
INTRODUCTION
Although the existence of circulating tumor DNA (ctDNA) was first reported more than 30 years ago [1], interest in the practical application of ctDNA has only recently accelerated with the development of technologies suited to detecting and measuring this analyte [2-4]. Tumor monitoring with ctDNA may offer a non-invasive approach to assess microscopic residual disease, response to therapy, and tumor molecular profiles in the context of intratumoral heterogeneity and clonal evolution [5, 6].
For colorectal cancer (CRC) patients, there have been recent advances in screening, surgical techniques, and use of chemotherapeutic and biologic agents, which have significantly prolonged survival [7-9]. A deeper understanding of this disease, particularly the importance of KRAS mutational status in conferring resistance to therapies directed against the epidermal growth factor receptor (EGFR), has also enabled molecularly-guided treatment strategies [9]. Still, improved methods for monitoring disease burden and tumor molecular profiles of CRC are needed to optimize detection strategies and use of existing therapies, as well as to accelerate development of new treatments. Conventional monitoring of CRC is primarily based on cross-sectional imaging and measurement of serum carcinoembryonic antigen (CEA). Both of these methods can be associated with false positives: inflammatory conditions such as diverticulitis or inflammatory bowel disease can lead to elevated levels of CEA [10-12], while benign conditions can mimic malignant lesions on imaging and thereby necessitate confirmatory biopsy [13, 14]. Imaging and CEA are also associated with false negatives, since subcentimeter lesions (e.g. at the periphery of ablated liver metastases) may not be detected by imaging [13, 14], and a subset of patients with advanced stage CRC may not show elevated levels of CEA [11]. For both methods, a significant further limitation is that neither provides information about the molecular profile of the disease. Improved tumor monitoring tools may be particularly important for patients with resectable metastatic disease, where a subset of patients can achieve long-term disease-free survival [15, 16]. Better assessment of residual disease and evolving changes in tumor molecular profiles may enable improved risk stratification and tailoring of perioperative therapy in metastatic CRC.
Multiple methods have been developed to enable the assessment of ctDNA in CRC, including digital PCR, ‘BEAMing’ (beads, emulsion, amplification, and magnetics) and other approaches based on PCR and next-generation sequencing [2-4]. Several studies have shown that for patients with identifiable KRAS mutations in their tumor tissue, the corresponding mutations can be detected in DNA isolated from plasma, and elevated ctDNA levels have been associated with decreased overall 2-year survival [4, 17, 18]. Furthermore, acquired resistance to EGFR-inhibitors due to emergence of mutations in KRAS, NRAS, and other genes can be monitored non-invasively by serial assessment of ctDNA [19, 20]; resistance mutations can be identified in plasma DNA up to ten months earlier than conventional imaging reveals recurrence [19].
To date, most studies have analyzed plasma samples from patients with advanced stage CRC, as the abundance of ctDNA has been found to increase with disease progression; ctDNA has not been reliably detected in a significant proportion of patients with early stage disease [3, 17, 18, 21]. For clinical applications such as assessment of residual disease, surveillance, and early detection, specialized techniques with high sensitivity and specificity will thus need to be developed for the detection and quantification of mutant alleles in these settings [2, 3]. The ability to reliably detect ctDNA in CRC patients with minimal residual disease would enhance the clinical utility of ctDNA, not only for CRC tumor monitoring, but also for therapeutic decision-making.
To address this need, we have developed a new assay for the detection of ctDNA across a panel of cancer-related mutations with high sensitivity and specificity based upon sequence-specific synchronous coefficient of drag alteration (SCODA) technology [22], which enables efficient enrichment of mutant DNA from plasma. To evaluate this new assay, we analyzed tissue and plasma samples from a cohort of patients with non-metastatic and metastatic CRC. Additionally, we performed an exploratory analysis of longitudinal ctDNA measurements in patients undergoing surgical resection for hepatic metastatic disease.
RESULTS
Development and characterization of multiplexed SCODA mutation enrichment and detection platform
To develop an assay with high sensitivity and specificity across a large panel of defined mutations, we engineered a multiplexed version of the previously described sequence-specific synchronous coefficient of drag alteration (SCODA) assay (Figure 1A) [22]. This methodology overcomes issues of low specificity introduced by error rates in conventional PCR and sequencing techniques by simultaneously enriching for multiple mutant DNA sequences based on repetitive transient hybridization (see Supplementary Material).
Initial implementation of this multiplexed SCODA mutation enrichment and detection assay focused on 46 mutations in 4 genes (BRAF, EGFR, KRAS, and PIK3CA), constituting a set of commonly encountered mutations of clinical relevance in colorectal and non-small cell lung cancers (Table 1) [15, 23-31]. For each individual mutation, the assay underwent a series of analytical characterization experiments for specificity and sensitivity performed on reference wild-type DNA samples supplemented with defined quantities of synthetic DNA molecules carrying the corresponding mutant sequences. Analysis of control samples (without synthetic mutant DNA) demonstrated very low background signal, below the single molecule limit for the vast majority of assessments (Supplementary Material). In experiments with defined inputs of mutant sequences, the assay was able to consistently detect 10 or fewer input molecules of mutant DNA (Figure 1B). These characterization experiments enabled definition of the limit of detection (LOD) for each of the 46 mutations in the assay, which, for most mutations, represents as little as one copy of mutant DNA in plasma samples (Table 1 and Supplementary Material). Assessment of the assay in a set of healthy volunteers without a history of cancer revealed no detectable mutant DNA sequences in 43 of 47 subjects, with the remaining 4 cases showing signal at or near the LOD (Supplementary Material).
Table 1: Genes, mutations, and Limit of Detection (LOD) of multiplexed SCODA mutation detection assay.
Mutation |
LOD |
BRAF V600D |
0.001% |
BRAF V600E (1799T>A) |
0.002% |
BRAF V600E (1799_1800TG>AA) |
0.001% |
BRAF V600K |
0.001% |
EGFR E746_A750del_2235 |
0.001% |
EGFR E746_A750del_2236 |
0.001% |
EGFR E746_A750>IP |
0.001% |
EGFR E746_P753>VS |
0.001% |
EGFR E746_S752>A |
0.001% |
EGFR E746_S752>D |
0.001% |
EGFR E746_S752>I |
0.001% |
EGFR E746_S752>V |
0.001% |
EGFR E746_T751>A |
0.001% |
EGFR E746_T751>I |
0.001% |
EGFR E746_T751>IP |
0.001% |
EGFR E746_T751>V |
0.001% |
EGFR E746_T751>VA |
0.001% |
EGFR E746_T751del |
0.001% |
EGFR K745_E749del |
0.001% |
EGFR L747_A750>P_2238 |
0.001% |
EGFR L747_A750>P_2239 |
0.001% |
EGFR L747_E749del |
0.001% |
EGFR L747_P753>Q |
0.001% |
EGFR L747_P753>S |
0.001% |
EGFR L747_S752>Q |
0.001% |
EGFR L747_S752del |
0.001% |
EGFR L747_T751>P |
0.001% |
EGFR L747_T751>Q |
0.001% |
EGFR L747_T751>S |
0.001% |
EGFR L747_T751del |
0.001% |
EGFR T790M |
0.020% |
EGFR L858R (2573T>G) |
0.001% |
EGFR L858R (2573_2574TG>GT) |
0.001% |
KRAS G12A |
0.003% |
KRAS G12C |
0.001% |
KRAS G12D |
0.020% |
KRAS G12R |
0.001% |
KRAS G12S |
0.018% |
KRAS G12V |
0.001% |
KRAS G13C |
0.008% |
KRAS G13D |
0.018% |
PIK3CA E542K |
0.023% |
PIK3CA E545K |
0.024% |
PIK3CA Q546K |
0.007% |
PIK3CA H1047L |
0.001% |
PIK3CA H1047R |
0.002% |
The table shows the specificity limit of detection (LOD) for the assay, i.e. the minimum mutational abundance that can be confidently detected. The specificity limit is determined by performing the assay over multiple runs on multiple lots of reference wild-type DNA samples to calculate the average and standard deviation of the wild-type signal. For each mutation, LOD is defined as this average background signal + 3 standard deviations; in cases where the calculated LOD would be less than 1 molecule, the LOD is set to 1 molecule. For example, for 300 ng of total input DNA, a LOD of 0.001% corresponds to 1 mutant molecule in 100,000 genome equivalents
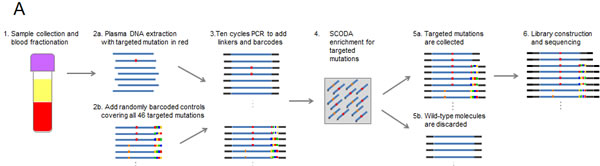
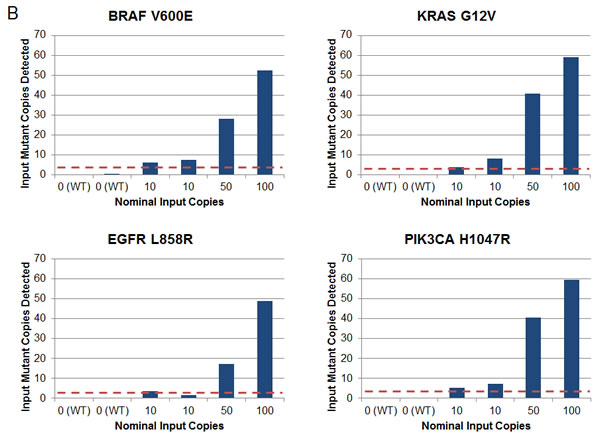
Figure 1: Multiplexed SCODA mutation enrichment and detection assay. A) Method. Following plasma preparation (1), plasma DNA is extracted (2a) and spiked with randomly barcoded internal controls (2b) representing every mutation in the panel, and amplified with a limited number of cycles in a multiplex PCR reaction (3). PCR primers are designed to amplify regions of the genome containing the 46 mutations of interest, and carry universal linker sequences and sample barcodes to enable subsequent sample processing and multiplexing. Amplified DNA and internal controls are enriched for the 46 mutations of interest by SCODA cyclic electrophoresis (4) in a gel containing hybridization probes against all mutations of interest. DNA enriched for mutant sequences (5a) is amplified through additional PCR as per conventional Illumina library construction, pooled with additional samples, and sequenced (6). B) Assay characterization. Each graph denotes results from samples created by titrating synthetic DNA carrying the specified mutation into reference wild-type (WT) DNA. Horizontal axes denote estimated input copies per sample, while the vertical axes denote the number of copies detected by the assay. Limit of Detection (LOD) is calculated as the greater of a single copy of mutant DNA or three standard deviations above the average background detected for each mutation in wild-type DNA samples. Input copies are limited to 10 and greater to limit the effect of sampling fluctuation in the titration. Each sample is 300 ng of DNA, such that 10 copies are equivalent to an allele fraction of ~ 0.01%.
Detection of plasma ctDNA in non-metastatic and metastatic colorectal cancer patients
As an initial application of this new mutation detection platform, we performed an exploratory analysis of tumor and pre-operative plasma samples from 38 CRC patients undergoing surgery (Table 2). This cohort included patients with non-metastatic (stage I-III, n=19) and metastatic (stage IV, n=19) disease. Median age was 63 years and 61% of patients were male. Among the stage IV patients, metastatic disease was most commonly confined to the liver (16/19). Over half (10/19) of the metastatic patients received preoperative systemic therapy, whereas none of the patients with non-metastatic disease received pre-operative treatment.
The multiplexed SCODA mutation detection assay was used to analyze extracted DNA from patient tumor tissue and pre-operative plasma samples for the presence of mutations in KRAS, PIK3CA, BRAF and EGFR as defined in our panel (Table 1). In tumor tissue, 68% of the cohort (26 of 38 patients) showed at least one detectable mutation from the panel, including 14 of 19 (74%) patients with metastatic disease and 12 of 19 (63%) patients with non-metastatic disease. The distribution of observed mutations was consistent with prior reports (Figure 2): 50% (19 of 38 patients) had a KRAS mutant tumor, 16% (6 of 38 patients) had a PIK3CA mutation, 8% (3 of 38 patients) showed a mutation in BRAF, and none showed an EGFR mutation. Of note, two patients harbored concurrent mutations in KRAS and PIK3CA. Importantly, the SCODA mutation detection platform demonstrated excellent concordance (18/19 cases tested, 95%) with a conventional quantitative PCR assay for KRAS performed on tumor tissue, a standard-of-care assessment for patients with metastatic CRC (Table 3). No discordances were observed when the conventional assay identified a KRAS mutation and the SCODA assay did not; the only discordance was a case where the SCODA assay found a very low KRAS mutant signal in tissue, at a level below the reported sensitivity for conventional KRAS PCR assays [32].
By comparison, the multiplexed SCODA mutation detection assay detected mutant DNA in the plasma of 53% (21 of 38) of CRC patients, including 20 of 26 patients (77%) found to have mutations in tumor tissue (Figure 2), with the identical allele detected in both tissue and plasma. Tissue/plasma concordance differed markedly between the metastatic and non-metastatic cases. In metastatic cases (stage IV), 14 of the 15 mutations (93%) identified in tumor tissue (14 patients) were also found in plasma. In contrast, for the non-metastatic patients (stages I-III), only 7 of the 13 mutations (54%) identified in tumor tissue samples (12 patients) could be detected in plasma. For the stage I patients, none of the three mutations observed in tissue were detectable in the corresponding plasma samples. Interestingly, in three of the patients with metastatic disease, a mutation was detected in plasma that was not observed in tissue; in all of those cases, the plasma-specific mutations were found in the PIK3CA gene.
Differences in the detection of ctDNA in metastatic and non-metastatic patients were not influenced by the amount of total cell-free DNA in the plasma, as levels of recovered cell-free DNA were not statistically different between these groups (p=0.096 by t-test), or between patients with and without detectable ctDNA (p=0.26 by t-test) (data not shown).
Table 2: Demographics and clinical characteristics of study patients.
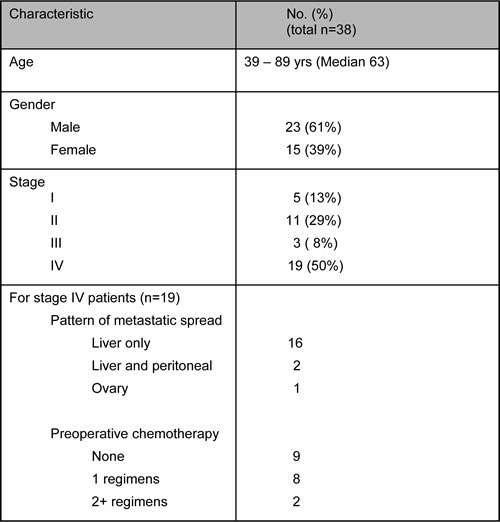
Table 3: KRAS mutational status of tumor tissue from metastatic colorectal cancer patients determined by multiplexed SCODA mutation detection assay vs. conventional quantitative PCR method.
Patient |
KRAS Mutation Status |
|
Multiplexed SCODA mutation detection |
Conventional quantitative PCR |
|
SUCRC12 |
G12D (41%) |
G12D |
SUCRC14 |
G13D (0.04%) |
WT |
SUCRC16 |
G12D (6.5%) |
G12D |
SUCRC21 |
G12V (4.75%) |
G12V |
P004 |
G12S (24%) |
G12S |
P008 |
G12D (17%) |
G12D |
A007 |
G12D (36%) |
G12D |
SUCRC24 |
G13D (1.7%) |
G13D |
SUCRC17 |
WT |
WT |
SUCRC26 |
WT |
WT |
SUCRC27 |
WT |
WT |
SUCRC18 |
G13D (3.8%) |
G13D |
SUCRC13 |
G12D (18%) |
G12D |
SUCRC20 |
G12D (0.285%) |
G12D |
P001 |
WT |
WT |
SUCRC15 |
WT |
WT |
SUCRC19 |
WT |
WT |
SUCRC22 |
WT |
Not analyzed |
SUCRC23 |
WT |
WT |
Note: WT, wild-type.
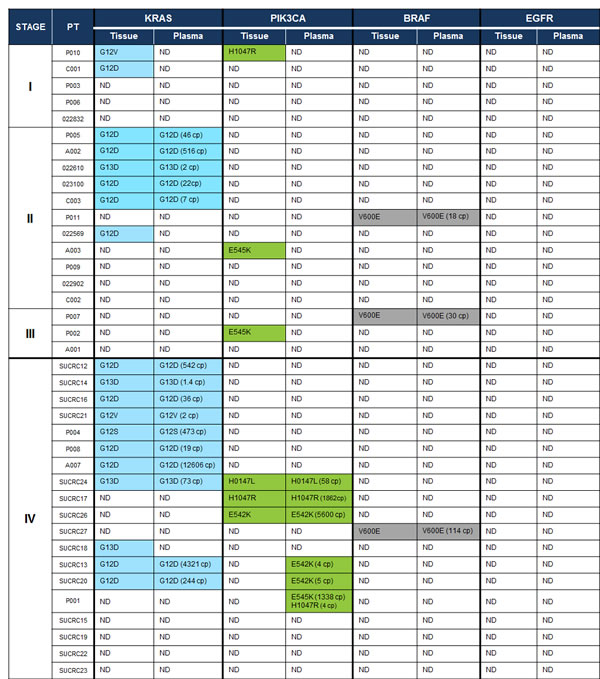
Figure 2: Mutations detected in tissue and plasma with the multiplexed SCODA mutation enrichment and detection assay. In highlighted cells, the detected mutation allele (or alleles) is specified, and for plasma, the number of detected copies (cp) of mutant DNA is also provided, normalized for a 5 mL input volume of plasma. ND, not detected.
Perioperative monitoring of ctDNA in stage IV colorectal cancer patients with potentially resectable disease
For the subset of patients with potentially resectable metastatic disease and detectable mutations by the SCODA assay in pre-operative plasma, we further analyzed post-operative plasma samples obtained on the fifth post-operative day and at routine follow-up visits in the context of CEA levels, cross-sectional imaging, and post-operative therapy. In particular, we sought to explore how ctDNA levels might be useful for this specific population of patients, potentially to inform the completeness of surgical resection and response to subsequent therapy, as well as to anticipate the recurrence of disease.
Figure 3 shows this longitudinal assessment of disease course for four illustrative patients undergoing surgical resection of metastatic disease. All four patients had detectable levels of ctDNA in pre-operative plasma, which declined following surgery, suggesting that ctDNA levels are generally reflective of disease burden. For patient SUCRC17 (Figure 3A), the plasma level of PIK3CA H1047R mutant DNA fell nearly 10-fold following surgery but remained detectable at high levels; this patient developed rapid disease recurrence on imaging, with concomitant rapidly rising ctDNA levels, despite a constantly normal CEA level. By contrast, for the other three patients (SUCRC14, SUCRC20, and SUCRC12; Figures 3B, 3C, and 3D, respectively), mutations detected in pre-operative plasma DNA were not detectable at the first assessment following surgery, presumably reflecting complete removal of metastatic lesions. The follow-up course for these three patients, however, revealed important differences in the relationships of ctDNA levels with clinical findings. Patient SUCRC14 had no detectable ctDNA in follow-up plasma at day 192, confirming a normal CEA value on an earlier follow-up, and demonstrated no evidence of disease recurrence; suspected progression of disease on imaging at day 132 was deemed a false positive after a biopsy showed no evidence of tumor (Figure 3B). Similarly, for patient SUCRC20, the two detectable mutations (KRAS G12D and PIK3CA E542K) in pre-operative plasma were not detected in the immediate post-operative period; however, follow-up assessments identified re-emergence of the KRAS G12D mutant DNA in plasma two months after surgery, while imaging and CEA levels suggested no evidence of recurrence. The patient ultimately developed progressive disease on imaging, as well as rising levels of CEA and ctDNA, despite the administration of systemic therapy (Figure 3C). Patient SUCRC12 had a detectable KRAS mutation in pre-operative plasma (G12D), which became undetectable following surgery (Figure 3D); however, a distinct KRAS mutation (G12V) became evident in plasma samples taken following surgery and also during follow up, 80 days later. No definitive imaging or CEA level correlate was identified for this signal, and following a course of capecitabine, the KRAS G12V mutant DNA was no longer detectable in plasma.
For the patients with two mutations found in ctDNA (SUCRC20 and SUCRC12), the dynamics of the mutations seen during follow-up may be indicative of clonal heterogeneity and evolution. For patient SUCRC20 in particular, the preoperatively analyzed plasma sample had concurrent mutations in KRAS and PIK3CA, but plasma samples taken during disease progression carried only the KRAS mutation; the absence of the PIK3CA mutation at these later timepoints may have implications for the selection of therapy, including the likelihood of response to targeted agents. It is also notable that, for patients SUCRC17, SUCRC20 and SUCRC12, elevated ctDNA levels appeared to anticipate disease progression better and at earlier time points than CEA levels. Although confirmation in additional large studies will be required, these observations suggest potential applications for ctDNA in the management of patients following resection of metastatic disease, with both prognostic and tumor monitoring utility, which should at least complement existing clinical tools.
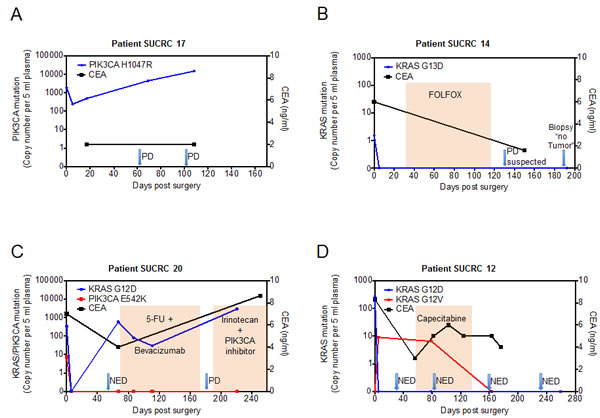
Figure 3: Perioperative dynamics of plasma mutation levels in patients with stage IV colorectal cancer. Panels A-D show plasma levels of detected mutations in the circulating tumor DNA (ctDNA, left y-axis) as well as CEA values (right y-axis) in patients that underwent surgery with the intent of complete metastasis resection. The value at time point 0 represents the preoperative ctDNA level. ctDNA levels (cp, copies) are normalized for 5 mL input volume of plasma. After blood draw, surgery was performed at day 0. Arrows indicate imaging assessments by computed tomography (CT). In panels B, C and D, colored shading indicates the administration of chemotherapy. PD, progressive disease; NED, no evidence of disease. CEA normal range is 0-5 ng/mL.
DISCUSSION
In this study, we introduced a novel multiplexed SCODA mutation detection platform that identifies ctDNA in patient plasma with high sensitivity and specificity. Application of this assay in an exploratory analysis of CRC patients revealed lower rates of ctDNA detection in plasma from non-metastatic patients, despite the analytical capabilities of our assay, suggesting a biological cause. By contrast, the assay reliably detected ctDNA in the plasma of patients with metastatic disease, and longitudinal assessments in metastatic patients undergoing surgical resection and chemotherapy provide a rationale for prognostic and tumor monitoring applications, which warrant further study in this patient population.
In some clinical settings, the measurement of plasma ctDNA can be very challenging due to the low level of signal and the high background level of circulating wild-type DNA. Through enrichment of mutant alleles, the SCODA method effectively removes background wild-type DNA, leading to a reduction in the false-positive rate and ultimately enabling detection of as little as single copies of mutant DNA. As such, this method is extremely sensitive (with a limit of detection as low as 0.001%) that meets – and in some cases exceeds – what has previously been reported for droplet digital PCR, BEAMing, and next generation sequencing approaches [3, 4, 33, 34]. Importantly, this level of sensitivity and specificity is achieved without previous knowledge of each patient’s tumor mutation profile from tissue, which, together with multiplexed assessment of a large and expandable panel of defined mutations, should readily enable generalized application without patient-specific assay development, as is required for other methods [3, 35].
Several groups have studied ctDNA in non-metastatic and metastatic CRC patients [3, 4, 17-19, 36]. Our observed concordance of 54% between tissue and plasma mutations in non-metastatic patients is consistent with previous reports [17, 18, 36]. Bettegowda et al. used a combination of methods, including BEAMing and SafeSeq [34, 37], to show that 49-78% of patients with localized disease had detectable ctDNA [17]. Lecomte et al. found that 63% of a group of stage I, II and III CRC patients had ctDNA detectable by mutant allele-specific amplification directed against KRAS and methylation-specific PCR for p16 [18]. Based on the technical performance of our assay, the absence of detectable ctDNA signal in the plasma of the early-stage patients appears to be likely related to differences in tumor burden, anatomy, or biology, rather than inadequate analytical sensitivity. Similar considerations have been described for circulating tumor cells, which are also much less abundant in early stage disease [38-42]. Associations of ctDNA levels with tumor burden, as evidenced by tumor stage and CEA for example, have been reported [3, 20], although ctDNA levels vary considerably within stage [17], suggesting additional contributory factors. In this regard, anatomic differences may also be explanatory, as early stage tumors may have less access to larger blood vessels owing to more limited depth of invasion of the bowel wall. Additionally, variation in tumor biology is also likely to underlie these differences, and may include properties of metastatic disease related to higher proliferative and apoptotic rate, and also lower efficiency of host mechanisms for clearing necrotic tumor cells [2, 43]. It is possible that higher sensitivity in early-stage disease could be achieved with a broader assessment of tumor molecular profiles, including early mutational events (e.g., APC and TP53 which may be shared across all subclones) as well as copy-number variations and patterns of hyper- and hypomethylation [3, 18, 44, 45]. Nevertheless, the available data suggests that other methods for assaying ctDNA (such as by broad epigenetic panels) will need to be clinically tested to robustly address clinical questions for patients with early-stage disease.
In our metastatic CRC patients, we found a high concordance rate (93%) between plasma and tumor mutations, which is consistent with previous reports [4, 17]. These data suggest that ctDNA may be a meaningful non-invasive surrogate for tissue biopsies for patients with metastatic disease. In addition, in three cases we detected a PIK3CA mutation in plasma DNA which was not present in the corresponding metastatic tissue sample, indicating that ctDNA may further provide insight into tumor heterogeneity and thus avoid tumor sampling bias [46]. During the longitudinal assessment of ctDNA levels in patients with resectable liver metastases from CRC, we observed two cases where two different mutations were detected in plasma DNA, presumably reflecting different metastatic subclones, of which one re-emerged following surgery. As such, ctDNA may offer valuable real-time insight into the clonal tumor evolution during the course of treatment, and thus offer the opportunity to direct therapy to the dominant subclone at any given point in a patient’s treatment course. Finally, our results additionally suggest that ctDNA levels following surgery for metastatic disease may be prognostic for disease recurrence. For patients with resectable metastatic disease, assessment of residual disease following surgery, along with the ability to anticipate recurrence and to gain insight into the molecular profile of the tumor tissue, may eventually enable optimization of post-operative therapy. All of these concepts warrant further study and will require further testing in larger patient cohorts.
Interpretation of these results should be caveated by the limitations of our study. Broader application of this technology in clinical practice will also require a larger panel of mutations with representation of additional genes relevant to CRC. In this regard, an expanded assay is currently under development. Furthermore, the patient cohort included in this exploratory study is relatively small, and therefore does not permit definitive conclusions. Nevertheless, this study represents proof of concept for the multiplexed SCODA assay for analyzing tumor mutations in plasma and is informative with respect to the technical challenges facing ctDNA assessment, particularly in early-stage disease, as well as for the detection of residual disease following surgical resection.
In this study, we present a multiplexed SCODA mutation detection assay for measuring plasma ctDNA, which has been characterized here for its ability to assess a panel of 46 mutations with very high sensitivity and specificity. Using this assay we provide evidence for ctDNA as a very promising non-invasive tool for real-time tumor molecular profiling in CRC, including the setting of resectable metastatic disease, where more accurate information about residual disease, prognosis and tumor molecular evolution should enable optimization of perioperative therapy. While our findings require further investigation, the technology platform we have described should enable studies designed to establish the utility of ctDNA in improving patient care across a variety of clinical settings.
MATERIALS & METHODS
Patients and sample collection
From September 2013 to September 2014, clinical data, tissue and plasma specimens were collected from a total of 38 patients with stage I, II, III, and IV colorectal cancer at multiple sites in the United States and Europe. Eligibility included histologically confirmed adenocarcinoma of the colon or rectum, availability of a plasma specimen drawn prior to surgery, and sufficient frozen tissue for molecular analysis. With the exception of 4 patients with advanced disease, all patients underwent surgery with curative intent, including 16 patients with resectable metastatic disease recruited at Stanford University Hospital. Additionally, plasma samples from 47 healthy donors were also evaluated. Blood samples were collected in Cell-Free DNA BCT tubes (Streck Inc., Omaha, NE) [47] pre-operatively, within 1-3 days prior to surgery. For a subset of patients undergoing curative resection of metastatic disease, additional blood collections were performed post-operatively and at follow-up visits. Tumor tissue was collected during surgery, transported on ice and stored at -80°C until further processing.
The study was conducted according to the ethics and Institutional Review Board standards of the participating hospitals and clinics. All patients provided written informed consent prior to being included into the study.
Mutation enrichment and detection
The methodology for mutation enrichment and detection used in this study is based on the sequence-specific synchronous coefficient of drag alteration (SCODA) technology [22]. This multiplexed SCODA mutation enrichment and detection platform is depicted schematically in Figure 1. Full details of the mutation enrichment and detection workflow, including data analysis, are provided in the Supplementary Material.
Briefly, plasma was prepared by conventional protocols from 5-10 mL of whole blood from each patient, from which DNA was extracted. Synthetic internal positive controls (IPCs) for each mutation, used to calculate the process yield, to enable quantitation of mutant DNA, and to monitor assay performance, were then added to each sample. The resulting DNA sample was then subjected to a multiplex PCR reaction (at low amplification to minimize PCR errors prior to enrichment) with primers carrying sample-specific barcodes (to enable sample multiplexing) as well as universal linker sequences which serve as priming sites during library construction.
The amplified sample was then enriched for mutant DNA by a multiplexed SCODA mutation enrichment method on a device specifically engineered for this application. Following enrichment, a sequencing library was constructed by PCR using primers complementary to the universal linkers, and tagged with adaptors (Illumina, San Diego, CA). The library for each sample was then quantified by quantitative PCR, samples were pooled into sets of six, each having a unique sample barcode, and sequenced on the MiSeq platform (Illumina, San Diego, CA).
Forward and reverse reads from the MiSeq FastQ files were merged to give a single read for each cluster, de-multiplexed according to sample barcode and then aligned to the amplicons within the panel. Clusters were assigned mutant status if they contained one of the 46 mutations in the multiplexed SCODA mutation detection assay. Clusters arising from genomic DNA and IPC molecules were distinguished by the presence of random identifier sequences in the IPCs. IPC molecules were then binned according to the random identifier sequences, and the number of clusters arising from each unique combination of identifiers (random identifier sequences, barcodes, mutations) was counted. From these distributions, we measured the average cluster yield for each mutation in each sample, which was used to calculate the number of input mutant copies from the number of mutant clusters observed. To make mutation calls, the number of input mutant copies was compared to the limit of detection for each mutation, defined as 3 standard deviations above the mean background signal (observed over multiple runs of reference wild-type samples). Positive mutant calls were made only where at least a single mutant copy was observed and the abundance of the mutation (percentage with respect to all input genomes) was greater than the limit of detection.
ACKNOWLEDGEMENTS
This study was supported by the John and Marva Warnock Research Fund and kind contributions from Vladimir and Natalie Ermakoff. The authors thank Haiyan Liu, Scott Dashner, Aparna Natarajan, Abdi Khalif, Laura Mai, and Gosuke Shibahara for excellent technical assistance.
REFERENCES
1. Swarup V, Rajeswari MR. Circulating (cell-free) nucleic acids--a promising, non-invasive tool for early detection of several human diseases. FEBS Lett. 2007; 581: 795-799.
2. Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011; 11: 426-437.
3. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA, Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14: 985-990.
4. Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, Del Rio M, Lamy PJ, Bibeau F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014; 20: 430-435.
5. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013; 501: 338-345.
6. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012; 481: 306-313.
7. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014; 64: 104-117.
8. Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011; 29: 2011-2019.
9. Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011; 22: 1535-1546.
10. Loewenstein MS, Zamcheck N. Carcinoembryonic antigen (CEA) levels in benign gastrointestinal disease states. Cancer. 1978; 42: 1412-1418.
11. Tanaka T, Tanaka M, Tanaka T, Ishigamori R. Biomarkers for colorectal cancer. Int J Mol Sci. 2010; 11: 3209-3225.
12. Litvka A, Cercek A, Segal N, Reidy-Lagunes D, Stadler ZK, Yaeger RD, Kemeny NE, Weiser MR, Pessin MS, Saltz L. False-positive elevations of carcinoembryonic antigen in patients with a history of resected colorectal cancer. J Natl Compr Canc Netw. 2014; 12: 907-913.
13. Selzner M, Hany TF, Wildbrett P, McCormack L, Kadry Z, Clavien PA. Does the novel PET/CT imaging modality impact on the treatment of patients with metastatic colorectal cancer of the liver?. Ann Surg. 2004; 240: 1027-1034; discussion 1035-1026.
14. Metser U, You J, McSweeney S, Freeman M, Hendler A. Assessment of tumor recurrence in patients with colorectal cancer and elevated carcinoembryonic antigen level: FDG PET/CT versus contrast-enhanced 64-MDCT of the chest and abdomen. AJR Am J Roentgenol. 2010; 194: 766-771.
15. Jones RP, Stattner S, Sutton P, Dunne DF, McWhirter D, Fenwick SW, Malik HZ, Poston GJ. Controversies in the oncosurgical management of liver limited stage IV colorectal cancer. Surg Oncol. 2014; 23: 53-60.
16. Lam VW, Spiro C, Laurence JM, Johnston E, Hollands MJ, Pleass HC, Richardson AJ. A systematic review of clinical response and survival outcomes of downsizing systemic chemotherapy and rescue liver surgery in patients with initially unresectable colorectal liver metastases. Ann Surg Oncol. 2012; 19: 1292-1301.
17. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6: 224ra224.
18. Lecomte T, Berger A, Zinzindohoue F, Micard S, Landi B, Blons H, Beaune P, Cugnenc PH, Laurent-Puig P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer. 2002; 100: 542-548.
19. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486: 532-536.
20. Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486: 537-540.
21. Marzese DM, Hirose H, Hoon DS. Diagnostic and prognostic value of circulating tumor-related DNA in cancer patients. Expert Rev Mol Diagn. 2013; 13: 827-844.
22. Thompson JD, Shibahara G, Rajan S, Pel J, Marziali A. Winnowing DNA for rare sequences: highly specific sequence and methylation based enrichment. PLoS One. 2012; 7: e31597.
23. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330-337.
24. Rizzo S, Bronte G, Fanale D, Corsini L, Silvestris N, Santini D, Gulotta G, Bazan V, Gebbia N, Fulfaro F, Russo A. Prognostic vs predictive molecular biomarkers in colorectal cancer: is KRAS and BRAF wild type status required for anti-EGFR therapy?. Cancer Treat Rev. 2010; 36 Suppl 3: S56-61.
25. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, et al. Aspirin Use, Tumor PIK3CA Mutation, and Colorectal-Cancer Survival. N Engl J Med. 2012; 367: 1596-1606.
26. Ogino S, Meyerhardt JA, Irahara N, Niedzwiecki D, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, Hantel A, Benson AB 3rd, Goldberg RM, Bertagnolli MM, Fuchs CS. KRAS mutation in stage III colon cancer and clinical outcome following intergroup trial CALGB 89803. Clin Cancer Res. 2009; 15: 7322-7329.
27. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11: 753-762.
28. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543-550.
29. Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E, Meldgaard P. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer. 2014; 120: 3896-3901.
30. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350: 2129-2139.
31. Sun Y, Ren Y, Fang Z, Li C, Fang R, Gao B, Han X, Tian W, Pao W, Chen H, Ji H. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010; 28: 4616-4620.
32. Zuo Z, Chen SS, Chandra PK, Galbincea JM, Soape M, Doan S, Barkoh BA, Koeppen H, Medeiros LJ, Luthra R. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol. 2009; 22: 1023-1031.
33. Taly V, Pekin D, Benhaim L, Kotsopoulos SK, Le Corre D, Li X, Atochin I, Link DR, Griffiths AD, Pallier K, Blons H, Bouche O, Landi B, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013; 59: 1722-1731.
34. Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006; 3: 551-559.
35. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW Jr, Alizadeh AA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014; 20: 548-554.
36. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102: 16368-16373.
37. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011; 108: 9530-9535.
38. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004; 351: 781-791.
39. Lucci A, Hall CS, Lodhi AK, Bhattacharyya A, Anderson AE, Xiao L, Bedrosian I, Kuerer HM, Krishnamurthy S. Circulating tumour cells in non-metastatic breast cancer: a prospective study. Lancet Oncol. 2012; 13: 688-695.
40. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007; 450: 1235-1239.
41. Sastre J, Maestro ML, Puente J, Veganzones S, Alfonso R, Rafael S, García-Saenz JA, Vidaurreta M, Martín M, Arroyo M, Sanz-Casla MT, Díaz-Rubio E. Circulating tumor cells in colorectal cancer: correlation with clinical and pathological variables. Annals of Oncology. 2008; 19: 935-938.
42. Iinuma H, Okinaga K, Egami H, Mimori K, Hayashi N, Nishida K, Adachi M, Mori M, Sasako M. Usefulness and clinical significance of quantitative real-time RT-PCR to detect isolated tumor cells in the peripheral blood and tumor drainage blood of patients with colorectal cancer. Int J Oncol. 2006; 28: 297-306.
43. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32: 579-586.
44. Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O’Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B, Diaz LA Jr, Velculescu VE. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012; 4: 162ra154.
45. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61: 759-767.
46. Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010; 16: 790-799.
47. Norton SE, Luna KK, Lechner JM, Qin J, Fernando MR. A new blood collection device minimizes cellular DNA release during sample storage and shipping when compared to a standard device. J Clin Lab Anal. 2013; 27: 305-311.