
Introduction
Due to a multistep nature of the tumorigenesis process whose progression and completion requires an extended period of time, incidence rates of many cancers increase with age [1-3]. Therefore, cancer is considered as a disease associated with aging [3-7]. In the generally accepted paradigm of the relationship between aging and cancer, they share common aetiology (i.e., an age-related progressive accumulation of cellular damage) and have coalescent mechanisms [4-9]. A body of evidence supports the validity of this paradigm. First, aging and cancer indeed have convergent underlying mechanisms. They include: 1) a longevity-defining signaling network that integrates the pro-aging AMP-activated protein kinase/target of rapamycin (AMPK/TOR), cAMP/protein kinase A (cAMP/PKA) and insulin/insulin-like growth factor 1 (IGF-1) signaling pathways and also incorporates a sirtuin-governed protein deacetylation module [4-14]; 2) a cytoprotective process of autophagy [8-9,15-23]; and 3) tricarboxylic acid cycle, respiration, and reactive oxygen species (ROS) production and detoxification in mitochondria [24-32]. Second, some of the proteins implemented in such convergent mechanisms could function as oncoproteins, whereas others act as tumor suppressor proteins [3-6,10-21,24-33]. Third, certain pharmacological and dietary interventions exhibit both anti-aging and anti-cancer effects by modulating the longevity signaling network that integrates the AMPK/TOR, cAMP/PKA and insulin/IGF-1 pathways and incorporates the sirtuin-governed protein deacetylation module [4-7,10-14,33-52].
The interplay between aging and cancer is more complex than only sharing common aetiology and having convergent mechanisms. In some situations aging and cancer can have antagonistic aetiologies and divergent mechanisms [5,8,9,53-65]. Indeed, the age-dependent accumulation of DNA damage and mutations in normal somatic cells (especially in adult stem and progenitor cells) triggers telomere shortening and/or a gradual rise in the expression of the INK4a/ARF locus encoding the p16INK4a and p14ARF/p19ARF tumor suppressor proteins [53-56,58,62]. Both these processes reduce the proliferative potential of normal somatic cells, thereby promoting cellular senescence, causing a decline in tissue regeneration and repair, impairing tissue homeostasis, and ultimately accelerating cellular and organismal aging [53-58]. While both telomere shortening and enhanced expression of INK4a/ARF display pro-aging effects in normal somatic cells, they exhibit potent anti-cancer effects in tumor cells by reducing their proliferative potential [53-62]. Hence, an anti-cancer intervention that can limit the excessive proliferation of tumor cells by inhibiting telomerase or activating expression of INK4a/ARF could have a pro-aging effect on cellular and organismal levels [5,8,9,55-59,62-65].
The complexity of the interplay between aging and cancer is further underscored by the recent findings implying that tumor cells in the epithelia of breast cancers can cause “accelerated aging” of adjacent normal fibroblasts by stimulating their intracellular ROS production [66-77]. In response to the resulting oxidative stress these fibroblasts establish a pro-aging pattern by activating aerobic glycolysis and autophagic degradation, thereby providing epithelial cancer cells within the tumor microenvironment with lactate, ketone bodies and glutamine [67,70,76-79]. These catabolic and anabolic substrates support proliferation of epithelial cancer cells and, thus, accelerate tumor growth, progression and metastasis [67,76,77]. Further emphasizing the complexity of the relationship between aging and cancer, this model of breast cancer as an “accelerated host aging” disease defines autophagy (a cytoprotective anti-aging cellular process [8,9,15-23]) within cancer-associated fibroblasts as a pro-cancer process that supports the growth of already established tumors [67,76,77]. In contrast, by preventing initiation of some cancers, autophagy operates as an anti-cancer process prior to tumor establishment [8,9,15-23,80-85].
We found that lithocholic acid (LCA), a bile acid, delays chronological aging of yeast [86] known to mimic aging of postmitotic mammalian cells (e.g., neurons) [87-89]. In yeast, LCA extends longevity not by attenuating the pro-aging AMPK/TOR and cAMP/PKA signaling pathways [86], both of which operate as pro-cancer pathways in mice and humans [4-14]. Furthermore, we revealed that the longevity-extending effect of LCA in chronologically aging yeast depends on autophagy (Kyryakov et al., manuscript in preparation), a cytoprotective anti-aging process that in different situations can play either an anti-cancer or a pro-cancer role [8,9,15-23,80-85]. Moreover, LCA extends yeast chronological life span by altering the age-related dynamics of mitochondria-confined respiration and ROS production [86], both of which are integrated into convergent mechanisms underlying aging and cancer [24-32]. Additionally, we found that in chronologically aging yeast LCA slows down telomere shortening (Iouk et al., unpublished data), a pro-aging process that exhibits an anti-cancer effect [53-62]. Besides, we demonstrated that LCA not only extends the chronological life span of quiescent yeast but also hinders cellular quiescence and senescence by delaying replicative aging of yeast (Lindsay et al., manuscript in preparation), which mimics aging of mitotically active mammalian cells [87-89]. In sum, these findings imply that in yeast models of aging of mitotically active and inactive mammalian cells LCA extends longevity by modulating several processes integrated into either convergent or divergent mechanisms underlying aging and cancer.
We therefore sought to examine if LCA exhibits an anti-tumor effect in cultured human cancer cells by activating certain anti-cancer processes that may play an essential role in cellular aging. As a model for assessing such effect of LCA, we choose several cell lines of the human neuroblastoma (NB) tumor. NB is the most commonly diagnosed extra-cranial solid tumor among children [90-92]. All NBs originate from primordial neuroblast cells that eventually differentiate into the adrenal medulla and early sympathetic nervous system [92-95]. In over 70% of all cases studied so far, NBs metastasize to other tissues [90-95]. Although most patients diagnosed with non-metastasizing NBs have been reported to be cured, only less than 40% of those with cancerous migration to other tissues survive despite rigorous chemotherapeutic and surgical treatment [92]. The recurrent high-risk NB patients with an often fatal prognosis frequently contain the following two genetic abnormalities: 1) amplification of the MYCN gene [96-98] encoding a transcription factor involved in growth, cell metabolism and division [99]; and 2) deletion of the short arm of chromosome 1p [100], which reduces expression of numerous tumour-suppressing genes [101-104]. One of the main challenges of combating high-risk recurring NBs is the use of non-toxic agents that not only prevent tumor metastasis, but also eliminate the primary tumor. Currently, NBs are treated with a combination of the chemotherapeutic drugs that cause apoptotic death of malignant cancer cells by inducing DNA strand intercalation (doxorubicin, cisplatin, cyclophosphamide and topotecan), DNA strand breaks (epipodophyllotoxins) or mitotic inhibition (vincristine) [105-111]. Noteworthy, all these anti-NB drugs stimulate the release of significant amount of ROS from mitochondria [107-111].
Here we show that LCA has a potent anti-tumor effect in four lines of cultured human NB cells. We demonstrate that this anti-aging compound kills three of them by causing apoptotic cell death. Our mechanistic studies of the apoptosis-based death of NB cell cultures exposed to LCA provide evidence that this bile acid triggers both the intrinsic and extrinsic apoptotic death pathways by binding to the cell surface and initiating the converging intracellular cascades activating caspases-3, -6, -8 and -9. We propose a model for a mechanism underlying a potent and specific anti-tumor effect of LCA in cultured human NB cells. We also show that LCA has a broad anti-tumor effect on cultured cancer cells derived from different tissues and organisms.
Results
LCA selectively kills cultured human NB cells
The MTT (3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide) assay is widely used to determine viability of cultured mammalian cells following drug treatment [112-116]. In this assay, only viable, metabolically active cells are able to reduce the yellow tetrazolium salt of MTT to form a purple formazan product. Thus, the percentage of viable cells in the MTT assay is calculated as the proportion of the cell population displaying a detectable level of redox potential. Using this assay, we found that LCA exhibits a potent and selective anti-tumor effect in several lines of cultured human NB cells. In fact, this bile acid killed the NB cell lines BE(2)-m17, SK-n-SH, SK-n-MCIXC and Lan-1 if used at concentrations that were not cytotoxic or only mildly cytotoxic (as in case of Lan-1) to primary cultures of human neurons (Fig. 1A).
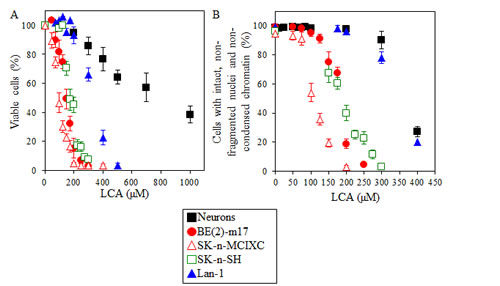
Figure 1: LCA selectively kills all four tested lines of cultured human NB cells - by causing an apoptosis-based death of BE(2)-m17, SK-n-SH and SK-n-MCIXC lines, but triggering a non-apoptotic death of Lan-1 line. In A, the percentage of viable cells was calculated as a portion of their population displaying a detectable level of redox potential which was monitored using the MTT (3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide)-based CellTiter 96 Non-Radioactive Cell Proliferation Assay. In B, the fluorescent dye Hoechst was used to visualize chromatin in various cell cultures and the percentage of viable non-apoptotic cells carrying intact, non-fragmented nuclei containing non-condensed chromatin was calculated; dead apoptotic cells carried fragmented nuclei containing condensed chromatin, a hallmark event of apoptotic death. A. LCA kills NB cell lines BE(2)-m17, SK-n-SH, SK-n-MCIXC and Lan-1 if used at concentrations that are not cytotoxic or only mildly cytotoxic (as in case of Lan-1) to primary cultures of human neurons. B. The NB cell line SK-n-MCIXC is the most sensitive to LCA-induced apoptotic death; two other lines, BE(2)-m17 and SK-n-SH, exhibit much higher sensitivity to LCA-induced apoptotic death than primary cultures of human neurons or the NB cell line Lan-1. Data are presented as means ± SD (n = 4-6).
Chromatin condensation and nuclear fragmentation are hallmark events of apoptosis that are not seen under any other modes of cell death [117-122]. We used the fluorescent dye Hoechst to visualize chromatin in various NB cell cultures and in primary cultures of human neurons exposed to LCA or remained untreated. For each cell culture, we calculated the percentage of viable non-apoptotic cells carrying intact, non-fragmented nuclei containing non-condensed chromatin. Dead apoptotic cells carried condensed and/or fragmented nuclei. Our data imply that if LCA is used at concentrations that are not cytotoxic to primary cultures of human neurons, it selectively kills the NB cell lines BE(2)-m17, SK-n-SH and SK-n-MCIXC by causing their apoptotic death. We found that SK-n-MCIXC is the most sensitive (as compared to other NB cell lines tested and especially as compared to primary cultures of human neurons) to the apoptotic death induced by LCA (Fig. 1B), as it is to the MTT-monitored cytotoxic effect of this bile acid (Fig. 1A). Furthermore, although BE(2)-m17 and SK-n-SH were less sensitive to LCA-induced apoptosis than SK-n-MCIXC, they exhibited much higher sensitivity to such form of cell death (Fig. 1B) - and to the MTT-monitored cytotoxic effect of LCA (Fig. 1A) - than primary cultures of human neurons or the NB cell line Lan-1. Importantly, although at the concentrations of LCA exceeding 200 µM the NB cell line Lan-1 was more sensitive to the MTT-monitored cytotoxic effect of LCA than primary cultures of human neurons (Fig. 1A), this cell line did not exhibit higher sensitivity to LCA-induced apoptotic cell death than primary neuron cultures (Fig. 1B). Thus, LCA selectively kills Lan-1 cells via a non-apoptotic cell death mechanism.
LCA sensitizes two NB cell lines to hydrogen peroxide-induced apoptotic cell death
Exogenously added hydrogen peroxide has been shown to cause mitochondria-controlled apoptotic cell death in a variety of cultured eukaryotic cells [123-128]. Our data imply that LCA protects primary cultures of human neurons, but not cultured human NB cell lines BE(2)-m17 or SK-n-MCIXC, against apoptosis induced in response to exogenously added 0.1 mM hydrogen peroxide (Fig. 2). In these experiments, chromatin condensation and nuclear fragmentation were visualized with the fluorescent dye Hoechst and were used as a measure of the efficacy of hydrogen peroxide-induced apoptotic cell death. Importantly, LCA greatly enhanced the susceptibility of BE(2)-m17 and SK-n-MCIXC to such a mitochondria-controlled form of cell death (compare Figs. 2B and 2C to Fig. 2A), even if it was used at concentrations that in the absence of hydrogen peroxide did not compromise their viability (compare Figs. 2B and 2C to Figs. 1A and 1B). Of note, while 75 μM LCA caused apoptotic death of all or most of the BE(2)-m17 and SK-n-MCIXC cells exposed to hydrogen peroxide, this concentration of LCA significantly increased the resistance of primary cultures of human neurons to the hydrogen peroxide-induced form of mitochondria-controlled apoptotic death (compare Fig. 2A to Figs. 2B and 2C). Based on these observations, it seems likely that an exposure of a mixed population of at least these two NB cell lines and non-cancerous neurons to low concentrations of simultaneously added hydrogen peroxide and LCA may concurrently 1) kill all or most of NB cells by causing their apoptotic cell death; and 2) promote the viability of non-cancerous neurons by increasing their resistance to hydrogen peroxide-induced apoptosis.
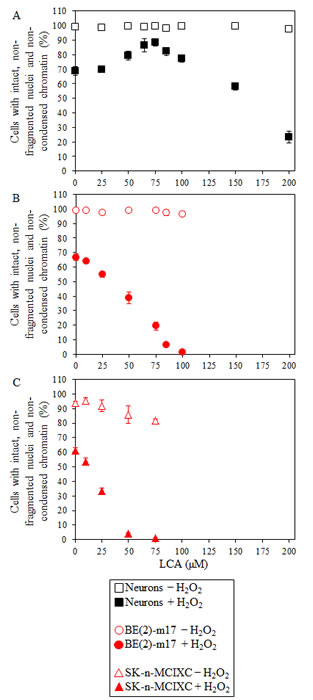
Figure 2: LCA increases sensitivity of cultured human NB cells to hydrogen peroxide-induced apoptotic cell death. A. If used at concentrations below 100 µM, LCA protects primary cultures of human neurons against mitochondria-controlled apoptosis induced in response to exogenously added 0.1 mM hydrogen peroxide. B and C. In contrast, if used at concentrations below 100 µM, LCA greatly enhances the susceptibility of cultured human NB cell lines BE(2)-m17 (B) and SK-n-MCIXC (C) to such hydrogen peroxide-induced form of cell death. While 75 μM LCA causes apoptotic death of all or most of the cells of BE(2)-m17 and SK-n-MCIXC exposed to 0.1 mM hydrogen peroxide (B and C, respectively), in this concentration it increases the resistance of primary cultures of human neurons to such form of cell death (A). Nuclear fragmentation and chromatin condensation were visualized with the fluorescent dye Hoechst and were used as a measure of the efficacy of hydrogen peroxide-induced apoptotic cell death. Each panel shows two datasets for the same cell type. One dataset is for cell aliquots treated with 0.1 mM hydrogen peroxide, whereas the other dataset is for cell aliquots remained untreated. For each cell culture, the percentage of viable non-apoptotic cells carrying intact, non-fragmented nuclei containing non-condensed chromatin was calculated. Data are presented as means ± SD (n = 3-5).
LCA triggers the intrinsic (mitochondrial) pathway of apoptotic death in two NB cell lines
As described above, LCA selectively kills the NB cell lines BE(2)-m17 and SK-n-MCIXC by causing apoptosis and enhances their susceptibility to a hydrogen peroxide-induced form of mitochondria-controlled apoptosis. We therefore sought to investigate if LCA kills these two NB cell lines by activating the intrinsic apoptotic cell death pathway. The key feature of such a pathway is mitochondrial outer membrane permeabilization (MOMP), which leads to the release of cytochrome c (and several other pro-apoptotic proteins) from the mitochondrial intermembrane space into the cytosol [129-131]. Following its efflux from mitochondria, cytochrome c binds an apoptotic protease-activating factor 1 (APAF-1) monomer whose oligomerization into the heptameric apoptosome complex recruits caspase-9 and ultimately activates this initiator caspase [119,131-133]. We found that LCA significantly increases caspase-9 activity in BE(2)-m17 and SK-n-MCIXC (Fig. 3). Furthermore, in both these NB cell lines LCA caused the fragmentation of a mitochondrial network (Figs. 4A and 4B). Such fragmentation is one of the earliest events of the mitochondrial pathway of apoptosis; it occurs before caspase-9 activation, around the point of MOMP and cytochrome c efflux from mitochondria [119,129-131,134,135]. Moreover, in both these cell lines LCA triggered the dissipation of the electrochemical potential across the inner mitochondrial membrane (Figs. 4C and 4D). The gradual dissipation and eventual loss of mitochondrial transmembrane potential is a hallmark event of mitochondria-dependent apoptotic cell death; it follows MOMP and occurs in both caspase-dependent and caspase-independent fashion [119,130,131,136-139]. Of note, the higher LCA-induced activity of caspase-9 (Fig. 3) and efficacies of both LCA-promoted mitochondrial fragmentation and LCA-triggered mitochondrial inner membrane depolarization (Figs. 4B and 4D) observed in SK-n-MCIXC cells, as compared to those seen in BE(2)-m17 cells, correlated with the higher sensitivity of SK-n-MCIXC (as compared to that of BE(2)-m17) to the cytotoxic, apoptotic cell death-inducing and hydrogen peroxide-sensitizing effects of LCA (Figs. 1 and 2).
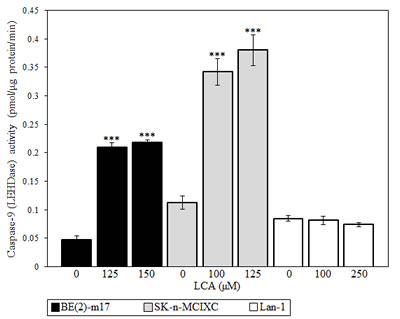
Figure 3: LCA significantly increases the activity of the initiator caspase-9 in cultured human NB cell lines BE(2)-m17 and SK-n-MCIXC. Specific activity of caspase-9 was measured as described in Materials and Methods and expressed in picomoles of fluorescent compound 7-amino-4-trifluoromethyl coumarin released per microgram of protein per minute, based on the linear range of the curve. Data are presented as means ± SD (n = 3-4); ***p<0.001.
Importantly, although LCA stimulated caspase-9 in BE(2)-m17 and SK-n-MCIXC cells, it did not increase caspase-9 activity in Lan-1 cells (Fig. 3). Furthermore, if used at a concentration (i.e., 150 μM or 175 μM) that caused mitochondrial fragmentation in most or all BE(2)-m17 and SK-n-MCIXC cells, LCA did not stimulate such fragmentation in Lan-1 cells (Fig. 4B). Moreover, if used at a concentration (i.e., 150 μM or 200 μM) that triggered mitochondrial inner membrane depolarization in most or all BE(2)-m17 and SK-n-MCIXC cells, LCA did not cause such depolarization in Lan-1 cells (Fig. 4D). These findings further validate our conclusion (see above) that LCA kills Lan-1 cells via a non-apoptotic cell death mechanism.
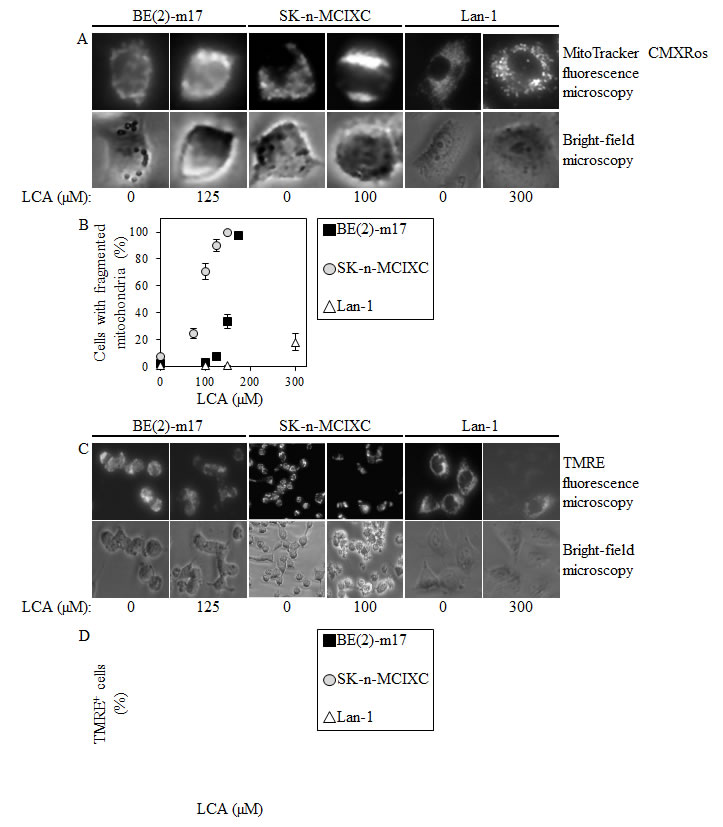
Figure 4: In cultured human NB cell lines BE(2)-m17 and SK-n-MCIXC (but not in Lan-1), LCA causes mitochondrial fragmentation and triggers the dissipation of the electrochemical potential across the inner mitochondrial membrane. A. Mitochondrial morphology in NB cells treated with LCA or remained untreated was visualized using MitoTracker Red CMXRos as described in Materials and Methods. Cells were viewed using fluorescence microscopy. B. The percentage of cells displaying fragmented mitochondria was calculated. C. The mitochondrial membrane potential (∆Ψ) in NB cells treated with LCA or remained untreated was measured using tetramethylrhodamine ethyl ester (TMRE) as described in Materials and Methods; the extent of reversible sequestration of TMRE by mitochondria is proportional to the value of ∆Ψ. Cells were incubated with 50 nM TMRE for 20 min and directly viewed using fluorescence microscopy. D. The percentage of TMRE-positive cells displaying a detectable level of ∆Ψ was calculated. Data in B and D are presented as means ± SD (n = 3-4).
LCA increases activities of caspases-3 and -6 in two NB cell lines
The apoptosome-driven activation of caspase-9 during mitochondria-controlled apoptosis initiates a stepwise activation of several executioner caspases. In one scenario, the caspase-9-dependent proteolytic activation of the executioner caspases-3 and -7 is followed by the caspase-3-driven proteolytic activation of the executioner caspases-2 and -6 and then by the caspase-6-enabled proteolytic activation of the executioner caspase-10; when all these caspases become activated, they complete the demolition phase of the mitochondria-controlled apoptotic program by cleaving their respective protein substrates and producing the morphological features characteristic of apoptotic cell death [119,130,131,140,141]. In another scenario, caspase-6 is activated independent of caspase-3 [142-151], either by caspase-1 [148] or through intramolecular self-cleavage [152-154], and does not induce apoptosis [143,144,149,152,155].
We found that LCA increases caspase-3 activity in BE(2)-m17 and SK-n-MCIXC (Fig. 5A) by promoting a proteolytic conversion of a zymogen pro-caspase-3 form into an active 17 kDa form (Fig. 5B). Importantly, the anti-tumor effect of LCA in these two cell lines was due in part to its ability to activate caspase-3. In fact, z-DEVD-fmk, a potent and specific inhibitor of caspase-3 (Fig. 5D), significantly reduced the efficacy with which LCA caused selective killing of BE(2)-m17 and SK-n-MCIXC cells (Fig. 5E). The observed incomplete reduction by z-DEVD-fmk of the anti-tumor efficacy of LCA in BE(2)-m17 and SK-n-MCIXC (Fig. 5E) suggests that the LCA-triggered, mitochondria-driven apoptotic death of both these cell lines could be due not only to a caspase-dependent mechanism, but also to a caspase-independent decline in mitochondrial function, which may lead to the observed LCA-dependent mitochondrial fragmentation and loss of mitochondrial transmembrane potential (Fig. 4). In BE(2)-m17 and SK-n-MCIXC cells, LCA also elevated caspase-6 activity (Fig. 6A) and promoted a proteolytic conversion of a zymogen pro-caspase-6 form of this caspase into its large (p20) and small (p10) subunits (Fig. 6B) known to assemble into an enzymatically active heterotetramer of two p20 and two p10 [152,156]. Of note, the different efficacies of the LCA-induced proteolytic activation of caspase-3 and caspase-6 seen in BE(2)-m17 and SK-n-MCIXC cells (Figs. 5 and 6) correlated with their dissimilar sensitivities to the cytotoxic, apoptosis-inducing and hydrogen peroxide-sensitizing effects of LCA (Figs. 1 and 2). Furthermore, even if used at a higher concentration (i.e., 250 μM) than the ones causing the highest extent of the LCA-induced proteolytic activation of caspase-3 and caspase-6 (i.e., 125 μM or 150 μM) in SK-n-MCIXC and BE(2)-m17 cells, LCA did not promote their proteolysis-driven activation in Lan-1 cells (Figs. 5 and 6), whose LCA-induced death was not due to apoptosis (see above).
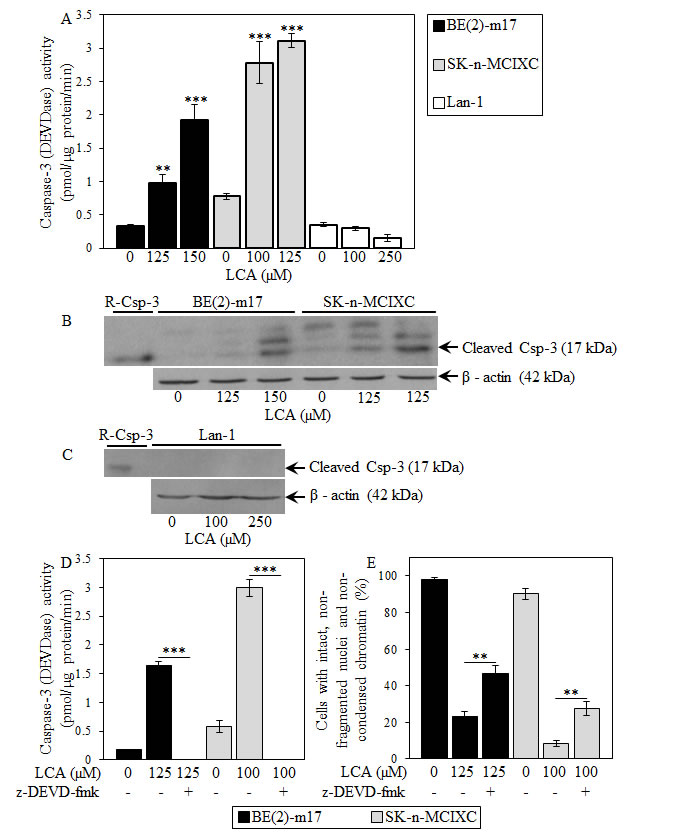
Figure 5: LCA increases activity of the executioner caspase-3 in cultured human NB cell lines BE(2)-m17 and SK-n-MCIXC (but not in Lan-1) by stimulating a proteolytic conversion of its zymogen pro-caspase-3 form into its enzymatically active 17 kDa form. The anti-tumor effect of LCA in BE(2)-m17 and SK-n-MCIXC cell lines is due in part to its ability to increase caspase-3 activity. A and D. Specific activity of caspase-3 was measured in NB cells treated with LCA and/or z-DEVD-fmk (a potent and specific inhibitor of caspase-3) or remained untreated as described in Materials and Methods and expressed in picomoles of fluorescent compound 7-amino-4-trifluoromethyl coumarin released per microgram of protein per minute. B and C. Pro-caspase-3 cleavage assay was carried out as described in Materials and Methods. E. The fluorescent dye Hoechst was used to visualize chromatin in NB cell cultures treated with LCA and/or z-DEVD-fmk or remained untreated. For each cell culture, the percentage of viable non-apoptotic cells carrying intact, non-fragmented nuclei containing non-condensed chromatin was calculated; dead apoptotic cells carried fragmented nuclei containing condensed chromatin, a hallmark event of apoptotic death. Data in A, D and E are presented as means ± SD (n = 3-4); **p<0.01; ***p<0.001.
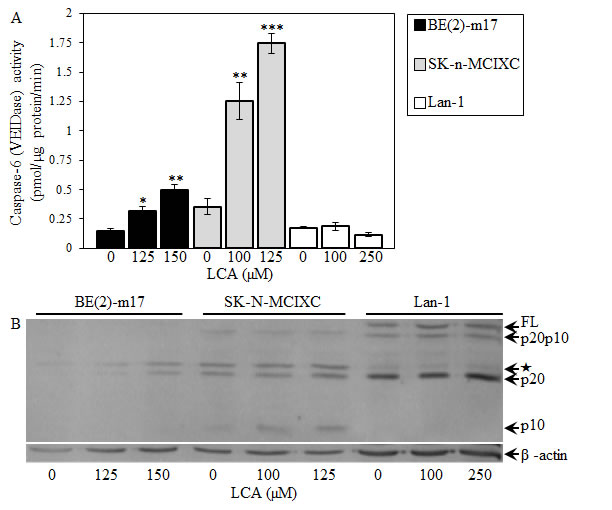
Figure 6: LCA increases activity of the executioner caspase-6 in cultured human NB cell lines BE(2)-m17 and SK-n-MCIXC (but not in Lan-1) by stimulating a proteolytic conversion of a zymogen pro-caspase-6 form into its large and small subunits known to assemble into an enzymatically active heterotetramer consisted of the two of each subunits. A. Specific activity of caspase-6 was measured as described in Materials and Methods and expressed in picomoles of fluorescent compound 7-amino-4-trifluoromethyl coumarin released per microgram of protein per minute. B. Pro-caspase-6 cleavage assay was carried out as described in Materials and Methods. The anti-human-caspase-6 mouse monoclonal IgG1 clone B93-4 antibodies raised against amino acids 271-285 (Pharmingen) were used; these antibodies specifically recognize the large (p20) and small (p10) subunits of an enzymatically active form of caspase-6. Abbreviations: FL, a full-length zymogen form of pro-caspase-6; p20p10, caspase-6 lacking the pro-domain; p20 and p10, large and small subunits (respectively) of caspase-6 lacking the pro- and linker-domains; , an unidentified protein band, perhaps p20-linker (p23), pro-p20 (p23) or pro-p20-linker subunits of caspase-6 [152]. Data in A are presented as means ± SD (n = 3-4); *p<0.05; **p<0.01; ***p<0.001.
LCA increases the activity of the initiator caspase-8 in two NB cell lines
The proteolytic cleavage of pro-caspase-3 and the resulting increase of caspase-3 activity can be triggered not only through the intrinsic (mitochondrial) apoptotic death pathway but also through the extrinsic (death receptor) pathway of apoptosis. This extrinsic pathway is initiated when specific extracellular death ligands cause the ligation of death receptors in the plasma membrane, thus triggering a process eventually leading to activation of the initiator caspase-8 [119,131]. In addition to its role in the proteolytic activation of caspase-3, activated in response to death receptor ligation caspase-8 can also cleave and activate the BH3-only protein BID, thereby causing MOMP and initiating the intrinsic pathway of apoptosis [119,131,157,158].
We found that LCA significantly increases the activity of caspase-8 in BE(2)-m17 and SK-n-MCIXC (Fig. 7). As it was observed for caspases-3, -6 and -9 (Figs. 5A, 6A and 3, respectively), the extent to which caspase-8 was activated by LCA in these two cell lines (Fig. 7) correlated with the degree of their sensitivity to the cytotoxic, apoptosis-inducing and hydrogen peroxide-sensitizing effects of LCA (Figs. 1 and 2). Importantly, unlike the stimulating effect of LCA on caspase-8 seen in BE(2)-m17 and SK-n-MCIXC cells, this bile acid did not stimulate caspase-8 activity in Lan-1 cells (Fig. 7). These findings suggest that LCA could kill both BE(2)-m17 and SK-n-MCIXC cell lines by activating not only the intrinsic apoptotic death pathway dependent on the initiator caspase-9, but also the extrinsic pathway of apoptotic death driven by the initiator caspase-8. We therefore propose that the LCA-dependent activation of caspase-8 seen in BE(2)-m17 and SK-n-MCIXC cells (Fig. 7) could be responsible not only for the direct proteolytic activation of caspase-3 (Figs. 5A and 5B), but also for MOMP (perhaps, due to BID cleavage by caspase-8) and the resulting initiation of the intrinsic apoptotic death pathway observed in these NB cells (Figs. 3 and 4). These caspase-8-driven events in BE(2)-m17 and SK-n-MCIXC cells are expected to enhance the extent of their demise in response to LCA treatment.
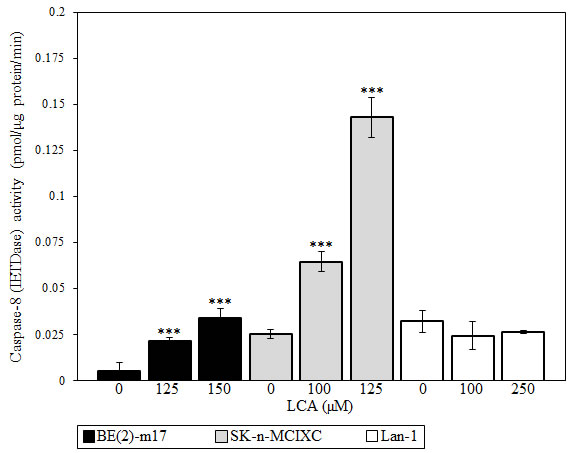
Figure 7: LCA increases activity of the initiator caspase-8 in cultured human NB cell lines BE(2)-m17 and SK-n-MCIXC (but not in Lan-1). Specific activity of caspase-8 was measured as described in Materials and Methods and expressed in picomoles of fluorescent compound 7-amino-4-trifluoromethyl coumarin released per microgram of protein per minute, based on the linear range of the curve. Data are presented as means ± SD (n = 3-4); ***p<0.001.
LCA reduces the activity of the inflammatory caspase-1 in two NB cell lines
In addition to its stimulatory effect on caspases-3, -6, -8 and -9, LCA significantly reduced caspase-1 activity in NB cell lines BE(2)-m17 and SK-n-MCIXC, but not in Lan-1 (Suppl. Fig. 1). Caspase-1 is an inflammatory caspase that drives the processing and unconventional secretion of the cytokines interleukin-1β and interleukin-18 [159-162]. Following their secretion by mammalian cells, cytokines promote the growth and proliferation of neighbouring cells in the same tissue [163-166]. Hence, it is tempting to speculate that by reducing caspase-1 activity in BE(2)-m17 and SK-n-MCIXC cells, and thereby impairing their ability to process and secrete cytokines, LCA could prevent growth and proliferation of neighbouring NB cells in culture.
LCA does not enter cultured NB cells
Our mass spectrometry-based measurement of cellular and extracellular levels of exogenously added LCA revealed that it does not enter BE(2)-m17, SK-n-MCIXC or Lan-1 cells (Suppl. Table 1). Thus, LCA selectively kills these cancer cells by binding to their surface and then initiating intracellular signaling cascades that cause their apoptotic (as in case of BE(2)-m17 and SK-n-MCIXC) or non-apoptotic (as in case of Lan-1) death. In BE(2)-m17 and SK-n-MCIXC cells, the intracellular signaling cascades triggered by LCA bound to their surface also reduce the activity of the inflammatory caspase-1.
LCA kills cultured human breast cancer (BC) and rat glioma (GL) cells
In addition to its high anti-tumor efficacy and specificity in several NB cell lines, LCA displayed an anti-tumor effect in two other types of cultured cancer cells. In fact, treatment of the drug-sensitive human BC cell line MCF7 with LCA demonstrated a dose-dependent killing effect, as measured by the MTT cell viability assay (Suppl. Fig. 2A). Treatment with 1 μg/ml doxorubicin demonstrated that these BC cells were highly drug sensitive (Suppl. Fig. 2A). Importantly, killing of MCF7 cells with LCA was associated with DNA damage, as visualized with antibodies against phosphorylated histone H2AX (Suppl. Fig. 2B). Once cells encounter DNA damage, histone H2AX phosphorylation at sites of damage has been shown to initiate the chromatin response required for DNA repair [167]. This is believed to serve as a docking site for DNA repair enzymes. At doses as low as 25 and 50 μM LCA, increased phosphorylated H2AX could be visualized in MCF7 cells (Suppl. Fig. 2B). Since most of these BC cells remain viable at 50 μM LCA (Suppl. Fig. 2A), DNA damage likely precedes their killing by LCA. We also assessed the ability of LCA to kill the cultured rat GL cell line F98. These highly proliferative and invasive cancer cells have increased resistance to chemo- and radiation therapy [168]. We found that at a concentration of 100 μM, LCA kills most F98 cells (Suppl. Fig. 3). Together, our findings imply that LCA has a broad anti-tumor effect on cultured cancer cells derived from different tissues and organisms.
Discussion
Mechanisms underlying an anti-tumor effect of LCA in human NB cell cultures
This study provides evidence that LCA, a bile acid that delays aging of quiescent and proliferating yeast, exhibits a potent and selective anti-tumor effect in cultured human NB cells. Our findings suggest a model for two mechanisms underlying such effect of LCA, as outlined below. Several aspects of this model remain hypothetical.
In NB cell lines BE(2)-m17 and SK-n-MCIXC, LCA triggers both the intrinsic and extrinsic pathways of apoptosis by binding to the cell surface and initiating the intracellular cascades that cause cell death (Fig. 8A). LCA binding to the cell surface leads to MOMP by activating caspase-8, which in turn causes MOMP - perhaps by cleaving and activating BID. LCA may also stimulate TGR5, its only known receptor on the cell surface (see below), which may then transmit a MOMP-activating signal from the plasma membrane to mitochondria via the cAMP/PKA signaling pathway. LCA-induced MOMP may result in efflux of cytochrome c from mitochondria, thereby causing apoptosome formation and the observed caspase-9 activation. Caspase-9 then activates caspase-3 by proteolytically processing pro-caspase-3. The proteolytic activation of caspase-3 is also carried out by caspase-8 operating in the LCA-induced extrinsic pathway of apoptosis. Active caspase-3 completes the demolition phase of the LCA-driven apoptotic program by cleaving its protein substrates. Furthermore, LCA binding to the cell surface leads to proteolytic activation of caspase-6; the mechanism underlying such activation (either self-cleavage or proteolytic processing by active caspase-3) and the involvement of caspase-6 in LCA-driven apoptosis remain to be established. Moreover, LCA binding to the cell surface causes inhibition of the inflammatory caspase-1. Such inhibition may contribute to the anti-tumor effect of LCA by attenuating the processing and secretion of the cytokines interleukin-1β and interleukin-18, thereby preventing growth and proliferation of neighbouring NB cells in culture.
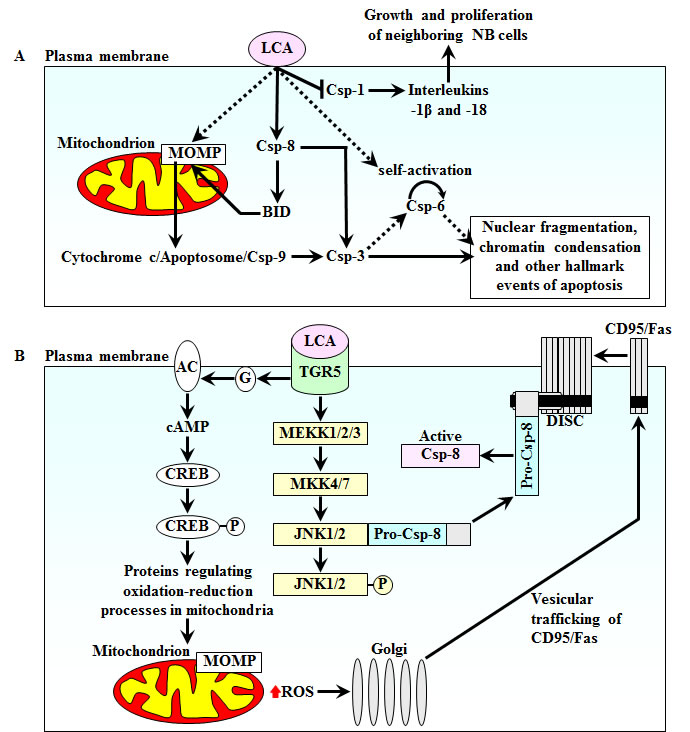
Figure 8: A model for a mechanism underlying an anti-tumor effect of LCA in cultured human NB cell lines BE(2)-m17 and SK-n-MCIXC. See text for details. Abbreviations: AC, adenylyl cyclase; BID, BH3-interacting domain death agonist; CD95/Fas (factor-activating Exo S), death receptor; CREB, cAMP-response element binding protein; Csp-1, -3, -6, -8 and -9, caspases-1, -3, -6, -8 and -9; DISC, death-inducing signaling complex; G, a G-protein; JNK, c-Jun-N terminal kinase; LCA, lithocholic acid; MEKK1/2/3, mitogen-activated protein kinase/ERK kinase kinase-1, -2 and -3; MKK4/7, mitogen-activated protein kinase kinase-4 and -7; MOMP, mitochondrial outer membrane permeabilization; PKA, protein kinase A; ROS, reactive oxygen species; TGR5, a member of the G-protein coupled receptor family.
Recent findings suggest the following cascade of events leading to the above described activation of both the intrinsic and extrinsic apoptotic pathways in response to LCA binding to the surface of BE(2)-m17 and SK-n-MCIXC cells. The only known LCA receptor on the cell surface in rodents and humans is TGR5, a member of the G-protein coupled receptor family [169,170]. LCA is the most potent natural agonist of TGR5, with EC50 of 530 nM [171,172]. Importantly, TGR5 has been shown to be expressed in neurons [170,173-175]. LCA-stimulated TGR5 could initiate three molecular cascades that trigger the intrinsic and extrinsic pathways of apoptotic cell death (Fig. 8B). First, it could initiate the cAMP/PKA signaling pathway known to response by altering oxidation-reduction processes in mitochondria and mitochondrial morphology [169,170,176-178]. The resulting changes in mitochondria may include such well-documented effects of LCA as MOMP activation and elevated ROS production [169,170,176-178]. Second, the elevated in response to LCA intracellular ROS could intensify vesicle-mediated trafficking of the CD95/Fas death receptor monomers from the Golgi to the plasma membrane, another well-documented effect of LCA in human cells [179,180]. Third, LCA-stimulated TGR5 could activate the MEKK1/2/3-MKK4/7 protein kinase cascade resulting in phosphorylation of c-Jun-N terminal kinase (JNK). Such LCA/TGR5-driven phosphorylation has been shown to release pro-caspase-8 from a complex that it forms with unphosphorylated JNK, thereby enabling its recruitment to the CD95/Fas death-inducing signaling complex consisting of the CD95/Fas death receptor trimers and the adaptor Fas-Associated Death Domain [176,181]. Following pro-caspase-8 dimerization and proteolytic processing, its active form could trigger both the intrinsic and extrinsic apoptotic death pathways (Fig. 8A).
Our findings imply that a different mechanism underlies the anti-tumor effect of LCA in cultured human NB cell line Lan-1. We propose such effect of LCA in Lan-1 is due to its ability to cause necrotic or some other non-apoptotic kind of death, which is not characterized by such major hallmark events of apoptosis as nuclear condensation and fragmentation, mitochondrial fragmentation and inner membrane depolarization, and activation of caspases-3, -6, -8 and -9.
A reason for the observed difference in the mechanisms by which LCA kills different NB cell lines (i.e., by triggering apoptotic cell death of BE(2)-m17 and SK-n-MCIXC, but by causing non-apoptotic cell death of Lan-1) remains to be established. All these cell lines exhibit amplified expression of the MYCN gene [96-98] and deletion of the short arm of chromosome 1p [100-104]. However, the level of an anti-apoptotic member of the B-cell lymphoma-2 (BCL-2) protein family containing four BCL-2 homology (BH) domains in BE(2)-m17 is significantly lower than that in Lan-1 [182]. The anti-apoptotic BCL-2 proteins prevent MOMP [183,184]. Therefore, it is conceivable that the lower efficacy of MOMP prevention in BE(2)-m17 makes this cell line more susceptible to LCA-induced apoptosis (which involves MOMP activation) than Lan-1. Furthermore, it is plausible that, due to the relatively high level of the anti-apoptotic BCL-2 protein with four BH domains in Lan-1, this cell line is more susceptible (as compared to BE(2)-m17) to a MOMP-independent kind of LCA-induced non-apoptotic cell death.
Another kind of dissimilarity between different NB cell lines consists in lower activity of caspase-1 in BE(2)-m17 and SK-n-MCIXC cells (as compared to that in Lan-1 cells) that have not been exposed to LCA; importantly, LCA further and significantly reduces the activity of this inflammatory caspase in BE(2)-m17 and SK-n-MCIXC, but not in Lan-1 (Suppl. Fig. 1). This observation suggests that certain pro-inflammatory alterations (including elevated activity of caspase-1) in NB cells may play an anti-apoptotic, cytoprotective role. It is conceivable therefore that the lower activities of caspase-1 in BE(2)-m17 and SK-n-MCIXC (especially following their exposure to LCA) make these NB cell lines more susceptible to LCA-induced apoptosis than Lan-1, in which a relatively high activity of caspase-1 is not reduced by LCA. Of note, recent findings in mice imply that certain mild pro-inflammatory alterations, including increased mitochondrial ROS and elevated expression of inflammatory cytokines, can extend animal longevity [185,186].
LCA modulates mitochondrial structure and
function to cause an anti-tumor effect in cultured human NB cells and an anti-aging effect in quiescent yeast
This study as well as our published [86] and recent unpublished data suggest that both an anti-tumor effect of LCA in cultured human NB cells and its longevity-extending effect in quiescent yeast are due to its ability to modulate mitochondria-confined processes playing essential roles in both cancer and aging. Interestingly, the effects of LCA on these mitochondrial processes seen in NB cell cultures are opposite of those observed in quiescent yeast and, for some processes, in primary cultures of human neurons. Indeed, while LCA enhances the susceptibility of NB cell lines BE(2)-m17 and SK-n-MC to mitochondria-controlled apoptotic cell death caused by exogenously added hydrogen peroxide (Fig. 2), it significantly increases the resistance of quiescent yeast [86] and primary cultures of human neurons (Fig. 2) to such a form of cell death. Furthermore, while LCA causes the fragmentation of a tubular mitochondrial network into individual mitochondria in BE(2)-m17 and SK-n-MC (Figs. 4A and 4B), it attenuates such mitochondrial fragmentation in quiescent yeast [86]. Moreover, LCA triggers the complete dissipation of the electrochemical potential across the inner mitochondrial membrane in BE(2)-m17 and SK-n-MC (Figs. 4C and 4D). In contrast, although exposure of yeast cells to LCA causes a reduction of such potential by 50% upon entry into a non-proliferative state [86], for a long time after becoming quiescent these cells maintain higher mitochondrial transmembrane potential than their quiescent counterparts that have not been exposed to LCA (Beach et al., manuscript in preparation). To the best of our knowledge, these findings provide the first example of a compound that can kill cancer cells and increase longevity of non-cancerous cells by causing quite opposite effects on the same kind of mitochondria-confined processes in these two different cell types. A key challenge for the future will be to understand why mitochondria in cancer and non-cancerous cells respond so differently to LCA exposure.
A broad anti-tumor effect of LCA
This study demonstrates that, in addition to the high anti-tumor efficacy and specificity exhibited by LCA in several NB cell lines, this bile acid displays an anti-tumor effect in human BC and rat GL cells. Future studies will examine if mechanisms underlying such broad anti-tumor effect of LCA are similar in cultured cancer cells that originate from different tissues and organisms.
Acknowledgements
We are grateful to Dr. Tatiana Iouk (Concordia University, Montreal) for sharing with us unpublished data on the ability of LCA to slow down telomere shortening in chronologically aging yeast. This study was supported by funding from the Canadian Institutes of Health Research (A.L. and T.A.A.H.), Natural Sciences and Engineering Research Council of Canada (V.I.T.), and Concordia University Chair Fund (V.I.T.). A.A.G. was supported by a doctoral scholarship from the Canadian Institutes of Health Research. A.B. was supported by a Master’s scholarship from the Fonds de la recherche en santé du Québec. V.I.T. is a Concordia University Research Chair in Genomics, Cell Biology and Aging.
Materials and Methods
Cell culture
Human NB cell lines were cultured in the following media: 1) SK-n-MCIXC and BE(2)-m17 cells in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium (DMEM) and Ham’s F-12 Nutrient Mixture (Invitrogen/Gibco BRL, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT); 2) SK-n-SH cells in Eagle’s Minimum Essential Medium (EMEM) (Invitrogen/Gibco BRL, Carlsbad, CA) supplemented with 10% FBS; and 3) Lan-1 cells in DMEM (Invitrogen/Gibco BRL, Carlsbad, CA) supplemented with 10% bovine calf serum (BCS) (HyClone, Logan, UT). Human primary neurons were prepared from fetal cerebrum tissue and cultured as previously described [187], in accordance with the Canadian Institute of Health Research regulations and as approved by the McGill University Institutional Review Board. MCF7 human BC and F98 rat GL cell lines from American Type Culture Collection were cultured in a 1:1 mixture of DMEM and Ham’s F-12 Nutrient Mixture (Sigma-Aldrich, St Louis, MI) supplemented with 10% FBS (Invitrogen/Gibco BRL, Carlsbad, CA) and 1× anti-mycotic/anti-biotic (Invitrogen/Gibco BRL, Carlsbad, CA). All cell lines were cultured in a humidified atmosphere (5% CO2) at 37 °C.
Treatment of cells with LCA, hydrogen peroxide or z-DEVD-fmk
Stock solutions of LCA (Sigma-Aldrich, St Louis, MI) in varying concentrations were first made in 100% dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St Louis, MI). For treatment of cultured human NB, human BC and rat GL cells, these stock solutions were then diluted to the indicated final concentration of LCA (the final concentration of DMSO was always kept at 1%) in either a 1:1 mixture of DMEM and Ham’s F-12 Nutrient Mixture supplemented with 10% FBS (for SK-n-MCIXC and BE(2)-m17 cells), EMEM supplemented with 10% FBS (for SK-n-SH cells), DMEM supplemented with 10% BCS (for Lan-1 cells) or a 1:1 mixture of DMEM and Ham’s F-12 Nutrient Mixture supplemented with 10% FBS and 1X anti-mycotic/anti-biotic (for MCF7 and F98 cells). For treatment of human primary neurons with LCA, stock solutions of LCA in varying concentrations made in 100% DMSO were diluted in EMEM containing 0.225% sodium bicarbonate, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1% dextrose (all from Life Technologies, Gaithersburg, MD) and 5% FBS (HyClone, Logan, UT) to the indicated final concentration of LCA (the final concentration of DMSO was always kept at 1%). For treatment of cultured cancer cells or primary neurons with LCA, they were incubated for 48 hours in the presence LCA at the indicated final concentrations; control cells were treated with an empty DMSO vehicle only. For cell treatment with hydrogen peroxide, its 30% stock solution (Fisher Scientific, Waltham, MA) was diluted in sterile H2O and added directly to the cell cultures after 24 h of their pre-treatment with LCA or an empty DMSO vehicle only; cells were then incubated for 24 h. In experiments involving cell treatment with z-DEVD-fmk, this caspase-3 inhibitor was added to a final concentration of 5 μM simultaneously with LCA or an empty DMSO vehicle only.
Cell viability assays
The number of viable cells in cultures exposed to LCA was measured using the MTT (3-(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide)-based CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). In this assay, only viable, metabolically active cells were able to reduce the yellow tetrazolium salt of MTT to form a purple formazan product. This insoluble product was solubilized by the addition of a detergent. The resulting intracellular purple formazan was then detected spectrophotometrically using a 96-well plate reader at a wavelength of 570 nm. The signal was corrected to account for cellular debris using a wavelength of 630 nm. Chromatin in cells exposed to LCA and/or hydrogen peroxide and/or z-DEVD-fmk was visualized with the fluorescent dye Hoechst used at a final concentration of 4 μM in culture media and viewed using fluorescence microscopy. For each cell culture, the percentage of viable non-apoptotic cells carrying intact, non-fragmented nuclei containing non-condensed chromatin was calculated. Dead apoptotic cells carried fragmented nuclei containing condensed chromatin, a hallmark event of apoptotic death.
Visualization of mitochondria and measurement of the mitochondrial membrane potential by fluorescence microscopy
Mitochondrial morphology of cells treated with LCA was visualized using MitoTracker Red CMXRos (Molecular Probes, San Diego, CA) used at a concentration of 125 nM in the culture media. Cells were viewed using fluorescence microscopy, and the percentage of cells displaying fragmented mitochondria was calculated. The mitochondrial membrane potential (∆Ψ) was measured using tetramethylrhodamine ethyl ester (TMRE), a cell-permeant, cationic fluorescent dye. The extent of reversible sequestration of TMRE by mitochondria is proportional to the value of ∆Ψ. Cells were incubated with 50 nM TMRE for 20 min and directly viewed using fluorescence microscopy. The percentage of TMRE-positive cells displaying a detectable level of ∆Ψ was calculated.
Fluorescence microscopy
For all cells and fluorescent dyes, images were collected with a Nikon Eclipse (TE2000-U) inverted fluorescence microscope under 20 × magnification. A filter cube with an excitation wavelength of 330-380 nm was used to visualize Hoechst-stained cells. Cells stained with MitoTracker Red CMXRos or TMRE were visualized using filter cubes with excitations of 590-650 nm or 532-587 nm, respectively.
Caspase activity assays
Proteins were extracted from harvested cells using an ice-cold lysis buffer containing 50 mM HEPES, 0.1% CHAPS, 0.1 mM EDTA and protease inhibitors (0.1μg/ml TLCK, 0.5 μg/ml leupeptin, 38 mg/ml AEBSF, 0.1 μg/ml pepstatin A). The cell lysate was centrifuged at 13,000 × g for 5 min at 4oC to remove cell debris and any detergent-insoluble proteins. The supernatant was collected and frozen at -80oC. Extracts were thawed and protein concentrations were quantified with the Pierce BCA protein assay (Thermo Scientific, Waltham, MA). The extracts were then assayed for caspase activities using fluorogenic substrates specific for caspase-1 (10 μM Ac-YVAD-AFC), caspase-3 (5 μM Ac-DEVD-AFC), caspase-6 (10 μM Ac-VEID-AFC), caspase-8 (10 μM Ac-IETD-AFC) or caspase-9 (10 μM Ac-LEHD-AFC) in caspase reaction buffer (20 mM PIPES, pH 7.2, 30 mM NaCl, 10 mM DTT, 1 mM EDTA, 0.1% CHAPS, 10% sucrose). The time-dependent release of the fluorescent compound 7-amino-4-trifluoromethyl coumarin (AFC) was monitored using a Bio-Rad Fluoromark fluorometer (Hercules, CA) at an excitation wavelength of 390 nm and an emission wavelength of 538 nm. Measurements were recorded at 2 min intervals for 1 h. A standard curve of AFC fluorescence was used to calculate AFC (in picomoles) released in the reactions. Specific activities were expressed as picomoles of AFC released per microgram of protein per minute, based on the linear range of the curve.
Pro-caspase-3 and pro-caspase-6 cleavage assays
50 μg of protein from crude cellular extracts were resolved by SDS-PAGE in a 15% polyacrylamide gel and then transferred to a PVDF Immobilon-P membrane (Millipore, Bedford, MA). Following wash with 5% milk, blots were incubated with primary antibodies used at the following dilutions: 1:500 for anti-cleaved-caspase-3 p17 (Cell Signaling, Beverly, MA), 1:250 for anti-cleaved-caspase-6 p10 (Pharmingen, San Diego, CA) and 1:5,000 for anti-β-Actin (Sigma-Aldrich, St Louis, MI). Immunoreactivity was visualized with horseradish peroxidase-conjugated secondary antibodies (Amersham, Oakville, ON) that were used at dilutions 1:2,000 (for anti-rabbit) and 1:5,000 (for anti-mouse), respectively. Antigen-antibody complexes were detected by enhanced chemiluminescence using an ECL plus chemiluminescence Western blot detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ).
Histone H2AX phosphorylation assay
BC cells were removed from 100-mm tissue culture dishes using a rubber policeman and ice-cold phosphate-buffered saline (PBS). The cells were pelleted by centrifugation at 3,000 rpm at 4oC. The pellets were resuspended in ice-cold lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.1 mM EGTA, 0,1% NP-40, 1× mammalian cell anti-protease cocktail (Sigma-Aldrich, St Louis, MI)). The cells were lysed using multiple freeze-thaw cycles followed by pulse sonication on ice and centrifugation at 3,000 rpm for 5 min at 40 C to remove cell debris. Western blot analysis of these protein lysates was performed as previously described [188]. Briefly, equivalent amounts of protein (assessed using Bio-Rad Protein Reagent; Bio-Rad Laboratories, Hercules, CA) were resolved on 10% SDS-PAGE gels. Following electrophoresis the proteins were trans-blotted onto nitrocellulose membranes (Pall-VWR International, Radnor, PA). The membranes were blocked overnight at 4o C on a gyratory plate with 5% molecular grade skim milk powder (Bio-Rad Laboratories, Hercules, CA) in PBS containing 0.1% Tween-20 (PBST). Primary and secondary antibody incubations and subsequent washes were carried out in the same buffer. The antibody to phosphorylated H2AX was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The primary antibody for GAPDH was purchased from Sigma-Aldrich (St Louis, MI). Secondary HRP antibodies were purchased from Bio-Rad Laboratories (Hercules, CA). Blots were immunoprobed overnight with primary antibodies used at a 1:1,000 dilution. Secondary HRP antibody was applied at room temperature on a gyratory plate at a 1:10,000 dilution for 30 min. Following multiple washes, an enhanced chemiluminescence detection system (DuPont-NEN; Boston, MA) was used to detect the target antigen/antibody complexes. Blots were then stripped at 50o C for 30 min in a Tris-buffered 20% SDS/1% 2-mercaptoethanol stripping solution, washed and reprobed with GAPDH antibody to verify protein loading equivalency.
Mass spectrometric measurement of LCA
Mass spectrometry-based quantitative analysis of LCA was performed as previously reported [189]. In brief, lipids were extracted by a modified Bligh and Dyer method [189] from cells pelleted by centrifugation for 5 min at 16,000 × g at 4oC and from the supernatant of cultural medium. The extracted lipids were dried under nitrogen and resuspended in chloroform. Immediately prior to injection the extracted lipids were combined with a 2:1 methanol:chloroform mixture supplemented with 0.1% (v/v) ammonium hydroxide. The sample was injected directly into a Q-TOF 2 mass spectrometer (Waters, Milford, MA) using a nano-ESI spray source at 1 µl/min. Spectra were obtained in a negative-ion mode. The cone voltage was set to 30 v, the capillary voltage to 3.2 kv and the collision gas to 10 (arbitrary units). Acquired spectra were centroided using the Masslynx software then deconvoluted and deisotoped with Excel macros.
Statistical analysis
Statistical analysis was performed using Microsoft Excel’s (2010) Analysis ToolPack-VBA. All data are presented as the means ± SD. The p values were calculated using an unpaired two-tailed t test.
References
1. Boland CR, Ricciardiello L. How many mutations does it take to make a tumor? Proc Natl Acad Sci USA. 1999; 96: 14675-14677.
2. Karakosta A, Golias C, Charalabopoulos A, Peschos D, Batistatou A, Charalabopoulos K. Genetic models of human cancer as a multistep process. Paradigm models of colorectal cancer, breast cancer, and chronic myelogenous and acute lymphoblastic leukaemia. J Exp Clin Cancer Res. 2005; 24: 505-514.
3. Weinberg RA. (2007). The Biology of Cancer (New York: Garland Science, Taylor and Francis Group, LLC).
4. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5: 2087-2102.
5. Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007; 448: 767-774.
6. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging. 2009; 1: 281-288.
7. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092-2097.
8. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007; 130: 223-233.
9. Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007; 8: 715-722.
10. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005; 37: 19-24.
11. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006; 6: 184-192.
12. Harikumar KB, Aggarwal BB. Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle. 2008; 7: 1020-1035.
13. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009; 9: 563-575.
14. Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009; 196: 65-80.
15. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132: 27-42.
16. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. (2008). Autophagy fights disease through cellular self-digestion. Nature. 2008; 451: 1069-1075.
17. Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009; 3: 366-375.
18. Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009; 14: 376-391.
19. Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the pro-death and pro-survival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010; 6: 322-329.
20. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol. Cell. 2010; 40: 280-293.
21. Liang C, Jung JU. Autophagy genes as tumor suppressors. Curr Opin Cell Biol. 2010; 22: 226-233.
22. Turcotte S, Giaccia AJ. Targeting cancer cells through autophagy for anticancer therapy. Curr Opin Cell Biol. 2010; 22: 246-251.
23. Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011; 7: 9-17.
24. Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005; 5: 857-866.
25. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006; 25: 4675-4682.
26. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006; 312:1650-1653.
27. Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007; 39: 243-246.
28. Goffrini P, Ercolino T, Panizza E, Giachè V, Cavone L, Chiarugi A, Dima V, Ferrero I, Mannelli M. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Hum Mol Genet. 2009; 18: 1860-1868.
29. Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009; 325: 1139-1142.
30. Kaelin WG. Jr. SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell. 2009; 16: 180-182.
31. Bayley JP, Devilee P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr Opin Genet Dev. 2010; 20: 324-329.
32. Huber HJ, Dussmann H, Kilbride SM, Rehm M, Prehn JH. Glucose metabolism determines resistance of cancer cells to bioenergetic crisis after cytochrome-c release. Mol Syst Biol. 2011; 7: 470.
33. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21-35.
34. Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003; 54: 131-152.
35. Mai V, Colbert LH, Berrigan D, Perkins SN, Pfeiffer R, Lavigne JA, Lanza E, Haines DC, Schatzkin A, Hursting SD. Calorie restriction and diet composition modulate spontaneous intestinal tumorigenesis in Apc(Min) mice through different mechanisms. Cancer Res. 2003; 63: 1752-1755.
36. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004; 10: 594-601.
37. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005; 40: 685-693.
38. Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006; 5: 493-506.
39. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006; 66: 10269-10273.
40. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the anti-diabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007; 67: 6745-6752.
41. Shevah O, Laron Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: a preliminary report. Growth Horm IGF Res. 2007; 17: 54-57.
42. Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer. 2008; 15: 833-839.
43. Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008; 412: 211-221.
44. Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci USA. 2008; 105: 8215-8220.
45. Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res. 2008; 1: 369-375.
46. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009; 325: 201-204.
47. Enns LC, Morton JF, Treuting PR, Emond MJ, Wolf NS, Dai DF, McKnight GS, Rabinovitch PS, Ladiges WC. Disruption of protein kinase A in mice enhances healthy aging. PLoS One. 2009; 4: e5963.
48. Goodwin PJ, Ligibel JA, Stambolic V. (2009). Metformin in Breast Cancer: Time for Action. J Clin Oncol. 2009; 27: 3271-3273.
49. Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009; 458: 725-731.
50. Fontana L, Partridge L, Longo VD. Extending healthy life span - from yeast to humans. Science. 2010; 328: 321-326.
51. Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis. 2010; 31: 83-89.
52. Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci. 2010; 31: 89-98.
53. Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003; 3: 339-349.
54. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005; 120: 513-522.
55. Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007; 3: 640-649.
56. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8: 729-740.
57. Rodier F, Campisi J, Bhaumik D. Two faces of p53: aging and tumor suppression. Nucleic Acids Res. 2007; 35: 7475-7484.
58. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007; 8: 703-713.
59. Grimes A, Chandra SB. Significance of cellular senescence in aging and cancer. Cancer Res Treat. 2009; 41: 187-195.
60. Ohtani N, Mann DJ, Hara E. Cellular senescence: its role in tumor suppression and aging. Cancer Sci. 2009; 100: 792-797.
61. Kong Y, Cui H, Ramkumar C, Zhang H. Regulation of senescence in cancer and aging. J Aging Res. 2011; 2011: 963172.
62. Sharpless NE. Ink4a/Arf links senescence and aging. Exp Gerontol. 2004; 39: 1751-1759.
63. Pelicci PG. Do tumor-suppressive mechanisms contribute to organism aging by inducing stem cell senescence? J Clin Invest. 2004; 113: 4-7.
64. Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004; 113: 160-168.
65. Vergel M, Marin JJ, Estevez P, Carnero A. Cellular senescence as a target in cancer control. J. Aging Res. 2011; 2011: 725365.
66. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: Aerobic glycolysis in cancer-associated fibroblasts and the tumor stroma. Cell Cycle. 2009; 8: 3984-4001.
67. Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, Witkiewicz A, Lin Z, Balliet R, Howell A, Sotgia F. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther. 2010; 10:537-542.
68. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Knudsen ES, Sotgia F, Lisanti MP. Oxidative stress in cancer-associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010; 9: 3256-3276.
69. Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, Migneco G, Witkiewicz AK, Martinez-Cantarin MP, Flomenberg N, Howell A, Pestell RG, Lisanti MP, Sotgia F. Tumor cells induce the cancer-associated fibroblast phenotype via Caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010; 9: 2423-2433.
70. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Caro J, Lisanti MP, Sotgia F. Autophagy in cancer-associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010; 9: 3515-3533.
71. Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, Tsirigos A, Migneco G, Witkiewicz A, Balliet R, Mercier I, Wang C, Flomenberg N, Howell A, Lin Z, Caro J, Pestell RG, Sotgia F, Lisanti MP. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010; 9: 4297-4306.
72. Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, Sotgia F, Lisanti MP. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010; 9: 3485-3505.
73. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Loss of Stromal Caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle. 2010; 9: 2201-2219.
74. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, Wang C, Pestell RG, Martinez-Outschoorn UE, Howell A, Sotgia F, Lisanti MP. Transcriptional evidence for the “reverse Warburg effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer disease and “neuron-glia metabolic coupling”. Aging. 2010; 2: 185-199.
75. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: Aerobic glycolysis in cancer-associated fibroblasts and the tumor stroma. Cell Cycle. 2009; 8: 3984-4001.
76. Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, Lisanti MP. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011; 43: 1045-1051.
77. Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, Sotgia F. Accelerated aging in the tumor microenvironment: Connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011; in press.
78. Blagosklonny MV. Hypoxia-inducible factor: Achilles’ heel of antiangiogenic cancer therapy. Int J Oncol. 2001; 19: 257-262.
79. Blagosklonny MV. Antiangiogenic therapy and tumor progression. Cancer Cell. 2004; 5: 13-17.
80. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009; 15: 5308-5316.
81. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010; 330: 1344-1348.
82. White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010; 22: 212-217.
83. Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther. 2011; 11: 157-168.
84. Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011; 21: 113-119.
85. Yang ZJ, Chee CE, Huang S, Sinicrope F. Autophagy modulation for cancer therapy. Cancer Biol Ther. 2011; 11: 169-176.
86. Goldberg AA, Richard VR, Kyryakov P, Bourque SD, Beach A, Burstein MT, Glebov A, Koupaki O, Boukh-Viner T, Gregg C, Juneau M, English AM, Thomas DY, Titorenko VI. Chemical genetic screen identifies lithocholic acid as an anti-aging compound that extends yeast chronological life span in a TOR-independent manner, by modulating housekeeping longevity assurance processes. Aging. 2010; 2: 393-414.
87. Longo VD, Kennedy BK. (2006). Sirtuins in aging and age-related disease. Cell. 2006; 126: 257-268.
88. Lin SJ, Sinclair D. Molecular mechanisms of aging: insights from budding yeast. In: Guarente LP, Partridge L, Wallace DC, eds. Molecular Biology of Aging. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2008: 483-516.
89. Kaeberlein M. Lessons on longevity from budding yeast. Nature. 2010; 464: 513-519.
90. Astigarraga I, Lejarreta R, Navajas A, Fernandez-Teijeiro A, Imaz I, Bezanilla JL. Secondary central nervous system metastases in children with neuroblastoma. Med Pediatr Oncol. 1996; 27: 529-533.
91. Esiashvili N, Anderson C, Katzenstein HM. Neuroblastoma. Curr Probl Cancer. 2009; 33: 333-360.
92. Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010; 24: 65-86.
93. Blatt J, Fitz C, Mirro J Jr. Recognition of central nervous system metastases in children with metastatic primary extracranial neuroblastoma. Pediatr Hematol Oncol. 1997; 14: 233-241.
94. DuBois SG, Kalika Y, Lukens JN, Brodeur GM, Seeger RC, Atkinson JB, Haase GM, Black CT, Perez C, Shimada H, Gerbing R, Stram DO, Matthay KK. Metastatic sites in Stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol. 1999; 21: 181-189.
95. Matthay KK, Brisse H, Couanet D, Couturier J, Bénard J, Mosseri V, Edeline V, Lumbroso J, Valteau-Couanet D, Michon J. Central nervous system metastases in neuroblastoma: radiologic, clinical, and biologic features in 23 patients. Cancer. 2003; 98: 155-165.
96. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984; 224: 1121-1124.
97. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985; 313: 1111-1116.
98. Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Curr Opin Pediatr. 2005; 17: 7-13.
99. Lee, L.A. and Dang, C.V. (2006). Myc target transcriptomes. Curr Top Microbiol Immunol. 2006; 302: 145-167.
100. Gilbert F, Feder M, Balaban G, Brangman D, Lurie DK, Podolsky R, Rinaldt V, Vinikoor N, Weisband J. Human neuroblastomas and abnormalities of chromosomes 1 and 17. Cancer Res. 1984; 44: 5444-5449.
101. Fujita T, Igarashi J, Okawa ER, Gotoh T, Manne J, Kolla V, Kim J, Zhao H, Pawel BR, London WB, Maris JM, White PS, Brodeur GM. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008; 100: 940-949.
102. Look AT, Meyerson M, Peeper DS, Carter BD, Kaelin WG. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008; 22: 884-893.
103. Geli J, Kiss N, Kogner P, Larsson C. Suppression of RIZ in biologically unfavourable neuroblastomas. Int J Oncol. 2010; 37: 1323-1330.
104. Liu Z, Yang X, Li Z, McMahon C, Sizer C, Barenboim-Stapleton L, Bliskovsky V, Mock B, Ried T, London WB, Maris J, Khan J, Thiele CJ. CASZ1, a candidate tumor-suppressor gene, suppresses neuroblastoma tumor growth through reprogramming gene expression. Cell Death Differ. 2011; Jan 21, Epub. ahead of print, doi:10.1038/cdd.2010.187.
105. George RE, Diller L, Bernstein ML. Pharmacotherapy of neuroblastoma. Expert Opin Pharmacother. 2010; 11: 1467-1478.
106. Modak S, Cheung NK. Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer Treat. Rev. 2010; 36: 307-317.
107. Sodhi A, Gupta P. Increased release of hydrogen peroxide (H2O2) and superoxide anion (O-2) by murine macrophages in vitro after cis-platin treatment. Int J Immunopharmacol. 1986; 8: 709-714.
108. Tsang WP, Chau SP, Kong SK, Fung KP, Kwok TT. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life Sci. 2003; 73: 2047-2058.
109. Timur M, Akbas SH, Ozben T. The effect of Topotecan on oxidative stress in MCF-7 human breast cancer cell line. Acta Biochim. Pol. 2005; 52: 897-902.
110. Woiniak A, Drewa G, Woźniak B, Schachtschabel DO, Mila-Kierzenkowska C, Drewa T, Olszewska-Słonina D, Sopońska M. The effect of antitumor drugs on oxidative stress in B16 and S91 melanoma cells in vitro. Med Sci Monit. 2005; 11: BR22-29.
111. Strauss G, Westhoff MA, Fischer-Posovszky P, Fulda S, Schanbacher M, Eckhoff SM, Stahnke K, Vahsen N, Kroemer G, Debatin KM. 4-hydroperoxy-cyclophosphamide mediates caspase-independent T-cell apoptosis involving oxidative stress-induced nuclear relocation of mitochondrial apoptogenic factors AIF and EndoG. Cell Death Differ. 2008; 15: 332-343.
112. Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983; 65: 55-63.
113. Tada H, Shiho O, Kuroshima K, Koyama M, Tsukamoto K. An improved colorimetric assay for interleukin 2. J Immunol Methods. 1986; 93: 157-165.
114. Campling BG, Pym J, Galbraith PR, Cole SP. Use of the MTT assay for rapid determination of chemosensitivity of human leukemic blast cells. Leuk Res. 1988; 12: 823-831.
115. McHale AP, McHale L. Use of a tetrazolium based colorimetric assay in assessing photoradiation therapy in vitro. Cancer Lett. 1988; 41: 315-321.
116. Jover R, Ponsoda X, Castell JV, Gómez-Lechón MJ. Acute cytotoxicity of ten chemicals in human and rat cultured hepatocytes and in cell lines: Correlation between in vitro data and human lethal concentrations. Toxic In Vitro. 1994; 8: 47-54.
117. Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972; 26: 239-275.
118. Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980; 68: 251-305.
119. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008; 9: 231-241.
120. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El-Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnóczky G, Hardwick JM, Hengartner MO, Ichijo H, Jäättelä M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema JP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nuñez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T,
Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009; 16: 1093-1107.
121. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nuñez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009; 16: 3-11.
122. Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011; 10 :221-237.
123. Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 2007; 39: 443-455.
124. Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007; 47: 143-183.
125. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007; 12: 913-922.
126. Cheng WC, Leach KM, Hardwick JM. Mitochondrial death pathways in yeast and mammalian cells. Biochim Biophys Acta. 2008; 1783: 1272-1279.
127. Carmona-Gutierrez D, Eisenberg T, Büttner S, Meisinger C, Kroemer G, Madeo F. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 2010; 17: 763-773.
128. Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta. 2011; 1813: 238-259.
129. Jourdain A, Martinou JC. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol. 2009; 41: 1884-1889.
130. Parsons MJ, Green DR. Mitochondria in cell death. Essays Biochem. 2010; 47: 99-114.
131. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010; 11: 621-632.
132. Jiang X, Wang X. Cytochrome c-mediated apoptosis. Annu Rev Biochem. 2004; 73: 87-106.
133. Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008; 9: 532-542.
134. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008; 22: 1577-1590.
135. Karbowski M. Mitochondria on guard: role of mitochondrial fusion and fission in the regulation of apoptosis. Adv Exp Med Biol. 2010; 687: 131-142.
136. Mootha VK, Wei MC, Buttle KF, Scorrano L, Panoutsakopoulou V, Mannella CA, Korsmeyer SJ. A reversible component of mitochondrial respiratory dysfunction in apoptosis can be rescued by exogenous cytochrome c. EMBO J. 2001; 20: 661-671.
137. Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001; 153: 319-328.
138. Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell. 2009; 20: 4871-4884.
139. Kushnareva Y, Newmeyer DD. Bioenergetics and cell death. Ann NY Acad Sci. 2010; 1201: 50-57.
140. Slee EA, Adrain C, Martin SJ. Serial killers: ordering caspase activation events in apoptosis. Cell Death Differ. 1999; 6: 1067-1074.
141. Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999; 144: 281-292.
142. Orth K, O’Rourke K, Salvesen GS, Dixit VM. Molecular ordering of apoptotic mammalian
CED-3/ICE-like proteases. J Biol Chem. 1996; 271: 20977-20980.
143. Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997; 388: 300-304.
144. Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997; 16: 6914-6925.
145. Grossmann J, Mohr S, Lapentina EG, Fiocchi C, Levine AD. Sequential and rapid activation of select caspases during apoptosis of normal intestinal epithelial cells. Am J Physiol. 1998; 274: G1117-G1124.
146. Miyashita T, Nagao K, Krajewski S, Salvesen GS, Reed JC, Inoue T, Yamada M. Investigation of glucocorticoid-induced apoptotic pathway: processing of caspase-6 but not caspase-3. Cell Death Differ. 1998; 5: 1034-1041.
147. Cowling V, Downward J. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ. 2002; 9: 1046-1056.
148. Guo H, Pétrin D, Zhang Y, Bergeron C, Goodyer CG, LeBlanc AC. Caspase-1 activation of caspase-6 in human apoptotic neurons. Cell Death Differ. 2006; 13: 285-292.
149. LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J Biol Chem. 1999; 274: 23426-23436.
150. Allsopp TE, McLuckie J, Kerr LE, Macleod M, Sharkey J, Kelly JS. Caspase 6 activity initiates caspase 3 activation in cerebellar granule cell apoptosis. Cell Death Differ. 2000; 7: 984-993.
151. Doostzadeh-Cizeron J, Yin S, Goodrich DW. Apoptosis induced by the nuclear death domain protein p84N5 is associated with caspase-6 and NF-kappa B activation. J Biol Chem. 2000; 275: 25336-25341.
152. Klaiman G, Champagne N, LeBlanc AC. Self-activation of Caspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. Biochim Biophys Acta. 2009; 1793: 592-601.
153. Lee AW, Champagne N, Wang X, Su XD, Goodyer C, Leblanc AC. Alternatively spliced caspase-6B isoform inhibits the activation of caspase-6A. J Biol Chem. 2010; 285: 31974-31984.
154. Wang XJ, Cao Q, Liu X, Wang KT, Mi W, Zhang Y, Li LF, LeBlanc AC, Su XD. Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 2010; 11: 841-847.
155. Gray DC, Mahrus S, Wells JA. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell. 2010; 142: 637-646.
156. Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, Robertson N, Armstrong RC, Wang L, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. The Ced-3/interleukin 1beta converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2alpha are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996; 271: 27099-27106.
157. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998; 944: 491-501.
158. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998; 94: 481-490.
159. Kersse K, Vanden Berghe T, Lamkanfi M, Vandenabeele P. A phylogenetic and functional overview of inflammatory caspases and caspase-1-related CARD-only proteins. Biochem Soc Trans. 2007; 35: 1508-1511.
160. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009; 27: 519-550.
161. Keller M, Rüegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell’ 2009; 132: 818-831.
162. Yi CH, Yuan J. The Jekyll and Hyde functions of caspases. Dev Cell. 2009; 16: 21-34.
163. Feagins LA. Role of transforming growth factor-β in inflammatory bowel disease and colitis-associated colon cancer. Inflamm Bowel Dis. 2010; 16: 1963-1968.
164. Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010; 49: 1-9.
165. Donath MY, Shoelson SE. (2011). Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011; 11: 98-107.
166. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011; 70, Suppl. 1: i104-108.
167. Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010; 9: 3602-3610.
168. Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neurooncol. 2009; 94: 299-312.
169. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008; 7: 678-693.
170. Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011; 54: 1263-1272.
171. Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun. 2002; 298:714-719.
172. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278: 9435-9440.
173. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006; 439: 484-489.
174. Keitel V, Görg B, Bidmon HJ, Zemtsova I, Spomer L, Zilles K, Häussinger D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia. 2010; 58: 1794-1805.
175. Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, Bunnett NW, Corvera CU. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. 2010; 22: 814-825.
176. Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009; 50: 1509-1520.
177. Wachs FP, Krieg RC, Rodrigues CM, Messmann H, Kullmann F, Knüchel-Clarke R, Schölmerich J, Rogler G, Schlottmann K. Bile salt-induced apoptosis in human colon cancer cell lines involves the mitochondrial transmembrane potential but not the CD95 (Fas/Apo-1) receptor. Int J Colorectal Dis. 2005; 20: 103-113.
178. Merrill RA, Dagda RK, Dickey AS, Cribbs JT, Green SH, Usachev YM, Strack S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011; 9: e1000612.
179. Sodeman T, Bronk SF, Roberts PJ, Miyoshi H, Gores GJ. Bile salts mediate hepatocyte apoptosis by increasing cell surface trafficking of Fas. Am J Physiol Gastrointest Liver Physiol. 2000; 278: G992-G999.
180. Katona BW, Anant S, Covey DF, Stenson WF. Characterization of enantiomeric bile acid-induced apoptosis in colon cancer cell lines. J Biol Chem. 2009; 284: 3354-3364.
181. Yang JI, Yoon JH, Myung SJ, Gwak GY, Kim W, Chung GE, Lee SH, Lee SM, Kim CY, Lee HS. Bile acid-induced TGR5-dependent c-Jun-N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem Biophys Res Commun. 2007; 361: 156-161.
182. Hanada M, Krajewski S, Tanaka S, Cazals-Hatem D, Spengler BA, Ross RA, Biedler JL, Reed JC. Regulation of Bcl-2 oncoprotein levels with differentiation of human neuroblastoma cells. Cancer Res. 1993; 53: 4978-4986.
183. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008; 9: 231-241.
184. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010; 11: 621-632.
185. Wang D, Malo D, Hekimi S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1α in long-lived Mclk1+/- mouse mutants. J Immunol. 2010; 184: 582-590.
186. Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011; Epub ahead of print.
187. LeBlanc AC, Koutroumanis M, Goodyer CG. Protein kinase C activation increases release of secreted amyloid precursor protein without decreasing Aβ production in human primary neuron cultures. J Neurosci. 1998; 18: 2907-2913.
188. Davies GF, Roesler WJ, Juurlink BH, Harkness TAA. Troglitazone overcomes doxorubicin-resistance in resistant K562 leukemia cells. Leuk Lymphoma. 2005; 46: 1199-1206.
189. Bourque SD, Titorenko VI. A quantitative assessment of the yeast lipidome using electrospray ionization mass spectrometry. J Vis Exp. 2009; 30: doi:10.3791/1513.