
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is a malignant disease characterized by the accumulation of monoclonal B cells that have escaped their regulated life cycle. CLL is the most common adult leukemia in the Western world, and though it allows longer survival than most leukemias, this disease creates a poor quality of life and currently remains incurable [1]. Particular factors that contribute to the incurability of CLL are the inter- and intra-tumor heterogeneity that exists among CLL populations and the subclonal expansion of refractory CLL cells that can arise after initial chemotherapy treatment [2, 3]. Based on these underlying characteristics of CLL, many large-scale biomarker studies have focused on determining molecular features that would help provide potential predictive and prognostic power in treating this disease. These studies have typically relied on mRNA-based gene expression or DNA-based somatic mutation and CpG methylation analysis. Such studies have created the opportunity to discover genetic and epigenetic marks as potential prognostic tools. Examples of these discovered prognostic markers include somatic mutations in genes like SF3B1, NOTCH1, and MYD88 [4-6], as well as differential gene expression and DNA methylation changes in genes like ZAP70, LPL, CRY1, and LDOC1 [7-14]. Along with discovering these single gene prognostic markers in CLL, genome-wide DNA methylation and gene expression studies have also demonstrated genomic signatures that correspond with specific CLL subtypes, like IGVH mutation and CD38 expression status and have provided better comprehension of the molecular abnormalities that occur in this disease [3, 8, 11, 12, 15-17]. Nevertheless, though the understanding of CLL pathobiology has been greatly enhanced by these large-scale gene studies, the limiting aspect that still remains in RNA or DNA-based profiling is the inability to provide clarity to the altered protein expression and signaling landscape in CLL.
Understanding how pro-proliferative proteins are differentially altered in CLL has currently gained popularity based on recent studies that demonstrate the efficacy of targeting signaling proteins involved in the B-cell Receptor (BCR) pathway [18-22]. Most of these efficacious effects observed when targeting the BCR pathway have stemmed from the selective inhibition of either the Bruton’s Tyrosine Kinase (BTK) using the inhibitor Ibrutinib or the PI3K-delta (PI3Kδ) specific kinase using the inhibitor Idelalisib. Based on the recent success of these two inhibitors’ respective clinical trials, the outlook of treatment strategies for CLL has changed and motivation has shifted towards better understanding the specific protein events that drive CLL pathogenesis [23-27].
Though several proteins have individually been investigated when determining the oncogenic features of CLL, no current study has simultaneously examined numerous protein alterations on a comprehensive level. Based on this current void, our goal was to perform reverse-phase protein array (RPPA) analysis using 167 antibody probes on primary CLL lysates in order to determine the deregulated protein events that occur in CLL. From this study, we demonstrate that the AKT/mTOR-related proteins are altered in CLL, with significant alteration occurring in the downstream mRNA translational machinery proteins eukaryotic translation factor 4G (eIF4G) and the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). These results and corresponding ex vivo treatment results with the dual PI3K/mTOR inhibitor NVP-BEZ235 help demonstrate the potential dependence of mRNA translation in CLL survival, as well as revealing mRNA translation as a potential therapeutic target in CLL.
RESULTS
Comprehensive analysis of RPPA probe intensities reveals a common protein signature among CLL patients
To gain a better understanding of the expression profile of CLL at the protein level, we collected 18 CLL patient and 6 healthy CD19+ B-cell lysates for RPPA analysis. The CLL lysates collected for this study are comprised of samples containing varying IGVH mutation, CD38 expression, FISH, ZAP70 expression, treatment, and Rai stage status in order to determine whether these clinical parameters demonstrate any large-scale protein signatures (Table 1). From the collected array data, we gained an initial understanding of how the clinical features overlapped with the corresponding molecular signature determined by non-supervised hierarchical clustering. Based on the clustered dendrogram and the heatmap patterns, there seems to be a clear separation in molecular profiles between CLL patients and healthy donors samples, demonstrating an overall transformation at the protein level between the cancer and normal phenotype. However, the contrast between CLL patient subtypes is less stark as there seems to be a less clear separation within the unsupervised hierarchical clustering patterns of the clinical subfeatures ascribed to the patient samples (Figure 1A). Thus, unsupervised clustering would suggest that an underlying protein signature that distinguishes itself from normal CD19+ B-cells is common among CLL patient samples.
Table 1: Clinical characteristics of CLL patient samples analyzed using reverse-phase protein array (RPPA).
Sample ID |
IGVH Status |
CD38 Expression |
ZAP70 Status |
11q Deletion |
13q Deletion |
17p Deletion |
Trisomy 12 |
Treatment |
Rai Stage |
CLL5918 |
UNMUTATED |
LOW |
ZAP70+ |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
0 |
CLL5984 |
UNMUTATED |
HIGH |
N/A |
NORMAL |
NORMAL |
NORMAL |
NORMAL |
TREATED |
1 |
CLL6389 |
MUTATED |
LOW |
ZAP70+ |
NORMAL |
NORMAL |
NORMAL |
NORMAL |
UNTREATED |
4 |
CLL6536 |
UNMUTATED |
HIGH |
ZAP70- |
NORMAL |
NORMAL |
NORMAL |
12+ |
UNTREATED |
0 |
CLL6950 |
MUTATED |
LOW |
N/A |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
0 |
CLL8404 |
UNMUTATED |
LOW |
ZAP70- |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
0 |
CLL8714 |
N/A |
HIGH |
N/A |
NORMAL |
NORMAL |
17p- |
NORMAL |
UNTREATED |
2 |
CLL8751 |
UNMUTATED |
LOW |
N/A |
NORMAL |
13q- |
NORMAL |
NORMAL |
TREATED |
3 |
CLL8755 |
MUTATED |
LOW |
ZAP70+ |
11q- |
NORMAL |
NORMAL |
NORMAL |
TREATED |
1 |
CLL8757 |
UNMUTATED |
LOW |
ZAP70+ |
NORMAL |
NORMAL |
17p- |
NORMAL |
UNTREATED |
0 |
CLL8816 |
N/A |
HIGH |
ZAP70+ |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
1 |
CLL8830 |
MUTATED |
LOW |
ZAP70+ |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
0 |
CLL8919 |
MUTATED |
LOW |
ZAP70- |
NORMAL |
NORMAL |
NORMAL |
NORMAL |
TREATED |
3 |
CLL9128 |
UNMUTATED |
LOW |
ZAP70- |
11q- |
13q- |
NORMAL |
NORMAL |
TREATED |
3 |
CLL9193 |
MUTATED |
LOW |
ZAP70- |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
2 |
CLL10372 |
UNMUTATED |
HIGH |
N/A |
NORMAL |
13q- |
NORMAL |
12+ |
UNTREATED |
1 |
CLL10907 |
MUTATED |
LOW |
N/A |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
1 |
CLL11449 |
UNMUTATED |
LOW |
N/A |
NORMAL |
13q- |
NORMAL |
NORMAL |
UNTREATED |
4 |
By further investigating the RPPA intensities of the antibody probes, we were able to determine through correlation comparison that eIF4G, SYK, RBM15, and GAB2 expression is higher in CLL than in healthy donor B-cells, whereas Annexin I and cleaved caspase 7 is lower in CLL compared to healthy donors (Figure 1B). In fact, based on log-fold/rank comparison of all probes represented in the RPPA panel, eIF4G overexpression is the most upregulated protein event in our CLL cohorts, whereas the pro-apoptotic protein events Annexin I expression and cleaved-caspase 7 are the most downregulated events in CLL [28-31] (Figure 1C). These extreme protein expression changes are examples that highlight the aberrant changes within the CLL protein signature.
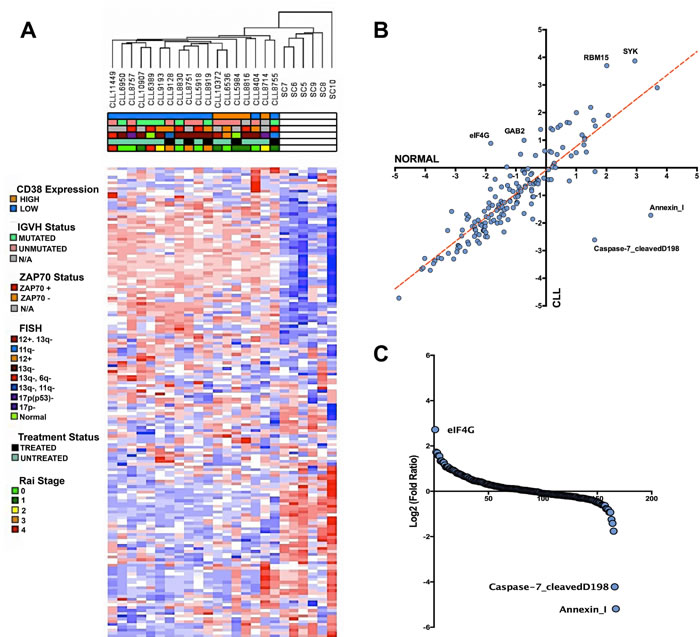
Figure 1: Altered protein landscape common among CLL patients. A. Non-supervised hierarchical clustering of normalized RPPA probe intensities using Euclidean distance. Heatmap demonstrates the separation of the CLL protein landscape and normal B-cell landscape without any statistical separation. Based on the clinical parameters, the altered protein expression is generally ubiquitous among CLL patients. B. Scatter plot comparing the average intensity of all 167 probes in CLL versus the average probe intensity in healthy B-cells C. Rank ordering plot based on the transformed (log2) fold ratio between CLL expression and normal B-cell expression. Rank ordering plot demonstrates the contrast between eIF4G upregulation, diminished cleaved caspase-7, and Annexin I downregulation from other probes in the RPPA dataset.
Supervised protein & pathway analysis in CLL reveals upregulation of mRNA translation machinery proteins
To systematically investigate the specific protein events that significantly correspond with the CLL phenotype, we further analyzed the protein arrays using ANOVA to determine which antibodies had significant differential probe intensities between CLL and normal B-cells samples. Based on this analysis, we determined that 58 distinct antibody probe intensities separate the CLL phenotype from the normal B-cell phenotype (Fold change > 1.25 or < -1.25, FDR p-value<0.05) with 38 probes being upregulated and 20 being downregulated in CLL (Figure 2A). The differentially altered proteins and phosphoproteins identified include proteins that have previously been associated with CLL, specifically the SYK and LCK kinase proteins and the Notch-1 signaling activator [32-41]. Paradoxically, we also identified the pro-apoptotic proteins BIM and BAK to be upregulated in our RPPA dataset. Nevertheless, the protein profile demonstrated by RPPA analysis helps provide a larger frame of reference for the molecular pathology of CLL as BIM overexpression in CLL commonly correlates with the overexpression of its direct anti-apoptotic antagonist BCL2, thus counteracting pro-apoptotic function of BIM [36]. A similar counteraction also occurs for the pro-apoptotic functions of BAK due to the fact that its pro-apoptotic binding partner BAX [42-44] is significantly downregulated in CLL patients. By identifying protein expression patterns of BIM, BCL2, BAK, and BAX as well as identifying the phosphorylation of the pro-apoptotic BAD at serine 112 (Table 2; Figure 2A), RPPA analysis helps provide a comprehensive illustration of how the anti-apoptotic characteristics of CLL are mediated among the BCL2 protein family.
Table 2: Expression profile of BH3-family proteins in CLL RPPA dataset.
Protein |
Apoptotic |
CLL Fold Ratio |
Bim |
Pro |
2.53 |
Bcl-2 |
Anti |
2.52 |
Bak |
Pro |
1.47 |
Bad_pS112 |
Anti |
1.34 |
Mcl1 |
Pro/Anti |
1.08 |
Bcl-xL |
Anti |
1.03 |
Bid |
Pro |
-1.22 |
Bax |
Pro |
-1.26 |
Supervised hierarchical analysis using ANOVA further revealed differential expression of several proteins implicated in cancer, including BRAF, STAT5A, and the Wnt pathway-related protein DVL3 [45-48]. Nonetheless, several of the proteins that were differentially altered in our array dataset are specifically involved in the AKT/mTOR signaling pathway [49, 50]. This signaling cascade is highly important in several types of cancers due to the fact that AKT and mTOR can regulate numerous tumorigenic events including cell proliferation, cell cycle progression, apoptotic resistance, autophagy, nutrient-dependent growth, and protein translation initiation [51-58]. Interestingly, the expression of total AKT and total mTOR were both significantly upregulated in CLL compared to healthy donor B-cells. These two events observed in our CLL samples are specifically noteworthy as upregulation of other kinases at the protein level have previously been identified in CLL, including the critical LCK, SYK, and BTK kinase proteins [23, 32, 35]. We also identified overexpression of scaffolding proteins that are traditionally involved in the AKT/mTOR pathway including IRS1, RICTOR, and GAB2, which have been implicated as oncogenic contributors in other cancer types [59-65]. With overexpression of these AKT/mTOR-related signaling proteins, we also observed higher phosphoprotein levels for PDK1 at serine 241, 4E-BP1 at serine 65 and threonine 70, p70S6K at threonine 389, BAD at serine 112, and PRAS40 at threonine 246 in CLL samples as compared to normal donors (Figure 2A).
To help determine the specific molecular mechanisms that are altered in CLL, we interrogated the total proteins from the differentially expressed probes in the RPPA dataset and observed overrepresentation of signaling events regulating the PI3K/AKT pathway, with pathways specifically involved in the mRNA translation machinery of eukaryotic cells (Figure 2B, Supplementary File S2). Overrepresentation of these pathways biologically corresponds with the phosphorylation of the translation regulator 4E-BP1 at serine 65 and overexpression of eIF4G, AKT, mTOR, and SF2/ASF, a splicing protein recently implicated as a translational regulator [28, 50, 66, 67]. The upregulation of these protein alterations therefore highlights the possibility of mRNA translation as a tumorigenic mechanism in CLL (Figure 2C).
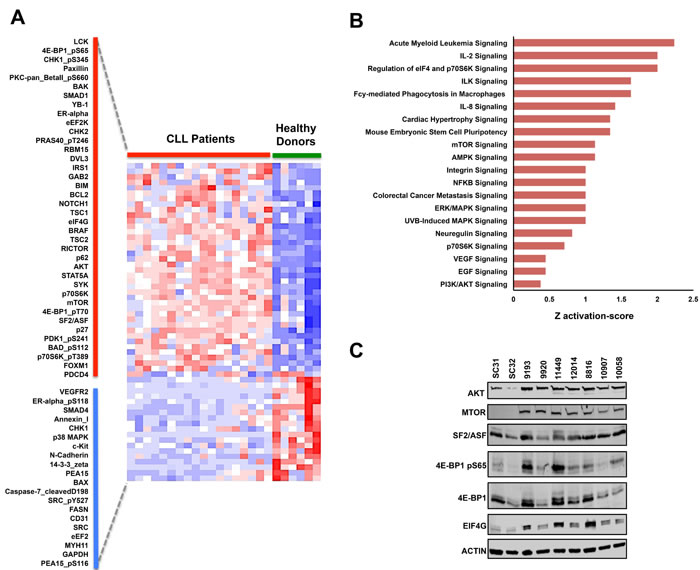
Figure 2: Supervised RPPA analysis reveals potential oncoproteins in CLL. A. Supervised hierarchical clustering analysis of RPPA results using an ANOVA FDR p-value threshold lower than 0.05 and a fold difference threshold of greater than 1.25 and less than -1.25. Based on this threshold, 58 probes were differentially altered in CLL with 38 probes being upregulated (along red bar) and 20 probes being downregulated (along blue bar). B. Ontology analysis of the differently expressed probes using Ingenuity Pathway Analysis (IPA) C. Immunoblot validation of differentially modified proteins that are involved in mRNA translation regulation.
4E-BP1 serine 65 phosphorylation occurs independently of AKT phosphorylation in CLL
We then compared the distinct phosphorylation events of the AKT/mTOR pathway to reveal the overall phosphorylation patterns of AKT/mTOR substrates in CLL. By normalizing the phosphoprotein levels to the internal levels of the corresponding total protein, we observed 4E-BP1 at serine 65 had higher phosphorylation levels in CLL compared to healthy donors. However, the high phosphorylation at this site did not correspond with other AKT/mTOR related phosphorylation sites, including AKT threonine 308, AKT serine 473, p70S6K threonine 389, and mTOR serine 2448, which were all relatively hypophosphorylated (Figure 3A). These phosphorylation patterns are quite interesting due to the fact that 4E-BP1 phosphorylation typically is dependent upon AKT phosphorylation [68, 69].
Because these B-cells were obtained from the peripheral blood and significant phosphorylation of AKT typically occurs through microenvironment interactions in the lymph node, we wanted to determine how AKT activation compares with 4E-BP1 serine 65 phosphorylation during ex vivo stimulation. To accomplish this feat, we performed a side-by-side activation of CLL B-cells cells and healthy donor B-cells with 10ug/ml anti-IgM for 1, 2, and 4 hours to determine the precise AKT threonine 308/4E-BP1 serine 65 phosphorylation pattern in CLL. Based on our results, AKT phosphorylation was higher in both CLL and normal B-cells during IgM stimulation. However, where healthy donors demonstrated increased phosphorylation of 4E-BP1 during IgM stimulation, CLL cells demonstrated consistently high and unchanging levels of 4E-BP1 phosphorylation, regardless of AKT activation (Figure 3C). Based on the results, it seems 4E-BP1 serine 65 phosphorylation in normal B-cells is dependent upon AKT phosphorylation, whereas 4E-BP1 sustains a constant phosphorylated state independent of AKT activation in CLL B-cells. This result would suggest that a disconnect takes place between AKT activation and 4E-BP1 phosphorylation during CLL transformation.
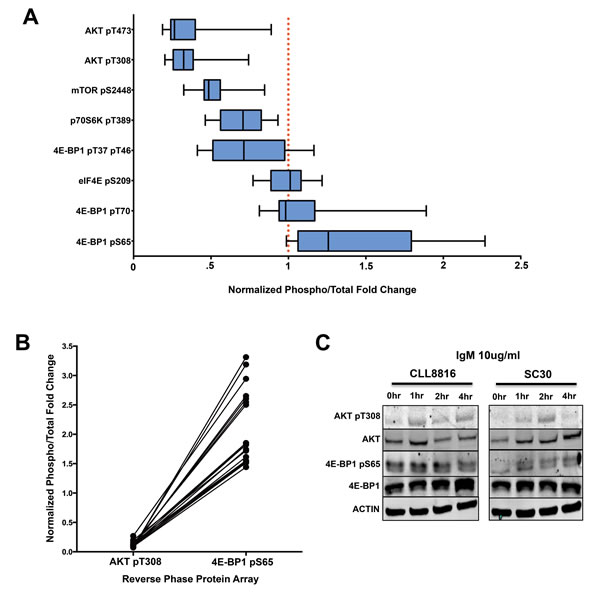
Figure 3: 4E-BP1 serine 65 phosphorylation is independent of AKT/mTOR activation in CLL. A. Normalized phosphorylation fold changes of AKT/mTOR substrates in CLL samples from RPPA (error bars = 95% CI). B. Paired comparison of CLL samples demonstrating the disconnection between AKT T308 phosphorylation and 4E-BP1 serine 65 phosphorylation. C. IgM activation of CLL sample 8816 and normal donor SC30. 4E-BP1 seems to be constitutively phosphorylated in CLL regardless of AKT pT308 activation, whereas the phosphorylation of 4E-BP1 in normal B-cells is dependent upon IgM-mediated AKT activation.
Increased CLL apoptosis corresponds with eIF4G cleavage and decreased 4E-BP1 serine 65 phosphorylation
With the gained understanding of 4E-BP1 phosphorylation and eIF4G overexpression in CLL, we wanted to test whether targeting the effects of these proteins would affect CLL viability and survival. With this rationale in mind, we compared the inhibitory capability of the recently developed BTK inhibitor Ibrutinib and the PI3Kδ inhibitor Idelalisib against the NVP-BEZ235, a dual PI3K/mTOR kinase inhibitor that has demonstrated ability to attenuate both 4E-BP1 phosphorylation and eIF4G activity in AML [23, 69-71].
To compare these three inhibitors, we first performed an IC50 proliferation assay for each of the three inhibitors in the MEC1 CLL cell line. From this experiment, we determined NVP-BEZ235 to have a lower IC50 value (Figure 4A). We then co-cultured 15 CLL patient B-cells with Huh7.5 cells, an adenocarcinoma cell line that provides a proper microenvironment for survival and viability of CLL cells [64], and treated the primary cells with each inhibitor for 48 hours at their maximum effective doses of 10uM [23, 70, 72]. Based on Annexin V/DAPI measurements, NVP-BEZ235 was able to significantly lower cell viability compared to Ibrutinib and Idelalisib. This lowered viability correlated with a potent induction of apoptosis in CLL cells as NVP-BEZ235 caused a significant increase in Annexin V+/DAPI+ cell death compared with side-by-side treatment of Ibrutinib and Idelalisib (Figure 4B).
We then wanted to determine if any differential 4E-BP1 and eIF4G alterations occurred when treating CLL cells with the three inhibitors. To observe any differential effects, we co-cultured CLL cells with HS-5 stromal cells and performed a side-by-side treatment with Ibrutinib, Idelalisib, and NVP-BEZ235. After 6 hours, we observed little distinction in AKT phosphorylation (T308 and S473) and AKT-dependent mTOR S2448 phosphorylation when comparing the three treatments (Supplementary Figure S1). However, we did observe a significant difference in 4E-BP1 phosphorylation when comparing the three treatments as NVP-BEZ235 significantly diminished 4E-BP1 serine 65 phosphorylation compared to Ibrutinib and Idelalisib, both of which caused little change in 4E-BP1 phosphorylation (Figure 4C).
Interestingly, this decrease in 4E-BP1 phosphorylation also correlated with greater cleavage of the eIF4G protein. This event previously characterized in immortalized lymphoma cell lines treated with chemotherapy [65], currently has not been characterized in clinical CLL samples. However, by treating CLL patient B-cells with the 3 inhibitors, we observed the lowest levels of the 220kDa eIF4G expression with NVP-BEZ235 treatment, while simultaneously observing increased detection of the protein fragments that correspond in size with the previously identified C-terminal fragment of apoptotic cleavage of eIF4G (C-FAG) (Figure 4C) [73, 74]. This observation implies that the mRNA translation is ultimately disrupted during apoptotic induction due to the enhanced apoptotic cleaving of the eIF4G translation initiator protein. Finally, we collectively compared the semi-quantitative 4E-BP1 phosphorylation and eIF4G cleavage levels with the Annexin V+/DAPI+ cells in each treatment and determined that greater apoptotic induction corresponds with greater 4E-BP1 dephosphorylation and eIF4G cleavage, suggesting that attenuating the protein events that upregulate translational initiation can impede cell survival mechanisms in CLL (Figure 4D).
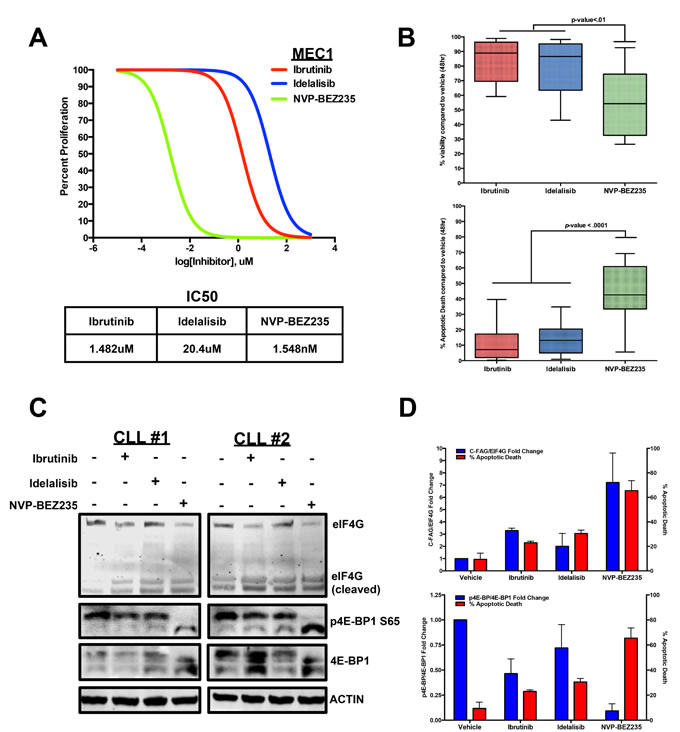
Figure 4: CLL apoptosis corresponds with eIF4G cleavage and 4E-BP1 dephosphorylation. A. Dose-dependent treatment analysis of Ibrutinib, Idelalisib, and NVP-BEZ235 in the MEC1 CLL cell line to compare the IC50 for each inhibitor. B. Annexin V/DAPI apoptotic assay comparing 10uM Ibrutinib, 10uM Idelalisib, and 10uM NVP-BEZ235 treatment in CLL cells co-cultured with Huh7.5 cells (n = 15). Based on the results, NVP-BEZ235 was able to cause more apoptotic death during the 48hr period. C. At 6 hours, 10uM NVP-BEZ235 causes greater dephosphorylation of 4E-BP1 as well as greater cleavage of eIF4G in HS-5 co-cultured CLL cells compared to 10 uM Ibrutinib and 10uM Idelalisib. D. Decreasing 4E-BP1 phosphorylation and increasing eIF4G cleavage correspond with higher apoptotic death in primary CLL samples (errors bars = S.E.M.).
DISCUSSION
By conducting RPPA analysis on primary CLL samples using 167 different antibody probes, we have been able to provide a comprehensive view of specific protein signaling and expression changes that take place in CLL. Specifically, we were able to determine from our selected samples that CLL contains a common protein expression signature that is seemingly more consistent among the CLL subtypes characterized by different genetic abnormalities. This common protein signature coincides with previous results demonstrating general overexpression of BCR-related signaling proteins, like BTK, regardless of a patient’s clinical subtype [21, 23, 24].
By further analyzing our clinical RPPA dataset through differential expression analysis between CLL and healthy B-cells, we revealed upregulation of proteins that are commonly associated with CLL pathogenesis, including apoptotic resistance (e.g. BCL2 upregulation, BAX downregulation, cleaved caspase-7 downregulation) and PI3K/AKT/mTOR pathway proteins (e.g. AKT, mTOR, IRS1, GAB2, p70S6K) [29, 31, 36, 39], while also identifying overexpression of oncogenic proteins that are not well classified in CLL (e.g. BRAF, STAT5A, DVL3). It is also interesting to note that upregulation of certain proteins were somewhat counterintuitive, specifically TSC1, TSC2, and PDCD4 as these proteins are typically associated with tumor suppression [75, 76]. However, these proteins are also negatively regulated by phosphorylation, and their phosphorylation states are not known based on our array study. Nevertheless, the differential protein expression patterns discovered in our RPPA samples provide further clarity and insight into how certain molecular mechanisms are altered in CLL.
Understanding the expression and activation states of numerous proteins simultaneously allows for better knowledge of how to target these oncogenic events in CLL. Within the realm of the AKT/mTOR pathway, we specifically determined that proteins involved in mRNA translational regulation were being upregulated in CLL patient samples, revealing a potential characteristic of CLL oncogenesis. These aberrant events regarding protein translation include overexpression of the translation initiator eIF4G and phosphorylation of the cap-dependent translation regulator 4E-BP1, an event directly regulated by mTOR activity [68]. These two events have previously been identified as drivers of other cancer types, like breast cancer and AML, yet neither event currently has been described as oncogenic factors in CLL [28, 69].
Nevertheless, the intriguing aspect of this translational machinery upregulation was its uncoupling from BCR-mediated AKT activation. Though total AKT and mTOR are upregulated in our RPPA dataset, the PI3K/AKT-related phosphorylation activity for these proteins appears to be negligible. This type of result is expected as the RPPA CLL patient samples were collected from the peripheral blood and are not in their active state, which occurs in the lymph node. However, 4E-BP1 serine 65 phosphorylation, an event that typically is regulated by the cascading phosphorylation of the PI3K/AKT/mTOR pathway [68], was hyperphosphorylated regardless of AKT activation as demonstrated by IgM stimulation comparisons. This result would suggest that 4E-BP1 phosphorylation, a typical indicator of translational activation, is potentially needed to help maintain survival of dormant CLL cells in the peripheral blood.
Currently, we do not know the specific mechanisms that mediate sustained 4E-BP1 phosphorylation in the absence of AKT activation. One possibility could be the dysregulation of phosphatases that mediate dephosphorylation of 4E-BP1. One particular phosphatase, PPM1G, has previously been identified as a 4E-BP1-specific phosphatase [77]. However, investigation into publically available gene expression and mutation datasets suggests that PPM1G is not highly altered in CLL patients [4, 78]. Nevertheless, dysregulation of other phosphatases could play a significant role in sustaining 4E-BP1 phosphorylation in CLL. However, we did observe in our RPPA dataset overexpression of the splicing protein SF2/ASF. SF2/ASF has previously been identified as an oncogenic mediator of mTOR-related translation activation in the absence of AKT activation [66, 79, 80]. Specifically, this protein has been shown to facilitate the phosphorylation of 4E-BP1, potentially by scaffolding together the cap-dependent mRNA translation complex and mTOR complex 1 [67]. Therefore, SF2/ASF may potentially mediate phosphorylation of 4E-BP1 in CLL B-cells even in the absence of AKT phosphorylation. Nevertheless, further studies are still required for determining the precise mechanisms that maintain 4E-BP1 phosphorylation in CLL.
This potential disconnect between AKT activation and 4E-BP1 activation provided rationale for investigating the effectiveness of simultaneously targeting both upstream and downstream of the PI3K/AKT/mTOR pathway in CLL. We examined this concept by determining the inhibitory capability of the dual PI3K/mTOR inhibitor NVP-BEZ235. From our results, we saw that 4E-BP1 and eIF4G were significantly altered in CLL cells treated with NVP-BEZ235 when compared to Ibrutinib and Idelalisib, two drugs that target upstream BCR-related effectors involved in PI3K/AKT activation [20, 21]. The inhibitory results of NVP-BEZ235 in CLL correspond with the effectiveness of NVP-BEZ235 treatment in other AKT/mTOR-driven cancer types, as well as with previous data demonstrating suppression of eIF4G activity and 4E-BP1 phosphorylation in AML [69, 71].
Thus, with the coordinating results of our RPPA and inhibitor treatment comparisons in CLL patient samples, we demonstrate a rationale for considering mTOR-mediated mRNA translation as a therapeutic target in CLL. This general idea of targeting the mTOR pathway with rapamycin-based inhibitors has previously been investigated in CLL. However, current understanding of the rapamycin-induced negative feedback loop, which causes increased AKT signaling, provides explanation of why treating with single-agent rapalogs may have been ineffective in the clinical setting [81, 82]. More recent evidence has demonstrated the effectiveness of using the eIF4-mimetic 4EG-1 as a means to sensitize CLL cells towards apoptosis in pre-clinical studies, subsequently underlining the critical role of mRNA translation in CLL pathogenesis [83]. Based on our corresponding results that demonstrate the overexpression of eIF4G, the phosphorylation of 4E-BP1, and the subsequent reversal of both events in NVP-BEZ235-mediated CLL apoptosis, we provide further insight into the potential interplay between mRNA translation activation and CLL survival as well as demonstrate a proper strategy for effectively targeting this aberrant molecular mechanism in CLL.
Materials and METHODS
Reverse Phase Protein Array & Immunoblotting
Peripheral blood samples from CLL patients were obtained with patient consent and approved for use by the Institutional Review Board of Georgia Regents University. Peripheral blood samples from normal donors were purchased from a local blood bank. Once blood samples were obtained, CD19+ B-cells were isolated using the RosetteSepTM B-cell isolation kit (STEMCELL Technologies, Vancouver, Canada). The isolated B-cells were then prepared in RIPA buffer supplemented with proteasome inhibitor and phosphatase inhibitor (Pierce, Rockford, IL, USA). The lysates were prepared to provide 1-1.5mg/ml of total protein lysate. RPPA analysis of the prepared lysates was then performed as previously described [49]. For this particular study, 118 total and 49 phospho-specific antibodies were used to determine expression and phosphorylation levels of the corresponding proteins (list of antibodies in Supplementary File S1). Validation immunoblotting for selected probes was performed by running 30ug of B-cell lysates on 4-20% gradient polyacrylamide gels (Bio-Rad, Hercules, CA, USA), transferred onto PVDF membranes (EMD Millipore, Billerica, MA, USA), and probed using corresponding primary antibodies from the RPPA study. Primary antibodies were detected with either an anti-mouse 680 red fluorescent antibody or an ant-rabbit 800 green fluorescent antibody (Pierce, Rockford, IL, USA). Immunoblot detection was performed using an LI-COR Odyssey (LI-COR Biosciences, Lincoln, NE, USA).
Cell culture, reagents, and kinase inhibitors
All cell culture experiments were maintained in RPM1640 Medium with 1% Penn/Strep (Hyclone, Waltham, MA, USA) and 10% Human Serum (Mediatech, Manassas, VA, USA). For co-culture experiments, primary CLL cells were seeded at 1X107 cells per well of a 6-well dish and co-cultured with either Huh7.5 cells or HS-5 cells at a density of 1X106 cells per well of a 6-well dish. NVP-BEZ235, Ibrutinib (PCI-32765), and Idelalisib (GS-1101) were obtained from Selleck Biochem (Houston, TX, USA). Inhibitors were reconstituted in DMSO in order to treat primary CLL cells at a 10uM concentration.
Cell proliferation, cell viability and apoptosis
Cell proliferation of the MEC1 CLL cell line was measured using the PrestoBlue Cell Viability reagent (Invitrogen, Waltham, MA, USA). Briefly, 25,000 MEC1 cells were treated with Ibrutinib, Idelalisib, or NVP-BEZ235 for 72 hours with a log-fold increase in concentration for each inhibitor. After 72 hours, MEC1 cells were incubated with PrestoBlue reagent for 2 hours and measured on a Spectramax Plus fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA) in order to determine an IC50 for each inhibitor in MEC1.
Cell viability and apoptosis of inhibitor-treated primary CLL cells was assessed by Annexin V+/DAPI apoptotic assay using an Annexin V-FITC antibody (Biolegend, San Diego, CA, USA) and DAPI staining (Sigma-Aldrich, St. Louis, MO, USA). Annexin V and DAPI staining of primary cells were measured using an LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Statistical analysis
Hierarchical clustering analysis for RPPA was performed using Partek Genomics Suite 6.6 (St. Louis, MO, USA) and pathway analysis was performed using Ingenuity Pathway Analysis (Qiagen, Hilden, Germany). Statistical measures for individual protein analysis and apoptotic studies were assessed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA).
Acknowledgments
The authors would like to acknowledge Judith Giri at the GRU Cancer Center Tumor Bank for coordinating proper obtainment of patient blood samples. The authors would also like to acknowledge the MD Anderson Cancer Center RPPA Core Facility for their technical service and support in performing the RPPA. This work was supported by NIH Grant R21CA185833 and the GRU Cancer Center Translational Science Award. H.S. is a Georgia Research Alliance Distinguished Cancer Scientist.
conflicts of interest
The authors declare no competing financial interests for this work.
References
1. Furman RR. Prognostic markers and stratification of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2010; 2010:77-81.
2. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA, Vartanov A, Fernandes SM, Saksena G, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013; 152:714-726.
3. Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, Stevenson K, Sougnez C, Wang L, Li S, Kotliar D, Zhang W, Ghandi M, Garraway L, Fernandes SM, Livak KJ, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014; 26:813-825.
4. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, Lopez-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012; 44:47-52.
5. Cortese D, Sutton LA, Cahill N, Smedby KE, Geisler C, Gunnarsson R, Juliusson G, Mansouri L and Rosenquist R. On the way towards a ‘CLL prognostic index’: focus on TP53, BIRC3, SF3B1, NOTCH1 and MYD88 in a population-based cohort. Leukemia. 2014; 28:710-713.
6. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, Bassaganyas L, Baumann T, Juan M, Lopez-Guerra M, Colomer D, Tubio JM, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011; 475:101-105.
7. Duzkale H, Schweighofer CD, Coombes KR, Barron LL, Ferrajoli A, O’Brien S, Wierda WG, Pfeifer J, Majewski T, Czerniak BA, Jorgensen JL, Medeiros LJ, Freireich EJ, Keating MJ and Abruzzo LV. LDOC1 mRNA is differentially expressed in chronic lymphocytic leukemia and predicts overall survival in untreated patients. Blood. 2011; 117:4076-4084.
8. Jantus Lewintre E, Reinoso Martin C, Montaner D, Marin M, Jose Terol M, Farras R, Benet I, Calvete JJ, Dopazo J and Garcia-Conde J. Analysis of chronic lymphotic leukemia transcriptomic profile: differences between molecular subgroups. Leuk Lymphoma. 2009; 50:68-79.
9. Eisele L, Prinz R, Klein-Hitpass L, Nuckel H, Lowinski K, Thomale J, Moeller LC, Duhrsen U and Durig J. Combined PER2 and CRY1 expression predicts outcome in chronic lymphocytic leukemia. Eur J Haematol. 2009; 83:320-327.
10. Hanoun M, Eisele L, Suzuki M, Greally JM, Huttmann A, Aydin S, Scholtysik R, Klein-Hitpass L, Duhrsen U and Durig J. Epigenetic silencing of the circadian clock gene CRY1 is associated with an indolent clinical course in chronic lymphocytic leukemia. PLoS One. 2012; 7:e34347.
11. Cahill N, Bergh AC, Kanduri M, Goransson-Kultima H, Mansouri L, Isaksson A, Ryan F, Smedby KE, Juliusson G, Sundstrom C, Rosen A and Rosenquist R. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia. 2013; 27:150-158.
12. Mansouri L, Gunnarsson R, Sutton LA, Ameur A, Hooper SD, Mayrhofer M, Juliusson G, Isaksson A, Gyllensten U and Rosenquist R. Next generation RNA-sequencing in prognostic subsets of chronic lymphocytic leukemia. Am J Hematol. 2012; 87:737-740.
13. Mansouri M, Sevov M, Fahlgren E, Tobin G, Jondal M, Osorio L, Roos G, Olivecrona G and Rosenquist R. Lipoprotein lipase is differentially expressed in prognostic subsets of chronic lymphocytic leukemia but displays invariably low catalytical activity. Leuk Res. 2010; 34:301-306.
14. Maloum K, Settegrana C, Chapiro E, Cazin B, Lepretre S, Delmer A, Leporrier M, Dreyfus B, Tournilhac O, Mahe B, Nguyen-Khac F, Lesty C, Davi F and Merle-Beral H. IGHV gene mutational status and LPL/ADAM29 gene expression as clinical outcome predictors in CLL patients in remission following treatment with oral fludarabine plus cyclophosphamide. Ann Hematol. 2009; 88:1215-1221.
15. Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, Speir E, Awan F, Tae H, Arthur G, Schnabel JL, Taylor KH, Wang X, Xu D, Ding HF, Munn DH, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012; 7:567-578.
16. Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, Barberan-Soler S, Papasaikas P, Jares P, Bea S, Rico D, Ecker S, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012; 44:1236-1242.
17. Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A and Rosenquist R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010; 115:296-305.
18. Agathangelidis A, Ntoufa S and Stamatopoulos K. B cell receptor and antigens in CLL. Adv Exp Med Biol. 2013; 792:1-24.
19. Stevenson FK, Krysov S, Davies AJ, Steele AJ and Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011; 118:4313-4320.
20. Robak T and Robak P. BCR signaling in chronic lymphocytic leukemia and related inhibitors currently in clinical studies. Int Rev Immunol. 2013; 32:358-376.
21. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012; 7:26-33.
22. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, Gibellini F, Njuguna N, Lee E, Stennett L, Raghavachari N, Liu P, McCoy JP, Raffeld M, Stetler-Stevenson M, Yuan C, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011; 117:563-574.
23. Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy JJ, Hamdy A, Johnson AJ and Byrd JC. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011; 117:6287-6296.
24. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013; 369:32-42.
25. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014; 370:997-1007.
26. Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, Spurgeon SE, Kahl BS, Bello C, Webb HK, Johnson DM, Peterman S, Li D, Jahn TM, Lannutti BJ, Ulrich RG, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014; 123:3390-3397.
27. Fiorcari S, Brown WS, McIntyre BW, Estrov Z, Maffei R, O’Brien S, Sivina M, Hoellenriegel J, Wierda WG, Keating MJ, Ding W, Kay NE, Lannutti BJ, Marasca R and Burger JA. The PI3-kinase delta inhibitor idelalisib (GS-1101) targets integrin-mediated adhesion of chronic lymphocytic leukemia (CLL) cell to endothelial and marrow stromal cells. PLoS One. 2013; 8:e83830.
28. Fukuchi-Shimogori T, Ishii I, Kashiwagi K, Mashiba H, Ekimoto H and Igarashi K. Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res. 1997; 57:5041-5044.
29. Li G, He S, Chang L, Lu H, Zhang H and Chiu J. GADD45alpha and annexin A1 are involved in the apoptosis of HL-60 induced by resveratrol. Phytomedicine. 2011; 18:704-709.
30. Zhu F, Wang Y, Zeng S, Fu X, Wang L and Cao J. Involvement of annexin A1 in multidrug resistance of K562/ADR cells identified by the proteomic study. Omics. 2009; 13:467-476.
31. King D, Pringle JH, Hutchinson M and Cohen GM. Processing/activation of caspases, -3 and -7 and -8 but not caspase-2, in the induction of apoptosis in B-chronic lymphocytic leukemia cells. Leukemia. 1998; 12:1553-1560.
32. Talab F, Allen JC, Thompson V, Lin K and Slupsky JR. LCK is an important mediator of B-cell receptor signaling in chronic lymphocytic leukemia cells. Mol Cancer Res. 2013; 11:541-554.
33. Spurgeon SE, Coffey G, Fletcher LB, Burke R, Tyner JW, Druker BJ, Betz A, DeGuzman F, Pak Y, Baker D, Pandey A, Hollenbach SJ, Sinha U and Loriaux MM. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013; 344:378-387.
34. Hoellenriegel J, Coffey GP, Sinha U, Pandey A, Sivina M, Ferrajoli A, Ravandi F, Wierda WG, O’Brien S, Keating MJ and Burger JA. Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia. 2012; 26:1576-1583.
35. Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholome K, Burger M, Chevalier N, Vallat L, Timmer J, Gribben JG, Jumaa H, Veelken H, Dierks C and Zirlik K. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009; 69:5424-5432.
36. Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD and Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007; 117:112-121.
37. Masood A, Chitta K, Paulus A, Khan AN, Sher T, Ersing N, Miller KC, Manfredi D, Ailawadhi S, Borrelo I, Lee KP and Chanan-Khan A. Downregulation of BCL2 by AT-101 enhances the antileukaemic effect of lenalidomide both by an immune dependant and independent manner. Br J Haematol. 2012; 157:59-66.
38. Paterson A, Mockridge CI, Adams JE, Krysov S, Potter KN, Duncombe AS, Cook SJ, Stevenson FK and Packham G. Mechanisms and clinical significance of BIM phosphorylation in chronic lymphocytic leukemia. Blood. 2012; 119:1726-1736.
39. Scarfo L and Ghia P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett. 2013; 155:36-39.
40. Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, Bartoli A, Coaccioli S, Screpanti I and Marconi P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009; 113:856-865.
41. Baldoni S, Sportoletti P, Del Papa B, Aureli P, Dorillo E, Rosati E, Ciurnelli R, Marconi P, Falzetti F and Di Ianni M. NOTCH and NF-kappaB interplay in chronic lymphocytic leukemia is independent of genetic lesion. Int J Hematol. 2013; 98:153-157.
42. Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ and Cheng EH. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010; 330:1390-1393.
43. Jiang M, Pabla N, Murphy RF, Yang T, Yin XM, Degenhardt K, White E and Dong Z. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J Biol Chem. 2007; 282:2636-2645.
44. Pohland T, Wagner S, Mahyar-Roemer M and Roemer K. Bax and Bak are the critical complementary effectors of colorectal cancer cell apoptosis by chemopreventive resveratrol. Anticancer Drugs. 2006; 17:471-478.
45. Kafka A, Tomas D, Beros V, Pecina HI, Zeljko M and Pecina-Slaus N. Brain metastases from lung cancer show increased expression of DVL1, DVL3 and beta-catenin and down-regulation of E-cadherin. Int J Mol Sci. 2014; 15:10635-10651.
46. Haddad BR, Gu L, Mirtti T, Dagvadorj A, Vogiatzi P, Hoang DT, Bajaj R, Leiby B, Ellsworth E, Blackmon S, Ruiz C, Curtis M, Fortina P, Ertel A, Liu C, Rui H, et al. STAT5A/B gene locus undergoes amplification during human prostate cancer progression. Am J Pathol. 2013; 182:2264-2275.
47. Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, Mateus C, Routier E, Kamsu-Kom N, Agoussi S, Eggermont AM, Desaubry L, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014; 513:105-109.
48. Cagnol S and Rivard N. Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual-specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene. 2013; 32:564-576.
49. Lu Y, Muller M, Smith D, Dutta B, Komurov K, Iadevaia S, Ruths D, Tseng JT, Yu S, Yu Q, Nakhleh L, Balazsi G, Donnelly J, Schurdak M, Morgan-Lappe S, Fesik S, et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene. 2011; 30:4567-4577.
50. Ringshausen I, Peschel C and Decker T. Mammalian target of rapamycin (mTOR) inhibition in chronic lymphocytic B-cell leukemia: a new therapeutic option. Leuk Lymphoma. 2005; 46:11-19.
51. Jung CH, Ro SH, Cao J, Otto NM and Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010; 584:1287-1295.
52. Annovazzi L, Mellai M, Caldera V, Valente G, Tessitore L and Schiffer D. mTOR, S6 and AKT expression in relation to proliferation and apoptosis/autophagy in glioma. Anticancer Res. 2009; 29:3087-3094.
53. Zeng H and Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol. 2013; 25:347-355.
54. Cornu M, Albert V and Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013; 23:53-62.
55. Cuconati A, Mills C, Goddard C, Zhang X, Yu W, Guo H, Xu X and Block TM. Suppression of AKT anti-apoptotic signaling by a novel drug candidate results in growth arrest and apoptosis of hepatocellular carcinoma cells. PLoS One. 2013; 8:e54595.
56. Rassidakis GZ, Feretzaki M, Atwell C, Grammatikakis I, Lin Q, Lai R, Claret FX, Medeiros LJ and Amin HM. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005; 105:827-829.
57. Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM and Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003; 23:7315-7328.
58. Whiteman EL, Cho H and Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002; 13:444-451.
59. Dunn GP, Cheung HW, Agarwalla PK, Thomas S, Zektser Y, Karst AM, Boehm JS, Weir BA, Berlin AM, Zou L, Getz G, Liu JF, Hirsch M, Vazquez F, Root DE, Beroukhim R, et al. In vivo multiplexed interrogation of amplified genes identifies GAB2 as an ovarian cancer oncogene. Proc Natl Acad Sci U S A. 2014; 111:1102-1107.
60. Bocanegra M, Bergamaschi A, Kim YH, Miller MA, Rajput AB, Kao J, Langerod A, Han W, Noh DY, Jeffrey SS, Huntsman DG, Borresen-Dale AL and Pollack JR. Focal amplification and oncogene dependency of GAB2 in breast cancer. Oncogene. 2010; 29:774-779.
61. Daly RJ, Gu H, Parmar J, Malaney S, Lyons RJ, Kairouz R, Head DR, Henshall SM, Neel BG and Sutherland RL. The docking protein Gab2 is overexpressed and estrogen regulated in human breast cancer. Oncogene. 2002; 21:5175-5181.
62. Hao Y, Zhao S and Wang Z. Targeting the protein-protein interaction between IRS1 and mutant p110alpha for cancer therapy. Toxicol Pathol. 2014; 42:140-147.
63. Porter HA, Perry A, Kingsley C, Tran NL and Keegan AD. IRS1 is highly expressed in localized breast tumors and regulates the sensitivity of breast cancer cells to chemotherapy, while IRS2 is highly expressed in invasive breast tumors. Cancer Lett. 2013; 338:239-248.
64. Im-aram A, Farrand L, Bae SM, Song G, Song YS, Han JY and Tsang BK. The mTORC2 component rictor contributes to cisplatin resistance in human ovarian cancer cells. PLoS One. 2013; 8:e75455.
65. McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, Wederell ED, Bally MB, Foster LJ and Dedhar S. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 2008; 68:1618-1624.
66. Karni R, Hippo Y, Lowe SW and Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008; 105:15323-15327.
67. Michlewski G, Sanford JR and Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell. 2008; 30:179-189.
68. Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N and Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998; 12:502-513.
69. Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, Pannetier M, Willems L, Park S, Macone A, Maira SM, Ifrah N, Dreyfus F, Herault O, Lacombe C, Mayeux P, et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010; 16:5424-5435.
70. Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ, Giese NA, Zhang X, Wei L, Byrd JC and Johnson AJ. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010; 116:2078-2088.
71. Shehata M, Schnabl S, Demirtas D, Tauber S, Hilgarth M, Bilban M, Vanura K, Porpaczy E, Fonatsch C, Stilgenbauer S, Schwarzmeier JD, Hubmann R, Gaiger A, Zielinski C, Maira S-M, Garcia-Echeverria C, et al. Effective Targeting of the PI3-K Pathway in CLL with NVP-BEZ235, a Novel Orally Available Dual PI3K/mTOR Inhibitor. ASH Annual Meeting Abstracts. 2008; 112:3166-.
72. Titanji K, Velu V, Chennareddi L, Vijay-Kumar M, Gewirtz AT, Freeman GJ and Amara RR. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Invest. 2010; 120:3878-3890.
73. Bushell M, Poncet D, Marissen WE, Flotow H, Lloyd RE, Clemens MJ and Morley SJ. Cleavage of polypeptide chain initiation factor eIF4GI during apoptosis in lymphoma cells: characterisation of an internal fragment generated by caspase-3-mediated cleavage. Cell Death Differ. 2000; 7:628-636.
74. Badura M, Braunstein S, Zavadil J and Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci U S A. 2012; 109:18767-18772.
75. Vikhreva PN, Shepelev MV and Korobko IV. mTOR-dependent transcriptional repression of Pdcd4 tumor suppressor in lung cancer cells. Biochim Biophys Acta. 2014; 1839:43-49.
76. Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL and Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003; 11:1457-1466.
77. Liu J, Stevens PD, Eshleman NE and Gao T. Protein phosphatase PPM1G regulates protein translation and cell growth by dephosphorylating 4E binding protein 1 (4E-BP1). J Biol Chem. 2013; 288:23225-23233.
78. Ferreira PG, Jares P, Rico D, Gomez-Lopez G, Martinez-Trillos A, Villamor N, Ecker S, Gonzalez-Perez A, Knowles DG, Monlong J, Johnson R, Quesada V, Djebali S, Papasaikas P, Lopez-Guerra M, Colomer D, et al. Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res. 2014; 24:212-226.
79. Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D and Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007; 14:185-193.
80. Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK and Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012; 19:220-228.
81. Wan X, Harkavy B, Shen N, Grohar P and Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007; 26:1932-1940.
82. Decker T, Sandherr M, Goetze K, Oelsner M, Ringshausen I and Peschel C. A pilot trial of the mTOR (mammalian target of rapamycin) inhibitor RAD001 in patients with advanced B-CLL. Ann Hematol. 2009; 88:221-227.
83. Willimott S, Beck D, Ahearne MJ, Adams VC and Wagner SD. Cap-translation inhibitor, 4EGI-1, restores sensitivity to ABT-737 apoptosis through cap-dependent and -independent mechanisms in chronic lymphocytic leukemia. Clin Cancer Res. 2013; 19:3212-3223.