano A. Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res. 2000; 60:372–382.
12. Cinti C, Claudio PP, Howard CM, Neri LM, Fu Y, Leoncini L, Tosi GM, Maraldi NM, Giordano A. Genetic alterations disrupting the nuclear localization of the retinoblastoma-related gene RB2/p130 in human tumor cell lines and primary tumors. Cancer Res. 2000; 60:383–389.
13. Tanaka N, Odajima T, Mimura M, Ogi K, Dehari H, Kimijima Y, Kohama G. Expression of Rb, pRb2/p130, p53, and p16 proteins in malignant melanoma of oral mucosa. Oral Oncol. 2001; 37:308–314.
14. Zamparelli A, Masciullo V, Bovicelli A, Santini D, Ferrandina G, Minimo C, Terzano P, Costa S, Cinti C, Ceccarelli C, Mancuso S, Scambia G, Bovicelli L, Giordano A. Expression of cell-cycle-associated proteins pRB2/p130 and p27kip in vulvar squamous cell carcinomas. Hum Pathol. 2001; 32:4–9.
15. Milde-Langosch K, Goemann C, Methner C, Rieck G, Bamberger AM, Loning T. Expression of Rb2/p130 in breast and endometrial cancer: correlations with hormone receptor status. Br J Cancer. 2001; 85:546–551.
16. Caputi M, Groeger AM, Esposito V, De Luca A, Masciullo V, Mancini A, Baldi F, Wolner E, Giordano A. Loss of pRb2/p130 expression is associated with unfavorable clinical outcome in lung cancer. Clin Cancer Res. 2002; 8:3850–3856.
17. Li Q, Sakurai Y, Ryu T, Azuma K, Yoshimura K, Yamanouchi Y, Ikehara S, Kawamoto K. Expression of Rb2/p130 protein correlates with the degree of malignancy in gliomas. Brain Tumor Pathol. 2004; 21:121–125.
18. D’Andrilli G, Masciullo V, Bagella L, Tonini T, Minimo C, Zannoni GF, Giuntoli RL 2nd, Carlson JA Jr, Soprano DR, Soprano KJ, Scambia G, Giordano A. Frequent loss of pRb2/p130 in human ovarian carcinoma. Clin Cancer Res. 2004; 10:3098–3103.
19. Russo G, Zamparelli A, Howard CM, Minimo C, Bellan C, Carillo G, Califano L, Leoncini L, Giordano A, Claudio PP. Expression of cell cycle-regulated proteins pRB2/p130, p107, E2F4, p27, and pCNA in salivary gland tumors: prognostic and diagnostic implications. Clin Cancer Res. 2005; 11:3265–3273.
20. Masciullo V, Berardengo E, Boglione A, Sgambato A, Bernardi A, Forni M, Linari A, Cito L, Scambia G, Comandone A, Giordano A. The retinoblastoma family member pRb2/p130 is an independent predictor of survival in human soft tissue sarcomas. Clin Cancer Res. 2008; 14:4775–4779.
21. Cito L, Indovina P, Forte IM, Pentimalli F, Di Marzo D, Somma P, Barone D, Penon A, Penon D, Ceccherini E, Micheli P, Saragoni L, Di Domenico M, Feola A, Roviello F, Mattioli E, et al. pRb2/p130 localizes to the cytoplasm in diffuse gastric cancer. J Cell Physiol. 2015; 230:802–805.
22. Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer. 2013; 13:585–595.
23. Macaluso M, Montanari M, Giordano A. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene. 2006; 25:5263–5267.
24. Indovina P, Marcelli E, Casini N, Rizzo V, Giordano A. Emerging roles of RB family: new defense mechanisms against tumor progression. J Cell Physiol. 2012; 228:525–535.
25. Pentimalli F, Cito L, Giordano A. (2010). Dysfunction of the RB Retinoblastoma Gene in Cancer: Humana Press.
26. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70.
27. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000; 21:485–495.
28. Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003; 3:130–138.
29. DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A. 1997; 94:7245–7250.
30. Attardi LD, Sage J. RB goes mitochondrial. Genes Dev. 2013; 27:975–979.
31. Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003; 421:942–947.
32. de Bruin A, Wu L, Saavedra HI, Wilson P, Yang Y, Rosol TJ, Weinstein M, Robinson ML, Leone G. Rb function in extraembryonic lineages suppresses apoptosis in the CNS of Rb-deficient mice. Proc Natl Acad Sci U S A. 2003; 100:6546–6551.
33. Wenzel PL, Wu L, de Bruin A, Chong JL, Chen WY, Dureska G, Sites E, Pan T, Sharma A, Huang K, Ridgway R, Mosaliganti K, Sharp R, Machiraju R, Saltz J, Yamamoto H, et al. Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2007; 21:85–97.
34. Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, Haigis K, Gulino A, Lees JA. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009; 15:184–194.
35. Khidr L, Chen PL. RB, the conductor that orchestrates life, death and differentiation. Oncogene. 2006; 25:5210–5219.
36. Delston RB, Harbour JW. Rb at the interface between cell cycle and apoptotic decisions. Curr Mol Med. 2006; 6:713–718.
37. Wells J, Yan PS, Cechvala M, Huang T, Farnham PJ. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene. 2003; 22:1445–1460.
38. Young AP, Longmore GD. Differential regulation of apoptotic genes by Rb in human versus mouse cells. Oncogene. 2004; 23:2587–2599.
39. Evan G, Littlewood T. A matter of life and cell death. Science. 1998; 281:1317–1322.
40. Adegbola O, Pasternack GR. Phosphorylated retinoblastoma protein complexes with pp32 and inhibits pp32-mediated apoptosis. J Biol Chem. 2005; 280:15497–15502.
41. Lu Z, Bauzon F, Fu H, Cui J, Zhao H, Nakayama K, Nakayama KI, Zhu L. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat Commun. 2014; 5:3463.
42. Jiang Y, Saavedra HI, Holloway MP, Leone G, Altura RA. Aberrant regulation of survivin by the RB/E2F family of proteins. J Biol Chem. 2004; 279:40511–40520.
43. Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A. 2006; 103:6332–6337.
44. Raj D, Liu T, Samadashwily G, Li F, Grossman D. Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis. 2008; 29:194–201.
45. Shehata HH, Abou Ghalia AH, Elsayed EK, Ziko OO, Mohamed SS. Detection of survivin protein in aqueous humor and serum of retinoblastoma patients and its clinical significance. Clin Biochem. 2009; 43:362–366.
46. Ishii H, Igarashi T, Saito T, Nakano T, Mori M, Ohyama H, Miyamoto T, Saito Y, Oh H. Retinoblastoma protein expressed in human non-Hodgkin’s lymphoma cells generates resistance against radiation-induced apoptosis. Am J Hematol. 1997; 55:46–48.
47. Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, Lowe SW, Knudsen ES. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007; 117:218–228.
48. Zagorski WA, Knudsen ES, Reed MF. Retinoblastoma deficiency increases chemosensitivity in lung cancer. Cancer Res. 2007; 67:8264–8273.
49. Sharma A, Comstock CE, Knudsen ES, Cao KH, Hess-Wilson JK, Morey LM, Barrera J, Knudsen KE. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer Res. 2007; 67:6192–6203.
50. Biasoli D, Kahn SA, Cornelio TA, Furtado M, Campanati L, Chneiweiss H, Moura-Neto V, Borges HL. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013; 4:e767.
51. Almasan A, Yin Y, Kelly RE, Lee EY, Bradley A, Li W, Bertino JR, Wahl GM. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci U S A. 1995; 92:5436–5440.
52. Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, Feramisco JR, Wang JY, Knudsen ES. RB-dependent S-phase response to DNA damage. Mol Cell Biol. 2000; 20:7751–7763.
53. Mayhew CN, Perkin LM, Zhang X, Sage J, Jacks T, Knudsen ES. Discrete signaling pathways participate in RB-dependent responses to chemotherapeutic agents. Oncogene. 2004; 23:4107–4120.
54. Haas-Kogan DA, Kogan SC, Levi D, Dazin P, T’Ang A, Fung YK, Israel MA. Inhibition of apoptosis by the retinoblastoma gene product. EMBO J. 1995; 14:461–472.
55. Haupt Y, Rowan S, Oren M. p53-mediated apoptosis in HeLa cells can be overcome by excess pRB. Oncogene. 1995; 10:1563–1571.
56. McConkey DJ, Goodrich D, Bucana C, Klostergaard J. The human retinoblastoma gene product suppresses ceramide-induced apoptosis in human bladder tumor cells. Oncogene. 1996; 13:1693–1700.
57. Berry DE, Lu Y, Schmidt B, Fallon PG, O’Connell C, Hu SX, Xu HJ, Blanck G. Retinoblastoma protein inhibits IFN-gamma induced apoptosis. Oncogene. 1996; 12:1809–1819.
58. Choi KS, Eom YW, Kang Y, Ha MJ, Rhee H, Yoon JW, Kim SJ. Cdc2 and Cdk2 kinase activated by transforming growth factor-beta1 trigger apoptosis through the phosphorylation of retinoblastoma protein in FaO hepatoma cells. J Biol Chem. 1999; 274:31775–31783.
59. Masselli A, Wang JY. Phosphorylation site mutated RB exerts contrasting effects on apoptotic response to different stimuli. Oncogene. 2006; 25:1290–1298.
60. Chau BN, Pan CW, Wang JY. Separation of anti-proliferation and anti-apoptotic functions of retinoblastoma protein through targeted mutations of its A/B domain. PLoS One. 2006; 1:e82.
61. Dou QP, An B, Will PL. Induction of a retinoblastoma phosphatase activity by anticancer drugs accompanies p53-independent G1 arrest and apoptosis. Proc Natl Acad Sci U S A. 1995; 92:9019–9023.
62. Morana SJ, Wolf CM, Li J, Reynolds JE, Brown MK, Eastman A. The involvement of protein phosphatases in the activation of ICE/CED-3 protease, intracellular acidification, DNA digestion, and apoptosis. J Biol Chem. 1996; 271:18263–18271.
63. Wang H, Grand RJ, Milner AE, Armitage RJ, Gordon J, Gregory CD. Repression of apoptosis in human B-lymphoma cells by CD40-ligand and Bcl-2: relationship to the cell-cycle and role of the retinoblastoma protein. Oncogene. 1996; 13:373–379.
64. Day ML, Foster RG, Day KC, Zhao X, Humphrey P, Swanson P, Postigo AA, Zhang SH, Dean DC. Cell anchorage regulates apoptosis through the retinoblastoma tumor suppressor/E2F pathway. J Biol Chem. 1997; 272:8125–8128.
65. Wang RH, Liu CW, Avramis VI, Berndt N. Protein phosphatase 1alpha-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001; 20:6111–6122.
66. Glozak MA, Rogers MB. Retinoic acid- and bone morphogenetic protein 4-induced apoptosis in P19 embryonal carcinoma cells requires p27. Exp Cell Res. 2001; 268:128–138.
67. Popowski M, Ferguson HA, Sion AM, Koller E, Knudsen E, Van Den Berg CL. Stress and IGF-I differentially control cell fate through mammalian target of rapamycin (mTOR) and retinoblastoma protein (pRB). J Biol Chem. 2008; 283:28265–28273.
68. De Leon G, Sherry TC, Krucher NA. Reduced expression of PNUTS leads to activation of Rb-phosphatase and caspase-mediated apoptosis. Cancer Biol Ther. 2008; 7:833–841.
69. Dou QP, Lui VW. Failure to dephosphorylate retinoblastoma protein in drug-resistant cells. Cancer Res. 1995; 55:5222–5225.
70. Wallace M, Coates PJ, Wright EG, Ball KL. Differential post-translational modification of the tumour suppressor proteins Rb and p53 modulate the rates of radiation-induced apoptosis in vivo. Oncogene. 2001; 20:3597–3608.
71. Lentine B, Antonucci L, Hunce R, Edwards J, Marallano V, Krucher NA. Dephosphorylation of threonine-821 of the retinoblastoma tumor suppressor protein (Rb) is required for apoptosis induced by UV and Cdk inhibition. Cell Cycle. 2012; 11:3324–3330.
72. An B, Jin JR, Lin P, Dou QP. Failure to activate interleukin 1beta-converting enzyme-like proteases and to cleave retinoblastoma protein in drug-resistant cells. FEBS Lett. 1996; 399:158–162.
73. An B, Dou QP. Cleavage of retinoblastoma protein during apoptosis: an interleukin 1 beta-converting enzyme-like protease as candidate. Cancer Res. 1996; 56:438–442.
74. Tan X, Wang JY. The caspase-RB connection in cell death. Trends Cell Biol. 1998; 8:116–120.
75. Diederich L, Fotedar A, Fotedar R. Proteolytic cleavage of retinoblastoma protein upon DNA damage and Fas-mediated apoptosis. Cell Biol Toxicol. 1998; 14:133–140.
76. Browne SJ, MacFarlane M, Cohen GM, Paraskeva C. The adenomatous polyposis coli protein and retinoblastoma protein are cleaved early in apoptosis and are potential substrates for caspases. Cell Death Differ. 1998; 5:206–213.
77. Fattman CL, Delach SM, Dou QP, Johnson DE. Sequential two-step cleavage of the retinoblastoma protein by caspase-3/-7 during etoposide-induced apoptosis. Oncogene. 2001; 20:2918–2926.
78. Katsuda K, Kataoka M, Uno F, Murakami T, Kondo T, Roth JA, Tanaka N, Fujiwara T. Activation of caspase-3 and cleavage of Rb are associated with p16-mediated apoptosis in human non-small cell lung cancer cells. Oncogene. 2002; 21:2108–2113.
79. Tan X, Martin SJ, Green DR, Wang JY. Degradation of retinoblastoma protein in tumor necrosis factor- and CD95-induced cell death. J Biol Chem. 1997; 272:9613–9616.
80. Chau BN, Borges HL, Chen TT, Masselli A, Hunton IC, Wang JY. Signal-dependent protection from apoptosis in mice expressing caspase-resistant Rb. Nat Cell Biol. 2002; 4:757–765.
81. Boutillier AL, Trinh E, Loeffler JP. Caspase-dependent cleavage of the retinoblastoma protein is an early step in neuronal apoptosis. Oncogene. 2000; 19:2171–2178.
82. Wagner S, Roemer K. Retinoblastoma protein is required for efficient colorectal carcinoma cell apoptosis by histone deacetylase inhibitors in the absence of p21Waf. Biochem Pharmacol. 2005; 69:1059–1067.
83. Bowen C, Spiegel S, Gelmann EP. Radiation-induced apoptosis mediated by retinoblastoma protein. Cancer Res. 1998; 58:3275–3281.
84. Bowen C, Birrer M, Gelmann EP. Retinoblastoma protein-mediated apoptosis after gamma-irradiation. J Biol Chem. 2002; 277:44969–44979.
85. Munro S, Carr SM, La Thangue NB. Diversity within the pRb pathway: is there a code of conduct? Oncogene. 2012; 31:4343–4352.
86. Korah J, Falah N, Lacerte A, Lebrun JJ. A transcriptionally active pRb-E2F1-P/CAF signaling pathway is central to TGFbeta-mediated apoptosis. Cell Death Dis. 2012; 3:e407.
87. Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, Walensky LD, Lees JA. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. 2013; 27:1003–1015.
88. Antonucci LA, Egger JV, Krucher NA. Phosphorylation of the Retinoblastoma protein (Rb) on serine-807 is required for association with Bax. Cell Cycle. 2014; 13:3611–3617.
89. Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996; 10:1621–1632.
90. LeCouter JE, Kablar B, Whyte PF, Ying C, Rudnicki MA. Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development. 1998; 125:4669–4679.
91. Harb G, Vasavada RC, Cobrinik D, Stewart AF. The retinoblastoma protein and its homolog p130 regulate the G1/S transition in pancreatic beta-cells. Diabetes. 2009; 58:1852–1862.
92. Liu DX, Nath N, Chellappan SP, Greene LA. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005; 19:719–732.
93. Tonini T, Gabellini C, Bagella L, D’Andrilli G, Masciullo V, Romano G, Scambia G, Zupi G, Giordano A. pRb2/p130 decreases sensitivity to apoptosis induced by camptothecin and doxorubicin but not by taxol. Clin Cancer Res. 2004; 10:8085–8093.
94. Dingar D, Konecny F, Zou J, Sun X, von Harsdorf R. Anti-apoptotic function of the E2F transcription factor 4 (E2F4)/p130, a member of retinoblastoma gene family in cardiac myocytes. J Mol Cell Cardiol. 2012; 53:820–828.
95. Fusaro G, Wang S, Chellappan S. Differential regulation of Rb family proteins and prohibitin during camptothecin-induced apoptosis. Oncogene. 2002; 21:4539–4548.
96. Simpson DS, Mason-Richie NA, Gettler CA, Wikenheiser-Brokamp KA. Retinoblastoma family proteins have distinct functions in pulmonary epithelial cells in vivo critical for suppressing cell growth and tumorigenesis. Cancer Res. 2009; 69:8733–8741.
97. Bellan C, De Falco G, Tosi GM, Lazzi S, Ferrari F, Morbini G, Bartolomei S, Toti P, Mangiavacchi P, Cevenini G, Trimarchi C, Cinti C, Giordano A, Leoncini L, Tosi P, Cottier H. Missing expression of pRb2/p130 in human retinoblastomas is associated with reduced apoptosis and lesser differentiation. Invest Ophthalmol Vis Sci. 2002; 43:3602–3608.
98. Jori FP, Melone MA, Napolitano MA, Cipollaro M, Cascino A, Giordano A, Galderisi U. RB and RB2/p130 genes demonstrate both specific and overlapping functions during the early steps of in vitro neural differentiation of marrow stromal stem cells. Cell Death Differ. 2005; 12:65–77.
99. Pucci B, Claudio PP, Masciullo V, Bellincampi L, Terrinoni A, Khalili K, Melino G, Giordano A. pRb2/p130 promotes radiation-induced cell death in the glioblastoma cell line HJC12 by p73 upregulation and Bcl-2 downregulation. Oncogene. 2002; 21:5897–5905.
100. La Sala D, Macaluso M, Trimarchi C, Giordano A, Cinti C. Triggering of p73-dependent apoptosis in osteosarcoma is under the control of E2Fs-pRb2/p130 complexes. Oncogene. 2003; 22:3518–3529.
101. Derenzini M, Donati G, Mazzini G, Montanaro L, Vici M, Ceccarelli C, Santini D, Taffurelli M, Trere D. Loss of retinoblastoma tumor suppressor protein makes human breast cancer cells more sensitive to antimetabolite exposure. Clin Cancer Res. 2008; 14:2199–2209.
102. Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008; 10:R75.
103. Trere D, Brighenti E, Donati G, Ceccarelli C, Santini D, Taffurelli M, Montanaro L, Derenzini M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann Oncol. 2009; 20:1818–1823.
104. Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2012; 18:5110–5122.
105. Lehn S, Ferno M, Jirstrom K, Ryden L, Landberg G. A non-functional retinoblastoma tumor suppressor (RB) pathway in premenopausal breast cancer is associated with resistance to tamoxifen. Cell Cycle. 2011; 10:956–962.
106. Varma H, Conrad SE. Reversal of an antiestrogen-mediated cell cycle arrest of MCF-7 cells by viral tumor antigens requires the retinoblastoma protein-binding domain. Oncogene. 2000; 19:4746–4753.
107. Mack PC, Chi SG, Meyers FJ, Stewart SL, deVere White RW, Gumerlock PH. Increased RB1 abnormalities in human primary prostate cancer following combined androgen blockade. Prostate. 1998; 34:145–151.
108. Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, Morrissey C, Zhang X, Comstock CE, Witkiewicz AK, Gomella L, Knudsen ES, Nelson PS, Knudsen KE. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010; 120:4478–4492.
109. Knudsen KE, Arden KC, Cavenee WK. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998; 273:20213–20222.
110. Pollack A, Czerniak B, Zagars GK, Hu SX, Wu CS, Dinney CP, Chyle V, Benedict WF. Retinoblastoma protein expression and radiation response in muscle-invasive bladder cancer. Int J Radiat Oncol Biol Phys. 1997; 39:687–695.
111. Agerbaek M, Alsner J, Marcussen N, Lundbeck F, von der Maase H. Retinoblastoma protein expression is an independent predictor of both radiation response and survival in muscle-invasive bladder cancer. Br J Cancer. 2003; 89:298–304.
112. Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. 2003; 22:5897–5906.
113. Esuvaranathan K, Chiong E, Thamboo TP, Chan YH, Kamaraj R, Mahendran R, Teh M. Predictive value of p53 and pRb expression in superficial bladder cancer patients treated with BCG and interferon-alpha. Cancer. 2007; 109:1097–1105.
114. Cormio L, Tolve I, Annese P, Saracino A, Zamparese R, Sanguedolce F, Bufo P, Battaglia M, Selvaggi FP, Carrieri G. Altered p53 and pRb expression is predictive of response to BCG treatment in T1G3 bladder cancer. Anticancer Res. 2009; 29:4201–4204.
115. Addeo R, Casale F, Caraglia M, D’Angelo V, Crisci S, Abbruzzese A, Di Tullio MT, Indolfi P. Glucocorticoids induce G1 arrest of lymphoblastic cells through retinoblastoma protein Rb1 dephosphorylation in childhood acute lymphoblastic leukemia in vivo. Cancer Biol Ther. 2004; 3:470–476.
116. Kommoss S, du Bois A, Ridder R, Trunk MJ, Schmidt D, Pfisterer J, Kommoss F. Independent prognostic significance of cell cycle regulator proteins p16(INK4a) and pRb in advanced-stage ovarian carcinoma including optimally debulked patients: a translational research subprotocol of a randomised study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group. Br J Cancer. 2007; 96:306–313.
117. Ramos E, Llado L, Serrano T, Figueras A, Lastra R, Torras J, Figueras J, Rafecas A, Fabregat J. Utility of cell-cycle modulators to predict vascular invasion and recurrence after surgical treatment of hepatocellular carcinoma. Transplantation. 2006; 82:753–758.
118. Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008; 8:714–724.
119. Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res. 2010; 16:1094–1099.
120. McNeish IA, Bell SJ, Lemoine NR. Gene therapy progress and prospects: cancer gene therapy using tumour suppressor genes. Gene Ther. 2004; 11:497–503.
121. Lim MJ, Min SH, Lee JJ, Kim IC, Kim JT, Lee DC, Kim NS, Jeong S, Kim MN, Kim KD, Lim JS, Han SB, Kim HM, Heo DS, Yeom YI. Targeted therapy of DNA tumor virus-associated cancers using virus-activated transcription factors. Mol Ther. 2006; 13:899–909.
122. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003; 3:285–296.
123. Li B, Gordon GM, Du CH, Xu J, Du W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell. 2010; 17:469–480.
124. Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009; 9:738–748.
125. Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006; 444:61–66.
126. Kitagawa M, Aonuma M, Lee SH, Fukutake S, McCormick F. E2F-1 transcriptional activity is a critical determinant of Mdm2 antagonist-induced apoptosis in human tumor cell lines. Oncogene. 2008; 27:5303–5314.
127. Roig JM, Molina MA, Cascante A, Calbo J, Carbo N, Wirtz U, Sreedharan S, Fillat C, Mazo A. Adenovirus-mediated retinoblastoma 94 gene transfer induces human pancreatic tumor regression in a mouse xenograft model. Clin Cancer Res. 2004; 10:1454–1462.
128. Pentimalli F, Giordano A. Promises and drawbacks of targeting cell cycle kinases in cancer. Discov Med. 2009; 8:177–180.
129. Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009; 8:547–566.
130. Cirillo D, Pentimalli F, Giordano A. Peptides or small molecules? Different approaches to develop more effective CDK inhibitors. Curr Med Chem. 2011; 18:2854–2866.
131. Esposito L, Indovina P, Magnotti F, Conti D, Giordano A. Anticancer therapeutic strategies based on CDK inhibitors. Curr Pharm Des. 2013; 19:5327–5332.
132. Abate AA, Pentimalli F, Esposito L, Giordano A. ATP-noncompetitive CDK inhibitors for cancer therapy: an overview. Expert Opin Investig Drugs. 2013; 22:895–906.
133. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011; 11:558–572.
134. Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, Landa J, Qin LX, Rathbone DD, Condy MM, Ustoyev Y, Crago AM, Singer S, Schwartz GK. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013; 31:2024–2028.
135. Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, Yu JQ, Vallabhajosula S, Schoder H, English P, Neuberg DS, Martin P, Millenson MM, Ely SA, Courtney R, Shaik N, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012; 119:4597–4607.
136. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015; 14:130–146.
137. Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014; 16:207.
138. Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, Niesvizky R, Cho H, Ely S, Moore MA, Chen-Kiang S. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006; 66:7661–7667.
139. Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, Ramsey MR, Jin J, Wong KK, Su L, Zhou D, Sharpless NE. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010; 120:2528–2536.
140. Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, Ozawa T, James CD, Waldman T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010; 70:3228–3238.
141. Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012; 287:29075–29087.
142. Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008; 68:5519–5523.
143. Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs. 2013; 22:723–738.
144. Altenburg JD, Farag SS. The potential role of PD0332991 (Palbociclib) in the treatment of multiple myeloma. Expert Opin Investig Drugs. 2014; 24:261–271.
145. Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005; 23:9408–9421.
146. Giordano A, Rossi A, Romano G, Bagella L. Tumor suppressor pRb2/p130 gene and its derived product Spa310 spacer domain as perspective candidates for cancer therapy. J Cell Physiol. 2007; 213:403–406.
147. Bagella L, Sun A, Tonini T, Abbadessa G, Cottone G, Paggi MG, De Luca A, Claudio PP, Giordano A. A small molecule based on the pRb2/p130 spacer domain leads to inhibition of cdk2 activity, cell cycle arrest and tumor growth reduction in vivo. Oncogene. 2007; 26:1829–1839.
148. Giordano A, Bellacchio E, Bagella L, Paggi MG. Interaction between the Cdk2/cyclin A complex and a small molecule derived from the pRb2/p130 spacer domain: a theoretical model. Cell Cycle. 2007; 6:2591–2593.