
Introduction
UDP-glucose 6-dehydrogenase (UGDH) is a cytosolic and nuclear hexameric enzyme that catalyzes the conversion of UDP-glucose to UDP-glucuronic acid (UDP-GlcUA). UGDH plays a role in xenobiotic metabolism via the glucuronidation pathway, sugar metabolism, production of extracellular matrix (ECM) precursors, and proteoglycan (PG) synthesis, which suggests that it may be a potential therapeutic target for a variety of diseases (Figure 1) [1, 2]. In this review, the role of UGDH in tumor progression across various cancers and the on-going efforts to pharmacologically target UGDH are discussed. Multiple studies have demonstrated UGDH’s clinical relevance to the field of oncology, and this review summarizes the evidence implicating UGDH as a candidate biomarker of aggressive cancer phenotypes and/or a potential therapeutic target to mitigate tumor progression and enhance patient survival.
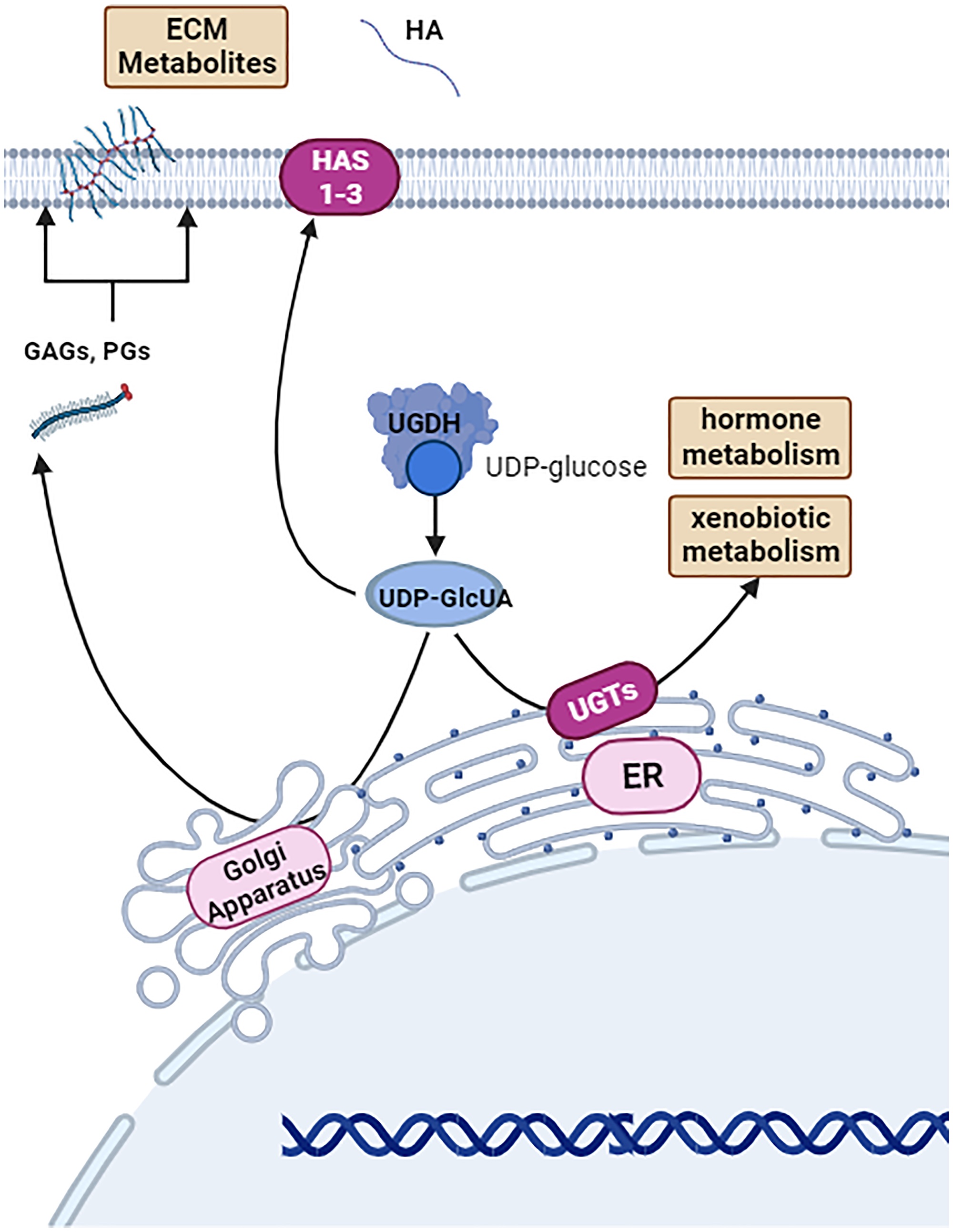
Figure 1: Basal function of UDP-glucose-6-dehydrogenase (UGDH).
UGDH is a cystolic and nuclear hexameric enzyme that metabolizes UDP-glucose to UDP-glucuronic acid (UDP-GlcUA) as a part of xenobiotic metabolism, sugar metabolism, and generating molecule precursors for the extracellular matrix. Image was created by https://www.biorender.com (2023).
The role of UGDH in oncology
The importance of UGDH in human cancer is a topic of recent interest, both from a mechanistic and prognostication standpoint [3–6]. This section will delineate what is known about the role of UDGH across different human cancers including lung, breast, esophageal, hepatocellular carcinoma (HCC), prostate, ovarian, colorectal and melanoma with a focus on mechanistic categories (summarized in Table 1).
Table 1: Studies that discuss the role of UGDH in oncology categorized by tumor type
| Study | Cancer type | Cell line studied | Mechanisms discussed | Primary findings |
|---|---|---|---|---|
| Paul et al., 2016 | Lung (NSCLC) | • NCI-H460 • NCO-H460R | • Chemotherapy (etoposide) resistance | • Etoposide treatment led to alteration of 83 proteins in NSCLC lung cancer cell lines (UGDH included) • UGDH not functionally a/w chemotherapeutic resistance |
| Wang et al., 2019 | Lung (adeno) | • A549 • H1299 • PC-9 | • EMT factor stabilization • Downstream signaling pathways | • Higher levels of activated (phosphorylated) UGDH associated with increased stability of EMT transcription factor SNAI1 • UGDH activation induced by EGF binding to EGFR • Inhibiting UGDH in vitro and in vivo decreased migratory and metastatic phenotypes; higher levels of pUGDH in patient associated with higher mortality • MEK inhibitor abrogated EGF induced pUGDH |
| Hagiuda et al., 2019 | Lung (adeno) | • LC-2 • A549 | • Cellular localization | • Nuclear localization of UGDH associated with more aggressive, migratory phenotypes • Worse survival in patient samples with nuclear localization of UGDH |
| Richter et al., 2021 | Lung (SCC*) Head and neck (SCC) | • NCI-H2170 (lung) • PCI-13.1 (H/n) | • Differential expression across cancer type | • UGDH upregulated in squamous cell cancer lung cancer • Levels of UGDH not different enough between SCC lung and head/neck cancer to distinguish them |
| Arnold et al., 2019 | Breast (TNBC) | • MDA-MB-231 | • EMC modulation • HA regulation • Lipid metabolism | • Higher levels of UGDH in more invasive malignant BC patient samples • Overexpression of EMT TF increased expression of UGDH • Depleting UDP-GlcUA inhibited mesenchymal phenotypes including cellular invasion and colony formation in vitro and metastatic phenotype in vivo • HA rescued UGDH KD phenotype • Fatty acid metabolism and PPAR-gamma pathway altered by UGDH KD |
| Teoh et al., 2021 | Breast (TNBC) | • 6DT1 | • UDP glucose metabolism • Downstream signaling pathways | • High UGDH expression associated with worse patient survival • UGDH KD decreased migratory and metastatic phenotype of BC cells in vitro and in vivo • High UGDH a/w TP53 mutations and copy number alterations in BRCA1 and PIK3CA |
| Vitale et al., 2021 | Breast (TNBC) | • MDA-MB-231 | • Chemotherapy (epirubicin resistance) • HA • ECM modulation | • Higher levels of UGDH correlated with worse prognosis in patients with TNBC who received chemo • UGDH KD associated with epirubicin resistance • UGDH KD resulted in increased epirubicin accumulation, increased apoptosis and positive modulation of autophagy • Epirubicin resistance potentially related to HA metabolism and ECM modulation |
| Lin et al., 2020 | Ovarian | • TOV21G | • EMT • Downstream signaling | • UGDH overexpressed in ovarian cancer tissue • UGDH KD decreased metastatic ability in vitro and in vivo • UGDH depletion down-regulated EMT markers and ERK/MAPK pathway |
| Liu et al., 2020 | Esophageal | • Human samples | • UGDH expression | • UGDH-AS1 (lncRNA) levels correlated with overall survival in patients • UGDH-AS1 had low expression level in samples • UGDH mRNA not a/w patient survival |
| Oyinlade et al., 2018 | Brain (GBM) | • U87 | • ECM modulation | • KLF4 upregulates UGDH expression by methylating CpGs • UGDH required for KLF4-induced cell migration • UGDH KD decreases GAG abundance, cell proliferation and migration in vitro and tumor growth/ migration in vivo • UGDH KD a/w decreased expression of ECM proteins tenascin C, brevican |
| Wei et al., 2009 | Prostate | • LNCaP C33 (low passage) • LNCaP C81 (high passage) | • Hormone metabolism • ECM modulation | • Dihydrotestosterone (DHT) increases UGDH expression in androgen-dependent cells • Increased metabolism of DHT in AD cells than non-AD cells • Increased DHT metabolism corresponded to slower cellular growth |
| Huang et al., 2010 | Prostate | • Human samples | • UGDH expression | • Higher UGDH expression in cancerous acini compared to noncancerous controls |
| Zimmer et al., 2016 | Prostate | • LNCaP (AD) • LNCaP 81 (CR) | • Hormone metabolism • ECM modulation | • CR tumor cells express higher levels of UGDH • AR-dependent expression of PSA and UGDH downregulated in CR cells • UDP-sugar flux increase through PG and GAG synthesis pathways rather than glucuronidation |
| Zimmer et al., 2021 | Prostate | • LNCaP (AD) • LNCaP (CR) | • Hormone metabolism • ECM modulation | • Overexpression of UGDH in AD cells blunted androgen-dependent gene expression, increased PG synthesis, and increased migratory phenotype • Overexpression of UGDH decreased growth suppression seen with enzalutamide • UGDH KD decreased PG production, restored AD, and sensitized cells to enzalutamide |
| Wang et al., 2010 | Colorectal Cancer (CRC) | • HCT-8 | • ECM modulation | • UGDH KD associated with decreased UDP-GlcUA and GAG production • Treatment with 4-MU decreased cell aggregation and motility in vitro • Cell aggregation and migration restored with exogenous HA |
| Shen et al., 2016 | Colorectal cancer (CRC) | • Human samples | • ECM modulation • Cell metabolism | • Assessed differentially expressed genes (DEGs) in CRC samples • UGDH identified in a network of genes functionally associated with metabolism-related functions |
| Deen et al., 2016 | Melanoma | • MV3 • C8161 | • ECM modulation • Glucose metabolism | • Recycling of HA synthesis enzymes controlled by cytosolic levels of UDP-GlcUA and UDP-GlcNAc • Lower levels of UDP-GlcUA and UDP-GlcNAc inhibits HA synthesis • Correlation between HA content in human melanoma samples |
| Fan et al., 2009 | Hepatocellular Carcinoma (HCC) | • LCI-D20 | • Drug mechanism (cell differentiation agent-II CDA-II) | • CDA-II suppresses growth and metastasis • 27 genes including UGDH differentially expressed in response to treatment with CDA-II • UGDH downregulated in response to CDA-II • Downstream genes c-myc, N-ras, and MMP-9 down-regulated |
UGDH as a prognostication marker of tumor progression
UGDH became an oncologic target of interest in the early 2000s primarily within breast (BC) and prostate cancer (PC) research [7, 8]. Several additional studies in BC have further established that higher levels of UGDH are associated with worse prognoses for patients (particularly those with triple negative breast cancer (TNBC) receiving chemotherapy [5]) and more invasive, metastatic phenotypes in BC samples [9, 10]. Similar findings are demonstrated in lung cancer; specifically in 2019, Wang et al., defined the role of UGDH in promoting the stability of epithelial-to-mesenchymal transition (EMT) factors in lung adenocarcinoma. They also reported that patients with tumors expressing higher levels of phosphorylated UGDH (specifically Y473) had lower median survival than those who did not [11]. While UGDH phosphorylation has not been mechanistically explored in other cancers or reported in normal physiology, this study did correlate phosphorylated UGDH to a metastatic and pro-EMT phenotype of lung adenocarcinoma. In contrast, higher levels of UGDH are not ubiquitously associated with worse prognoses in all human cancer patient samples; for example, within esophageal cancer, there are conflicting findings. Liu et al., 2020 explored the prognostic value of lncRNA and found that UGDH-AS1 had lower expression levels in esophageal cancer samples while Luo et al., 2021 did not find a correlation between mRNA levels of UGDH and prognosis/survival for patients with esophageal cancer [12, 13]. For prostate cancer, higher levels of UGDH have been observed in cancerous prostate acini than non-cancerous prostate tissue—a finding that established UGDH as a potential biomarker for PC [8].
UGDH regulation of the extracellular matrix (ECM) and hyaluronic acid production
UGDH catalyzes conversion of UDP-glucose to UDP-GlcUA to generate proteoglycans (PG) and glycosaminoglycans (GAGs) for the ECM. PGs and GAGs are building blocks of the extracellular matrix (ECM) and are critical metabolites in normal cellular structure and function such as wound healing, immune system processes, chondrogenesis, and embryonic development [2, 14, 15]. One GAG molecule of particular importance is hyaluronan or hyaluronic acid (HA) due to its abundance in the ECM and role in cell differentiation, survival, angiogenesis, and tumor formation [16, 17]. The importance of the ECM in tumor progression, growth, and migration has become increasingly complex as our understanding of the tumor microenvironment expands. The role of UGDH and its downstream product UDP-GlcUA in producing ECM precursors HA, proteoglycans (PGs) and other glycosaminoglycans (GAGs) is particularly salient to studies assessing tumor aggression and migratory capacity. These ECM precursors, specifically HA, have been implicated in worse patient prognoses, metastatic phenotypes, and higher proliferation/migratory capabilities of cancer cell lines in vitro. Independent of UGDH, high HA levels are considered a marker of malignancy in several types of solid tumors including melanoma, bladder, lung, prostate, breast, and colon [18–23]. HA is shown to regulate the tumor microenvironment by promoting cell adhesion, migration, and proliferation via signal transduction and interaction with different receptor signaling pathways. In doing so, HA promotes an invasive/metastatic phenotype through induction of EMT promoting pathways [24, 25]. As a necessary element in HA formation, UGDH knock down (UGDH KD) is shown to decrease HA formation and thus alter the associated aggressive tumor microenvironment phenotype [4, 9].
Within the field of breast cancer research, the role of HA in relation to UGDH has been primarily studied in TNBC. Arnold et al., 2019 demonstrated that depleting UDP-GlcUA (via knocking down UGDH) was sufficient to decrease HA production and thereby inhibit invasion, colony formation, and tumor growth both in vitro and in vivo. When HA was added back to these experiments, they were able to rescue 80 to 90% of the migratory phenotype, suggesting that these findings were significantly dependent upon HA production.
In a different study focused on the potential role of UGDH expression and HA metabolism in epirubicin resistance in TNBC, the authors found that while UGDH expression was correlated with worse prognosis, UGDH KD contributed to drug resistance. This was surprising as knocking down UGDH inhibited glucuronidation, which is responsible for the cellular elimination of epirubicin; and thereby increased intra-cellular epirubicin levels. Paradoxically, this did not increase cytotoxicity; rather, UGDH KD was associated with increased autophagy of cancer cells, which is involved in the development of epirubicin resistance [5]. The authors also observed that UGDH KD in combination with epirubicin treatment was associated with modulation of HA, HA synthesis (HAS) enzymes, and HA degrading enzymes synthases (HYAL). These enzymes are responsible for HA turnover; the authors found that more deposition and catabolism of HA resulted in a more resistant phenotype [5]. From this observation, they proposed that UGDH KD could produce an ECM with abnormal HA production and metabolism that ultimately favors treatment resistance. Thus, while there are demonstrated relationships between UGDH and HA in breast cancer in experimental settings, these relationships are less well studied in clinical practice.
The relationship of UGDH and HA metabolism has also been shown to play a role in tumor aggressiveness in melanoma, colorectal cancer, nasopharyngeal and primary brain tumors. In their 2016 study, Deen et al., established the importance of UD-GlcUA and UDP-GlcNAc levels to the intracellular movement and processing of hyaluronan synthases 1–3 (HAS1-3) for melanoma. Specifically, lower levels of UDP-GlcUA resulted in more HAS endocytosis and therefore inhibition of HA synthesis via a regulatory feedback cycle. UGDH was critical to this cycle as the authors demonstrated that decreasing its expression and activity correlated with lower levels of both UDP-GlcUA, and HA. Within tissue samples, the authors also correlated levels of hyaluronan and UGDH mRNA with different stages of melanoma development and thus suggested that UDP-sugar metabolism is critically linked with hyaluronan and may support progression of melanoma [26]. Similar phenotypic findings have been reported in colorectal cancer with UGDH KD effectively decreasing cell migration and motility in both transwell migration assays and 3-D collagen gels [27]. The authors were able to rescue the migratory phenotype with subsequent treatment of the colorectal cells with HA in vitro [27]. Within nasopharyngeal carcinoma, (a tumor with high metastatic potential due, in part, to high expression levels of Epstein-Barr virus latent membrane protein 2A (LMP2A)), LMP2A-induced higher expression of UGDH, subsequently increased GAG synthesis [28]. They were able to modulate this activation pathway by overexpressing or inhibiting specificity protein 1 (Sp1) upstream of LMP2A and UGDH [28].
The relationship of elevated GAG formation to more aggressive phenotypes in primary brain tumors (specifically glioblastoma multiforme (GBM)) had been well established when Oyinlade et al., 2018 directly linked upregulation of UGDH expression to increased GAG levels in GBM. Methylation of Kruppel-like factor 4 (KLF4) upregulated the expression of UGDH resulting in higher intra-tumoral levels of GAGs and thereby increased proliferation and migration of GBM cell lines. Subsequently, knocking down UGDH in vitro and in vivo abrogated this aggressive phenotype while decreasing the expression of specific ECM proteins (tenascin C, brevican) [4]. Thus, multiple cancer models demonstrate a direct link between UGDH activity, ECM precursor formation, and subsequent aggressive and metastatic oncologic phenotypes.
UGDH regulation of EMT in metastasis
Closely linked to HA production and ECM modulation is the role of UGDH in regulating genes responsible for the epithelial to mesenchymal transition (EMT). This process has been well studied in lung, breast, ovarian, brain, and colon cancer. In a landmark paper on the role of UGDH in lung cancer, Wang et al., 2019 directly linked UGDH with enhanced mRNA stability of the EMT factor SNAI1. This connection mechanistically explained the increased in vitro and in vivo tumor cell migration and metastasis associated with higher levels of UGDH in lung adenocarcinoma. Of note, this interaction was dependent on EGFR-mediated attachment of the RNA binding protein HuR to phosphorylated UGDH. Subsequently this phosphorylated UGDH was responsible for converting UDP-Glc (which prevents HuR from binding to mRNA) to UDP-GlcUA, allowing HuR to bind to and stabilize SNAI1 mRNA and promoting an aggressive EMT phenotype [11]. While not as clear mechanistically, UGDH KD has also been shown to decrease expression of EMT transcription factors SNAIL, SIP-1, and matrix mellatoprotease protein 2 (MMP2) in ovarian cancer cell models. In these ovarian cancer models, knocking down UGDH also decreased the activity of actin as a key migratory protein. While the mechanism for increased mRNA stability presented by Wang et al., 2019 provides potential mechanism for UGDH’s role in pro-EMT phenotypes, there are likely many other mechanisms contributing to this process across various cancers.
UGDH-mediated EMT gene expression is not shown to consistently modulate migratory phenotypes across all tumor types. For example, in breast cancer, Teoh et al., 2020 demonstrated that increased UGDH levels were significantly associated with more aggressive migratory phenotypes; however, many EMT genes were not transcriptionally inhibited by decreased UGDH expression [10]. Paradoxically, both fibronectin (Fn1) and Six1, glycoproteins in the extracellular matrix associated with more aggressive breast cancer phenotypes, were upregulated in UGDH KD. This suggests that EMT gene modulation was not responsible for the observed more aggressive tumorigenic phenotypes in this study. One study also suggested that rather than activating EMT, UGDH activation and subsequent ECM remodeling may be downstream of EMT initiation as the authors demonstrated that overexpressing EMT transcription factors SNAI1 and TW1ST induced UGDH expression [9]. This increased UGDH expression in turn increased flux through HA and UDP-sugar pathways [9]. They proposed that rather than it being the promoter of EMT, UGDH was a critical enzyme in the glucose metabolic reprogramming that accompanies the EMT process [9].
The role of UGDH in cancer biology
UGDH’s role in canonical cancer signaling pathways
To better identify mechanistic similarities of UGDH amongst various cancers, it is critical to understand the effect of modulating UGDH on downstream signaling pathways. While many of these relationships are not fully understood, several studies outlined previously have described connections between intracellular signaling enzymes such as MAPK, ERK, and AKT that provide a groundwork for our understanding of how UGDH affects these intracellular processes (Figure 2) [10, 28]. Furthermore, it has been well established that MAPKs are associated with cancer cell proliferation, survival, metastatic capacity, and motility [29].
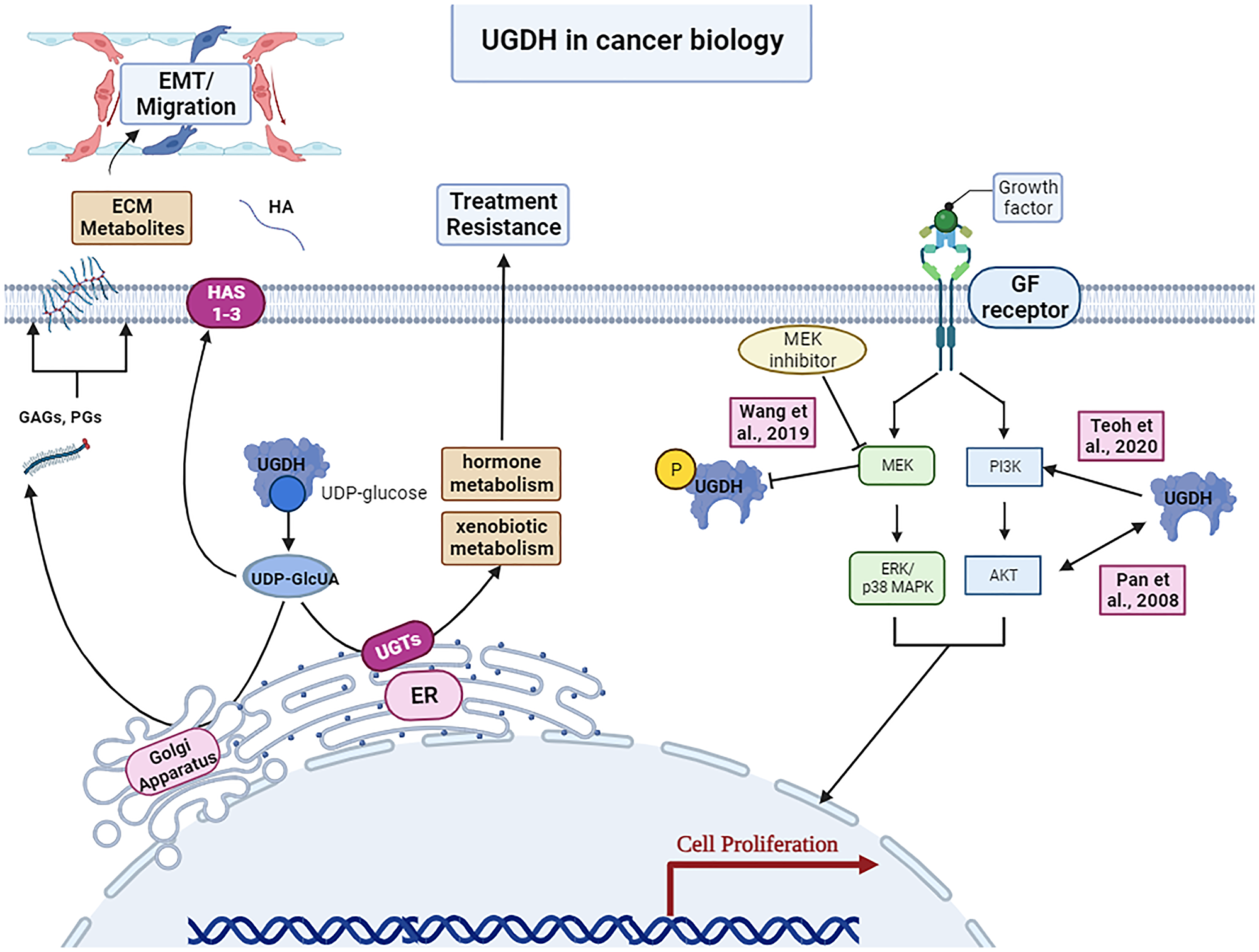
Figure 2: UGDH’s roles in cancer biology.
Upregulation of UGDH’s generation of ECM metabolites can contribute to epithelial mesenchymal transition and migration in metastasis. Hormone metabolism and xenobiotic metabolism by UGDH may promote treatment resistance. UGDH has also been shown to interact with the MEK/ERK and PI3K/AKT pathways of canonical growth factor signaling pathways in cancer biology. Image was created by https://www.biorender.com/ (2023).
Within lung cancer, Wang et al., 2019 explored the effects of inhibiting downstream targets within the EGFR induced MAPK pathway on the tumorigenic phenotype induced by phosphorylated UGDH (pUGDH). They found that the MEK inhibition but not p38 MAPK inhibition was able to abrogate EGF-induced pUGDH; additionally, knocking down UGDH did not affect ERK phosphorylation. Interestingly, in ovarian cancer, UGDH KD has been shown to negatively regulate levels of activated (phosphorylated) ERK with direct impacts on expression of MMP-2, MMP-9 and EMT-related factors further downstream [3]. These findings parallel those of Clarkin et al., 2011: while normal UGDH expression and function appear to be p38MAPK pathway dependent, stimuli activating UGDH (such as growth factors, induced overexpression, and phosphorylation as in Wang et al., 2019) shifts intracellular signaling towards a more MEK/ERK dependent mechanism [11, 14]. The AKT pathway has also been a target of interest within UGDH research. In breast cancer, Teoh et al., 2020 commented on an observed association between lower UGDH expression and copy number alterations in PIK3CA which produces PI3K, a component of the AKT signaling pathway. While they could not mechanistically explain this observation, it is nevertheless interesting as the AKT pathway has been shown to regulate UGDH expression in colorectal and nasopharyngeal cancer [28, 30]. Specifically, in nasopharyngeal cancer, Pan et al., 2008 demonstrated that UGDH expression induced PI3K/AKT and ERK activity [28] while Haggblad showed that AKT KD resulted in decreased expression of UGDH [30]. Thus, results of these studies both indicate the importance of these downstream signaling pathways and suggest that the relationship between them and UGDH is likely complex and bi-directional.
UGDH’s role in hormone metabolism in hormonally responsive cancers
Along with its relationship to downstream signaling pathways, UGDH has a key role in glucose/UDP-sugar metabolic pathways associated with cancer progression. As mentioned previously, both UGDH and HA levels have been connected to reprogramming glucose metabolism in breast cancer. Further, Arnold et al., 2019 demonstrated that changes in glucose metabolism via UGDH KD were also associated with differential essential of genes involved in fatty acid/lipid metabolism and PPAR signaling [9]. Within colon cancer, Shen et al., 2017 performed a study to identify differentially expressed genes (DEGs) in patient samples compared to non-cancerous controls and found that UGDH was key to a network of genes that regulated cancer cell metabolism (UGDH, ALDH1A1, FABP4, and MGLL). The specific mechanism was not identified; however, it was clear that expression of these genes along with several others involved in ECM regulation (COL1A1, COL1A2, and MMP9) were critical to the tumorigenesis of the CRC samples in this study [31]. Similarly, within melanoma, the synthesis and turnover of HA was directly linked to the downstream products of UDP-sugar metabolism—a relationship that together may be responsible for initiating and supporting the progression of melanoma [26].
In addition to the effect of UGDH on metabolic pathways, its previously discussed role in glucuronidation is important for regulating levels of intracellular hormones. Interestingly, the relationship between UGDH and hormones may be bi-directional as UGDH expression can be increased with exposure to certain endogenous hormones such as estradiol and dihydrotestosterone (DHT) [32]. Most studies exploring the relationship amongst UGDH, glucuronidation, and cancer have been conducted in hormonally responsive cancers such as prostate and breast. Specifically for prostate cancer, the leading hypothesis is that differential expression of UGDH can modulate how UDP-sugars flux through hormonal processing and ECM production pathways [32]. UGDH regulation of glucuronidation therefore could be a mechanism to promote androgen response deregulation and increase castration resistance within hormonally responsive prostate cancer. Mechanistically, Zimmer et al., 2016; 2021 have proposed that knocking down UGDH inhibits glucuronidation within cells and thereby decreases excretion of androgens, allowing for higher intracellular levels of this tumorigenic hormone. This is, in turn, accompanied by an increased flux of UDP-sugars through alternate UGDH mediated pathways within the cells to produce proteoglycans responsible for increased migratory phenotypes [32]. Indeed, the authors demonstrated that over-expressing UGDH in both androgen responsive and castrate resistant prostate cancer cell lines can induce androgen independent growth. Furthermore, loss of UGDH promoted androgen receptor dependent gene expression and restored androgen sensitivity to castrate resistant cells. This shift was accompanied by a decreased production of PGs [2]. These findings suggest that paradoxically, within certain hormonally responsive tumors, loss of UGDH may be associated with a more aggressive phenotype given its role in endogenous hormone processing. These findings highlight the importance of understanding the role of UGDH generally within in cancer but also emphasize how critical it is to study the nuances of UGDH function within specific cancers and environments.
UGDH as a therapeutic target
Given the well-established association between high levels of UGDH and aggressive, metastatic tumor phenotypes across many cancers, there is significant potential to use UGDH as both a prognostic marker and therapeutic target in clinical settings. As a catalyst of rate-limiting steps in several pro-tumorigenic pathways within cancer cells, UGDH, along with its substrates and downstream products, present promising targets for drug discovery.
There are multiple potential strategies to target either UGDH and/or up- or downstream pathways. A direct inhibitor of UGDH would be the most effective way to target its enzymatic activity; however, given the ubiquitous presence of UGDH in cells, specifically targeting the pro-tumorigenic effects of UGDH could be challenging. Indeed, there are no specific small molecule inhibitors of UGDH currently available in clinical settings. Alternatively, the UGDH pathway could be targeted by depleting substrates such as UDP-glucose or inhibiting its downstream products such as UDP-GlcUA or GAGs/HAs. In this section, we will discuss past and ongoing efforts to develop UGDH small molecule inhibitors along with therapeutic strategies to target substrates and downstream products (summarized in Table 2).
Table 2: Studies that discuss therapeutics targeting UGDH categorized by mechanism of action
| Therapeutic molecule | Associated study/studies | Classification | Mechanism of action |
|---|---|---|---|
| UDP-7-deoxy-α-D-gluco-hept- 6-ulopyranose (ketone 5) | Campbell and Tanner, 1999 | Direct competitive inhibitor of UGDH | Carbonyl functionality at C6 forms a carbon-carbon bond with UGDH and prevents second oxidation reaction (K i value of 6.7 uM) |
| UDP-a-D-xylose (UDX) | Neufeld 1965 Kadirvelraj et al., 2014 Sennett and Wood, 2012 | Allosteric inhibitor of UGDH | UDX binds to the active site of UGDH to increase the affinity between UGDH subunits and occlude binding sites for cofactor NAD+ to inactivate UGDH |
| Polyphenols (gallic acid, Quercetin) | Hwang et al., 2008 | Indirect target of UGDH (reduces activity of UGDH) | Inhibition of proliferation (exact mechanisms unknown) |
| Aphanizomenon flos-aquae (AFA) | Scoglio et al., 2016 | Antioxidant acting as a mixed-type inhibitor | Inhibits binding of both UDP-Glc and NAD+ |
| 5-hexyl-2-deoxyuridine (HUdR) | Lapis et al., 1987 Jeney et al., 1990 Timar et al., 1990 Pogany et al., 1990 Harisi et al., 2009 | Inhibition of UGDH substrate synthesis | Inhibits conversion of glucosamine to UDP-sugars to deplete UDP-glucose |
| 4-Methylumbelliferone (4-MU) OR Hymecromone | Yoshihara et al., 2005 Nakazawa et al., 2006 Lokeshwar et al., 2010 Arai et al., 2011 Twarock et al., 2011 Urakawa et al., 2012 Zhan et al., 2022 | Inhibition of UGDH downstream metabolites | Glucuronidation of 4-MU depletes cellular UDP-GlcUA to prevent HA synthesis |
Targetable structural properties of UGDH
Structurally, UGDH is most stable as a hexameric quaternary structure, as identified through X-ray crystallography [33–35]. Its most common form is a 57kDa hexamer assembled into a trimer of dimers, in which only three subunits are simultaneously active [1, 36, 37]. Enzymatic activity is controlled by induced fit responses that regulate subunit affinity and quaternary structure, and there is a single allosteric and active site [38]. While there is consensus that UGDH catalyzes UDP-glucose, the mechanism by which this catalysis occurs remains controversial and ambiguous despite extensive investigations. In the classic mechanism proposed by Ridley et al., the catalysis follows 4 steps. First, UDP-glucose is converted to an aldehyde intermediate, the UDP-α-D-gluco-hexodialdose (UDP-Glc-6-CHO) and NADH+ through a transfer of pro-R hydride to NAD+. The second step involves an attack of UDP-Glc-6-CHO by a cysteine residue in UGDH active site, leading to the formation of a thiohemiacetal intermediate that is covalently bound to the enzyme (Figure 3A). This reaction takes places immediately after the first oxidation, and it is the reason why the aldehyde intermediate isn’t released. In the third step, the thiohemiacetal is then oxidized to a thioester intermediate, resulting in the formation of a second NADH molecule. Finally, an irreversible hydrolysis of the thioester takes place to yield UDP-GlcUA [39]. Skeptics of this model argue that experimental evidence do not suggest the presence of a bound intermediate. Alternatively, it has been argued that UGDH converts to UDP-glucose to a Schiff base instead of an aldehyde intermediate [40], while more recent literature suggests a model in which the first oxidation step bypasses the aldehyde via an NAD+ dependent biomolecular nucleophilic substitution [41]. Despite these controversies regarding the first steps of this catalytic process, there is a consensus on the enzymatic steps after the first oxidation. The UGDH enzyme has a high affinity for allosteric inhibition. Due to its intrinsically disordered carboxy terminus (ID-tail), UGDH is highly regulated by allosteric binding of its downstream products including UDP-xylose (UDX), UDP-GluCA and other co-enzymes (NAD+), via feedback inhibition [36]. In most cases, UGDH is regulated by an atypical allosteric mechanism, in which UDP-Xyl competes with UDP-glucose for the active site, converting UGDH into an inactive state or inducing hysteresis or a “lagging” metabolism, which could have potential therapeutic implications [42].
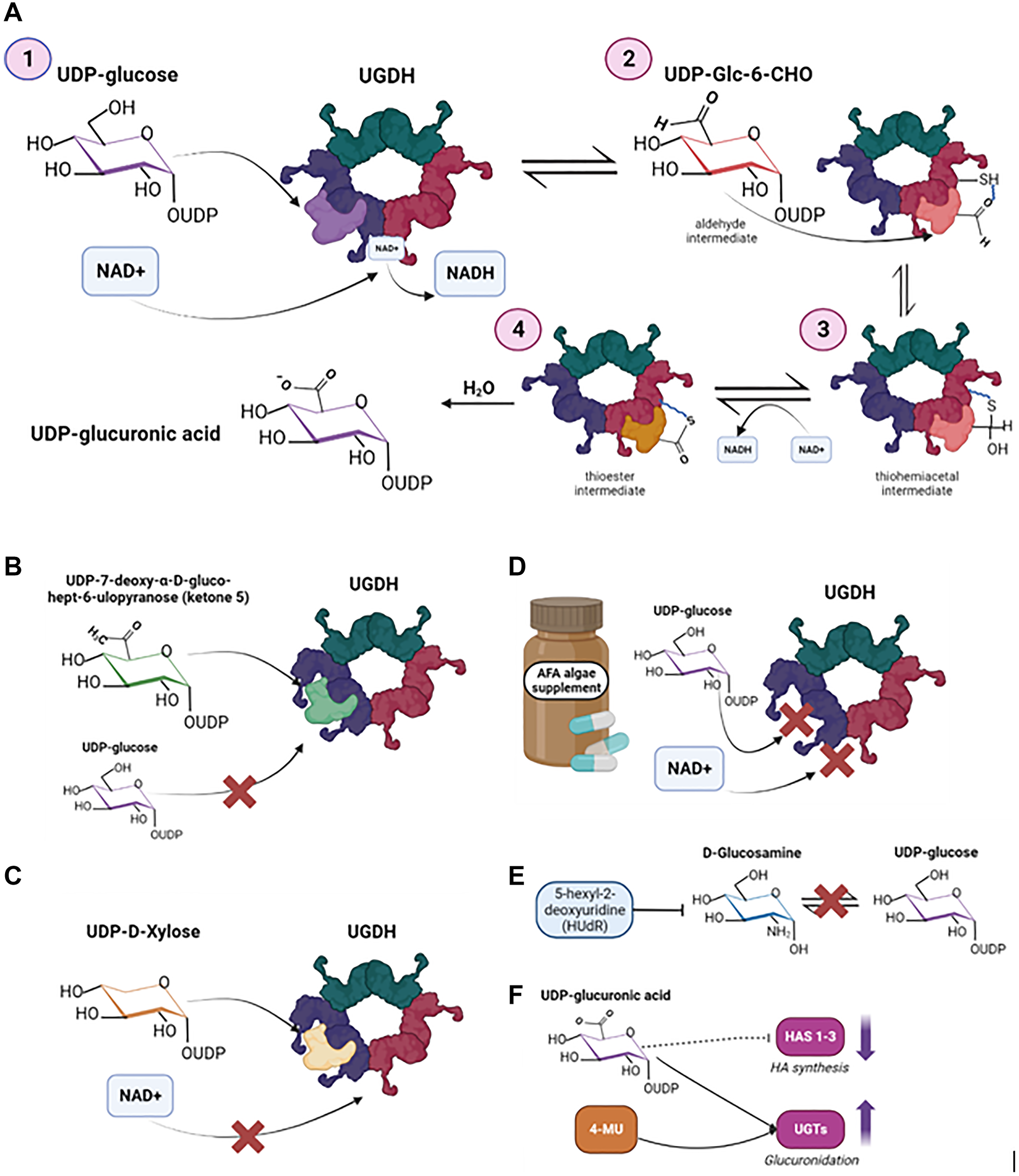
Figure 3: UGDH biochemical mechanism and therapeutic targets that exploit the structure of UGDH.
(A) Metabolism of UDP-6-glucose to UDP-GlcUA by UGDH proposed by Ridley et al., 1975. (B) UDP-7-deoxy-α-D-gluco-hept-6-ulopyranose (ketone 5) as a direct competitive inhibitor of UGDH, as proposed by Campbell and Tanner, 1999. (C) UDP-D-xylose (UDX) as an allosteric inhibitor of UGDH, preventing the binding and reduction of NAD+. Neufeld, 1965, introduced UDX as an endogenous feedback inhibitor of UGDH. (D) Aphanizomenon flos-aquae (AFA), an algae supplement that acts as a mixed-type inhibitor and prevents the binding of both UDP-glucose and NAD+, as discussed by Scoglio et al., 2016. (E) 5-hexyl-2-deoxyuridine (HUdR) as an inhibitor of UGDH’s substrate UDP-glucose, by preventing the conversion of glucosamine to UDP-sugars to ultimately prevent the biosynthesis of heparan sulfate. (F) 4-Methylumbelliferone (4-MU) or Hymecrome as a HA synthesis inhibitor, by depleting UDP-GlcUA. Image was created by https://www.biorender.com (2023).
Direct targets of UGDH
UDP-Glucose Analogues as inhibitors of UDGH
Certain aspects of UGDH’s enzymatic activity present potential targets for drug discovery. Specifically, targeting the bound aldehyde intermediate produced in the first step of UGDH’s enzymatic reaction has been previously explored [43]. Leveraging this step in the enzymatic activity of UGDH, Campbell and Tanner, 1999 designed UDP-glucose analogues that mimicked the bound aldehyde intermediate and thereby bound more tightly to UGDH than its natural substrate UDP-glucose. Using this strategy, they synthesized and tested UDP-7-deoxy-α-D-gluco-hept-6-ulopyranose (ketone 5) whose carbonyl functionality at C6 led to a formation of a carbon-carbon bond with UGDH, which prevented the second oxidation reaction from proceeding (Figure 3B). They found that this compound could act as a competitive inhibitor of UGDH with a K i value of 6.7 uM [43]. While this analogue was promising, there have not been follow-up studies to test its in vitro and in vivo efficacy or phenotypic impact.
UDP-a-D-xylose (UDX)
Apart from its roles in drug metabolism and ECM maintenance, UDP-GlcUA is a precursor for the biosynthesis of UDX, an endogenous feedback inhibitor of UGDH (Neufeld 1965). UDX differs from UDP-GlcUA as it lacks a C’ hydroxymethyl group. Thus, when UDX binds the UGDH enzyme in the active site, it triggers an allosteric switch that increases the affinity between the subunit interfaces, resulting in a hexamer that occludes the binding sites for the cofactor (NAD+) and the C5’hydroxymethyl on the substrate to inactivate the enzyme [44]. The crystal structure of UGDH bound to UDX was published in 2011 [45]. While UDX has not been formally studied as a therapeutic inhibitor of UGDH, the structural and enzymatic impacts of its binding to UGDH could be utilized to design mimetics of UDX for clinical use (Figure 3C).
Indirect therapeutic targets of UGDH
While there are not many, some studies have presented alternative strategies to inhibit UGDH activity in cancer cells. Specifically, Hwang et al., 2008 demonstrated the inhibitory effects of two polyphenols (Gallic acid and Quercetin) against UGDH in MCF-7 breast cancer cells [46]. They found that treating the cells with 300 uM of gallic acid reduced the specific activity of UGDH by 66% in comparison to control while treating with quercetin reduced the specific activity by 41%. While they were able to show that these compounds inhibited proliferation of MCF-7 cells, the study was limited as they could not directly link inhibition of UGDH to the anti-proliferation activities of these polyphenols. Additionally, the doses used in the study were 10-fold higher than what could realistically be achieved through diet. Despite these concerns, the structure of these polyphenols could be used for rational design of better compounds that could bind and inhibit UGDH is an effective manner and at lower doses.
Another study assessed the efficacy of the dietary supplement Aphanizomenon flos-aquae (AFA), which has strong antioxidant activity. They found out that AFA reduced UGDH activity in a dose-dependent manner and acted as a mixed-type inhibitor with respect to both UDP-Glc and NAD+ (Figure 3D). Phenotypically, they showed that AFA was also effective in reducing the colony formation capacity of PC-3 prostate cancer cells and FTC-133 thyroid cancer cells [47].
Inhibiting the UGDH pathway
Substrate inhibition upstream of UGDH
Along with directly inhibiting the enzymatic activity of UGDH, depleting its UDP-glucose substrate could be an effective strategy to target the UGDH pathway. 5-hexyl-2-deoxyuridine (HUdR) inhibits the conversion of glucosamine to UDP-sugars, which depletes UDP-glucose and subsequently reduces the biosynthesis of heparan sulfate (Figure 3E) [48, 49]. In the context of tumor biology, HUdR has been shown to reduce the migratory capacity of various tumor cells [50, 51] and has been tested in vivo as an anti-metastatic drug with efficacy in tumor cells with high metastatic potential [48, 52]. While these studies indirectly discuss UGDH as a therapeutic target, their findings demonstrate the potential anti-tumorigenic effects of targeting the availability of one of its substrates. Further work is necessary to follow up on the use of HUdR or similar therapies in the clinic.
Inhibition of UGDH products
Strategies that deplete UGDH’s downstream metabolite, UDP-GlcUA could mitigate the pro-metastatic, migratory phenotype induced by higher levels of UGDH. One of the most used drugs for this purpose is 4-MU, which results in depleted HA production downstream of UGDH. Mechanistically, glucuronidation of 4-MU depletes cellular UDP-GlcUA stores necessary for HA synthesis (Figure 3F) [18, 53–55]. 4-MU is non-toxic and non-polar which allows it to cross the lipidic intestinal barrier and thereby be administered orally. Studies have established the safety of 4-MU in humans when used as a choleretic and spasmolytic to increase bile production and thereby improve liver detoxification. Extensive in vitro and in vivo studies have shown that 4-MU reduces the proliferation, migration, invasion, and metastasis of multiple cancer cell types including pancreatic, prostate, melanoma, esophageal, breast, liver, bone and ovarian cancers [18, 53, 54, 56, 57]. Additionally, 4-MU inhibition of HA has been shown to increase access to drugs and immune infiltration to the tumor, which in turn prevent tumor growth and metastasis [55]. This was demonstrated by Nakazawa et al., 2006 who showed that, while 4-MU by itself did not cause cancer cell death or inhibition of proliferation of a pancreatic cancer cell line KP1-NL, 4-MU pretreatment increased the efficacy of an anti-cancer agent gemcitabine [55]. Additionally, 4-MU and UGDH knockdowns were shown to increase anti-tumor immune responses in GBM in vitro and in vivo by activating phagocytosis in tumor-residing macrophages, decreasing immune-suppressing regulatory T-cell activity, and increasing cytotoxic T-cell infiltration and activation [6]. This could mean that targeting the UGDH-HA pathway might expose therapeutic vulnerabilities of various cancers and lead to more efficient combination therapies involving both chemotherapies and immunotherapies.
In addition to the mechanism discussed above, recent literature has demonstrated potential direct effects of 4-MU on UGDH itself. Using limb bud micro mass cultures, Clarkin et al., 2011 found that 4-MU treatment reduced UGDH mRNA and HAS-2 expression in AS cells and produced modest suppression of UGDH protein levels. This was subsequently associated with decreased release of both HA and sulphated GAGs [14]. Further pharmacologic studies should be performed to establish the pharmacokinetics and mechanism of 4-MU inhibition of UGDH. Understanding the mechanism of inhibition will help in designing better UGDH antagonists.
Results showing that targeting HA by 4-MU leads to anti-cancer effects in vivo show promise that UGDH targeting could have similar anti-cancer treatment. Cancers expressing high levels of UGDH and/or HA could benefit from 4-MU treatment. Given the anti-metastatic potential of targeting UGDH-HA pathway, clinical trials should be established to test the efficacy of 4-MU as a stand-alone therapy or in combination with other chemotherapies or immunotherapies.
Conclusions
While much is known about the many roles of UGDH across both normal physiology and oncology, there is still significant work to be done to understand how it can best be harnessed in a clinical setting. Given the potential challenges of directly inhibiting UGDH, therapeutic strategies may extend to targeting downstream pathways and upstream substrates. Additionally, there has recently been more literature published on the potential role of UGDH in treatment resistance and immune modulation across various cancer types. Thus, while directly targeting UGDH may not be feasible as a standalone treatment strategy, it could be an important adjunct to current therapies. Furthermore, modulating its activity could help better understand mechanisms behind drug resistance and/or prevent resistance from developing. Therefore, UGDH is a promising enzyme of interest across many fields of oncology, and given its multi-faceted role in cellular functioning, it plays nuanced and complicated roles within tumorigenic pathways. While these multiple roles can provide challenges, they also provide significant opportunities for therapeutic targeting, prognostication, and drug development.
Abbreviations
4-MU: 4-Methylumbelliferone; EMT: epithelial mesenchymal transition; GAG: glycosaminoglycans; GBM: glioblastoma; HA: hyaluronic acid; HAS: hyaluronan synthase; PG: proteoglycans; UDP-GlcUA: UDP-glucuronic acid; UDX: UDP-xylose; UGDH: UDP-6 glucose dehydrogenase.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
Funding sources and disclosures from C. Rory Goodwin: Received grants the Robert Wood Johnson Harold Amos Medical Faculty Development Program, the Federal Food and Drug Administration, and the NIH 1R01DEO31053-01A1. Consultant for Stryker and Medtronic. The other authors do not have any relevant funding sources or other items to disclose.
References
1. Egger S, Chaikuad A, Kavanagh KL, Oppermann U, Nidetzky B. UDP-glucose dehydrogenase: structure and function of a potential drug target. Biochem Soc Trans. 2010; 38:1378–85. https://doi.org/10.1042/BST0381378. [PubMed].
2. Zimmer BM, Barycki JJ, Simpson MA. Integration of Sugar Metabolism and Proteoglycan Synthesis by UDP-glucose Dehydrogenase. J Histochem Cytochem. 2021; 69:13–23. https://doi.org/10.1369/0022155420947500. [PubMed].
3. Lin LH, Chou HC, Chang SJ, Liao EC, Tsai YT, Wei YS, Chen HY, Lin MW, Wang YS, Chien YA, Yu XR, Chan HL. Targeting UDP-glucose dehydrogenase inhibits ovarian cancer growth and metastasis. J Cell Mol Med. 2020; 24:11883–902. https://doi.org/10.1111/jcmm.15808. [PubMed].
4. Oyinlade O, Wei S, Lal B, Laterra J, Zhu H, Goodwin CR, Wang S, Ma D, Wan J, Xia S. Targeting UDP-α-D-glucose 6-dehydrogenase inhibits glioblastoma growth and migration. Oncogene. 2018; 37:2615–29. https://doi.org/10.1038/s41388-018-0138-y. [PubMed].
5. Vitale DL, Caon I, Parnigoni A, Sevic I, Spinelli FM, Icardi A, Passi A, Vigetti D, Alaniz L. Initial Identification of UDP-Glucose Dehydrogenase as a Prognostic Marker in Breast Cancer Patients, Which Facilitates Epirubicin Resistance and Regulates Hyaluronan Synthesis in MDA-MB-231 Cells. Biomolecules. 2021; 11:246. https://doi.org/10.3390/biom11020246. [PubMed].
6. Zhan D, Yalcin F, Ma D, Fu Y, Wei S, Lal B, Li Y, Dzaye O, Laterra J, Ying M, Lopez-Bertoni H, Xia S. Targeting UDP-α-d-glucose 6-dehydrogenase alters the CNS tumor immune microenvironment and inhibits glioblastoma growth. Genes Dis. 2022; 9:717–30. https://doi.org/10.1016/j.gendis.2021.08.008. [PubMed].
7. Huh JW, Choi MM, Yang SJ, Yoon SY, Choi SY, Cho SW. Inhibition of human UDP-glucose dehydrogenase expression using siRNA expression vector in breast cancer cells. Biotechnol Lett. 2005; 27:1229–32. https://doi.org/10.1007/s10529-005-0022-z. [PubMed].
8. Huang D, Casale GP, Tian J, Lele SM, Pisarev VM, Simpson MA, Hemstreet GP 3rd. Udp-glucose dehydrogenase as a novel field-specific candidate biomarker of prostate cancer. Int J Cancer. 2010; 126:315–27. https://doi.org/10.1002/ijc.24820. [PubMed].
9. Arnold JM, Gu F, Ambati CR, Rasaily U, Ramirez-Pena E, Joseph R, Manikkam M, San Martin R, Charles C, Pan Y, Chatterjee SS, Den Hollander P, Zhang W, et al. UDP-glucose 6-dehydrogenase regulates hyaluronic acid production and promotes breast cancer progression. Oncogene. 2020; 39:3089–101. https://doi.org/10.1038/s41388-019-0885-4. [PubMed].
10. Teoh ST, Ogrodzinski MP, Lunt SY. UDP-glucose 6-dehydrogenase knockout impairs migration and decreases in vivo metastatic ability of breast cancer cells. Cancer Lett. 2020; 492:21–30. https://doi.org/10.1016/j.canlet.2020.07.031. [PubMed].
11. Wang X, Liu R, Zhu W, Chu H, Yu H, Wei P, Wu X, Zhu H, Gao H, Liang J, Li G, Yang W. UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature. 2019; 571:127–31. https://doi.org/10.1038/s41586-019-1340-y. [PubMed].
12. Liu H, Zhang Q, Lou Q, Zhang X, Cui Y, Wang P, Yang F, Wu F, Wang J, Fan T, Li S. Differential Analysis of lncRNA, miRNA and mRNA Expression Profiles and the Prognostic Value of lncRNA in Esophageal Cancer. Pathol Oncol Res. 2020; 26:1029–39. https://doi.org/10.1007/s12253-019-00655-8. [PubMed].
13. Luo Q, He W, Mao T, Leng X, Wu H, Li W, Deng X, Zhao T, Shi M, Xu C, Han Y. MMS22L Expression as a Predictive Biomarker for the Efficacy of Neoadjuvant Chemoradiotherapy in Oesophageal Squamous Cell Carcinoma. Front Oncol. 2021; 11:711642. https://doi.org/10.3389/fonc.2021.711642. [PubMed].
14. Clarkin CE, Allen S, Kuiper NJ, Wheeler BT, Wheeler-Jones CP, Pitsillides AA. Regulation of UDP-glucose dehydrogenase is sufficient to modulate hyaluronan production and release, control sulfated GAG synthesis, and promote chondrogenesis. J Cell Physiol. 2011; 226:749–61. https://doi.org/10.1002/jcp.22393. [PubMed].
15. García-García MJ, Anderson KV. Essential role of glycosaminoglycans in Fgf signaling during mouse gastrulation. Cell. 2003; 114:727–37. https://doi.org/10.1016/s0092-8674(03)00715-3. [PubMed].
16. Caon I, Parnigoni A, Viola M, Karousou E, Passi A, Vigetti D. Cell Energy Metabolism and Hyaluronan Synthesis. J Histochem Cytochem. 2021; 69:35–47. https://doi.org/10.1369/0022155420929772. [PubMed].
17. Kobayashi T, Chanmee T, Itano N. Hyaluronan: Metabolism and Function. Biomolecules. 2020; 10:1525. https://doi.org/10.3390/biom10111525. [PubMed].
18. Arai E, Nishida Y, Wasa J, Urakawa H, Zhuo L, Kimata K, Kozawa E, Futamura N, Ishiguro N. Inhibition of hyaluronan retention by 4-methylumbelliferone suppresses osteosarcoma cells in vitro and lung metastasis in vivo. Br J Cancer. 2011; 105:1839–49. https://doi.org/10.1038/bjc.2011.459. [PubMed].
19. Heldin P, Basu K, Olofsson B, Porsch H, Kozlova I, Kahata K. Deregulation of hyaluronan synthesis, degradation and binding promotes breast cancer. J Biochem. 2013; 154:395–408. https://doi.org/10.1093/jb/mvt085. [PubMed].
20. Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front Immunol. 2015; 6:201. https://doi.org/10.3389/fimmu.2015.00201. [PubMed].
21. Pasonen-Seppänen S, Takabe P, Edward M, Rauhala L, Rilla K, Tammi M, Tammi R. Melanoma cell-derived factors stimulate hyaluronan synthesis in dermal fibroblasts by upregulating HAS2 through PDGFR-PI3K-AKT and p38 signaling. Histochem Cell Biol. 2012; 138:895–911. https://doi.org/10.1007/s00418-012-1000-x. [PubMed].
22. Rizzardi AE, Vogel RI, Koopmeiners JS, Forster CL, Marston LO, Rosener NK, Akentieva N, Price MA, Metzger GJ, Warlick CA, Henriksen JC, Turley EA, McCarthy JB, Schmechel SC. Elevated hyaluronan and hyaluronan-mediated motility receptor are associated with biochemical failure in patients with intermediate-grade prostate tumors. Cancer. 2014; 120:1800–9. https://doi.org/10.1002/cncr.28646. [PubMed].
23. Tammi MI, Oikari S, Pasonen-Seppänen S, Rilla K, Auvinen P, Tammi RH. Activated hyaluronan metabolism in the tumor matrix - Causes and consequences. Matrix Biol. 2019; 78-79:147–64. https://doi.org/10.1016/j.matbio.2018.04.012. [PubMed].
24. Bourguignon LY, Wong G, Earle C, Krueger K, Spevak CC. Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J Biol Chem. 2010; 285:36721–35. https://doi.org/10.1074/jbc.M110.162305. [PubMed].
25. Li L, Qi L, Liang Z, Song W, Liu Y, Wang Y, Sun B, Zhang B, Cao W. Transforming growth factor-β1 induces EMT by the transactivation of epidermal growth factor signaling through HA/CD44 in lung and breast cancer cells. Int J Mol Med. 2015; 36:113–22. https://doi.org/10.3892/ijmm.2015.2222. [PubMed].
26. Deen AJ, Arasu UT, Pasonen-Seppänen S, Hassinen A, Takabe P, Wojciechowski S, Kärnä R, Rilla K, Kellokumpu S, Tammi R, Tammi M, Oikari S. UDP-sugar substrates of HAS3 regulate its O-GlcNAcylation, intracellular traffic, extracellular shedding and correlate with melanoma progression. Cell Mol Life Sci. 2016; 73:3183–204. https://doi.org/10.1007/s00018-016-2158-5. [PubMed].
27. Wang TP, Pan YR, Fu CY, Chang HY. Down-regulation of UDP-glucose dehydrogenase affects glycosaminoglycans synthesis and motility in HCT-8 colorectal carcinoma cells. Exp Cell Res. 2010; 316:2893–902. https://doi.org/10.1016/j.yexcr.2010.07.017. [PubMed].
28. Pan YR, Vatsyayan J, Chang YS, Chang HY. Epstein-Barr virus latent membrane protein 2A upregulates UDP-glucose dehydrogenase gene expression via ERK and PI3K/Akt pathway. Cell Microbiol. 2008; 10:2447–60. https://doi.org/10.1111/j.1462-5822.2008.01221.x. [PubMed].
29. Aroui S, Aouey B, Chtourou Y, Meunier AC, Fetoui H, Kenani A. Naringin suppresses cell metastasis and the expression of matrix metalloproteinases (MMP-2 and MMP-9) via the inhibition of ERK-P38-JNK signaling pathway in human glioblastoma. Chem Biol Interact. 2016; 244:195–203. https://doi.org/10.1016/j.cbi.2015.12.011. [PubMed].
30. Häggblad Sahlberg S, Mortensen AC, Haglöf J, Engskog MK, Arvidsson T, Pettersson C, Glimelius B, Stenerlöw B, Nestor M. Different functions of AKT1 and AKT2 in molecular pathways, cell migration and metabolism in colon cancer cells. Int J Oncol. 2017; 50:5–14. https://doi.org/10.3892/ijo.2016.3771. [PubMed].
31. Shen X, Yue M, Meng F, Zhu J, Zhu X, Jiang Y. Microarray analysis of differentially-expressed genes and linker genes associated with the molecular mechanism of colorectal cancer. Oncol Lett. 2016; 12:3250–58. https://doi.org/10.3892/ol.2016.5122. [PubMed].
32. Zimmer BM, Howell ME, Wei Q, Ma L, Romsdahl T, Loughman EG, Markham JE, Seravalli J, Barycki JJ, Simpson MA. Loss of exogenous androgen dependence by prostate tumor cells is associated with elevated glucuronidation potential. Horm Cancer. 2016; 7:260–71. https://doi.org/10.1007/s12672-016-0268-z. [PubMed].
33. Lee HS, Son YJ, Chong SH, Bae JY, Leem CH, Jang YJ, Choe H. Computational analysis of the quaternary structural changes induced by point mutations in human UDP-glucose dehydrogenase. Arch Biochem Biophys. 2009; 486:35–43. https://doi.org/10.1016/j.abb.2009.03.017. [PubMed].
34. Huh JW, Robinson RC, Lee HS, Lee JI, Heo YS, Kim HT, Lee HJ, Cho SW, Choe H. Expression, purification, crystallization, and preliminary X-Ray analysis of the human UDP-glucose dehydrogenase. Protein Pept Lett. 2006; 13:859–62. https://doi.org/10.2174/092986606777841253. [PubMed].
35. Huh JW, Yang SJ, Hwang EY, Choi MM, Lee HJ, Kim EA, Choi SY, Choi J, Hong HN, Cho SW. Alteration of the quaternary structure of human UDP-glucose dehydrogenase by a double mutation. J Biochem Mol Biol. 2007; 40:690–96. https://doi.org/10.5483/bmbrep.2007.40.5.690. [PubMed].
36. Keul ND, Oruganty K, Schaper Bergman ET, Beattie NR, McDonald WE, Kadirvelraj R, Gross ML, Phillips RS, Harvey SC, Wood ZA. The entropic force generated by intrinsically disordered segments tunes protein function. Nature. 2018; 563:584–88. https://doi.org/10.1038/s41586-018-0699-5. [PubMed].
37. Rajakannan V, Lee HS, Chong SH, Ryu HB, Bae JY, Whang EY, Huh JW, Cho SW, Kang LW, Choe H, Robinson RC. Structural basis of cooperativity in human UDP-glucose dehydrogenase. PLoS One. 2011; 6:e25226. https://doi.org/10.1371/journal.pone.0025226. [PubMed].
38. Kadirvelraj R, Sennett NC, Polizzi SJ, Weitzel S, Wood ZA. Role of packing defects in the evolution of allostery and induced fit in human UDP-glucose dehydrogenase. Biochemistry. 2011; 50:5780–89. https://doi.org/10.1021/bi2005637. [PubMed].
39. Ridley WP, Houchins JP, Kirkwood S. Mechanism of action of uridine diphosphoglucose dehydrogenase. Evidence for a second reversible dehydrogenation step involving an essential thiol group. J Biol Chem. 1975; 250:8761–67. [PubMed].
40. Ordman AB, Kirkwood S. Mechanism of action of uridine diphoglucose dehydrogenase. Evidence for an essential lysine residue at the active site. J Biol Chem. 1977; 252:1320–26. [PubMed].
41. Chen J, Yang S. Catalytic mechanism of UDP-glucose dehydrogenase. Biochem Soc Trans. 2019; 47:945–55. https://doi.org/10.1042/BST20190257. [PubMed].
42. Beattie NR, Keul ND, Hicks Sirmans TN, McDonald WE, Talmadge TM, Taujale R, Kannan N, Wood ZA. Conservation of Atypical Allostery in C. elegans UDP-Glucose Dehydrogenase. ACS Omega. 2019; 4:16318–29. https://doi.org/10.1021/acsomega.9b01565. [PubMed].
43. Campbell RE, Tanner ME. UDP-Glucose Analogues as Inhibitors and Mechanistic Probes of UDP-Glucose Dehydrogenase. J Org Chem. 1999; 64:9487–92. https://doi.org/10.1021/jo991092h.
44. Kadirvelraj R, Custer GS, Keul ND, Sennett NC, Sidlo AM, Walsh RM Jr, Wood ZA. Hysteresis in human UDP-glucose dehydrogenase is due to a restrained hexameric structure that favors feedback inhibition. Biochemistry. 2014; 53:8043–51. https://doi.org/10.1021/bi500594x. [PubMed].
45. Sennett NC, Kadirvelraj R, Wood ZA. Cofactor binding triggers a molecular switch to allosterically activate human UDP-α-D-glucose 6-dehydrogenase. Biochemistry. 2012; 51:9364–74. https://doi.org/10.1021/bi301067w. [PubMed].
46. Hwang EY, Huh JW, Choi MM, Choi SY, Hong HN, Cho SW. Inhibitory effects of gallic acid and quercetin on UDP-glucose dehydrogenase activity. FEBS Lett. 2008; 582:3793–97. https://doi.org/10.1016/j.febslet.2008.10.010. [PubMed].
47. Scoglio S, Lo Curcio V, Catalani S, Palma F, Battistelli S, Benedetti S. Inhibitory effects of Aphanizomenon flos-aquae constituents on human UDP-glucose dehydrogenase activity. J Enzyme Inhib Med Chem. 2016; 31:1492–97. https://doi.org/10.3109/14756366.2016.1149478. [PubMed].
48. Jeney A, Timár J, Pogány G, Paku S, Moczár E, Mareel M, Otvös L, Kopper L, Lapis K. Glycosaminoglycans as novel target in antitumor therapy. Tokai J Exp Clin Med. 1990; 15:167–77. [PubMed].
49. Timár J, Pogány G, Balázs M, Szöllösi J, Ladányi A, Oláh J, Timár F, Lapis K, Jeney A. Modulation of membrane phenotype, matrix adhesion and microinvasiveness of metastatic tumour cells by HUdR. Cell Biochem Funct. 1990; 8:211–20. https://doi.org/10.1002/cbf.290080405. [PubMed].
50. Harisi R, Kenessey I, Olah JN, Timar F, Babo I, Pogany G, Paku S, Jeney A. Differential inhibition of single and cluster type tumor cell migration. Anticancer Res. 2009; 29:2981–85. [PubMed].
51. Pogány G, Jeney A, Timár J, Major J, Lapis K. Modulation of glycoconjugate biosynthesis by 5-hexyl-2’-deoxyuridine in highly metastatic Lewis lung carcinoma cells. Neoplasma. 1990; 37:501–10. [PubMed].
52. Lapis K, Timár J, Pál K, Jeney A, Timár F, Kopper L. Membrane Properties of Lewis Lung Tumor Cells with “Low” and “High” Metastatic Capacity: Anti-Metastatic Effect of a Glycosaminoglycan Biosynthesis Blocking Agent 5-Hexyl-2′-Deoxyuridine (HUdR). In: Cory JG, Szentivanyi A. (eds), Cancer Biology and Therapeutics. Springer: Boston, MA. 1987. https://doi.org/10.1007/978-1-4757-9564-6_5.
53. Urakawa H, Nishida Y, Wasa J, Arai E, Zhuo L, Kimata K, Kozawa E, Futamura N, Ishiguro N. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int J Cancer. 2012; 130:454–66. https://doi.org/10.1002/ijc.26014. [PubMed].
54. Yoshihara S, Kon A, Kudo D, Nakazawa H, Kakizaki I, Sasaki M, Endo M, Takagaki K. A hyaluronan synthase suppressor, 4-methylumbelliferone, inhibits liver metastasis of melanoma cells. FEBS Lett. 2005; 579:2722–26. https://doi.org/10.1016/j.febslet.2005.03.079. [PubMed].
55. Nakazawa H, Yoshihara S, Kudo D, Morohashi H, Kakizaki I, Kon A, Takagaki K, Sasaki M. 4-methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemother Pharmacol. 2006; 57:165–70. https://doi.org/10.1007/s00280-005-0016-5. [PubMed].
56. Lokeshwar VB, Lopez LE, Munoz D, Chi A, Shirodkar SP, Lokeshwar SD, Escudero DO, Dhir N, Altman N. Antitumor activity of hyaluronic acid synthesis inhibitor 4-methylumbelliferone in prostate cancer cells. Cancer Res. 2010; 70:2613–23. https://doi.org/10.1158/0008-5472.CAN-09-3185. [PubMed].
57. Twarock S, Freudenberger T, Poscher E, Dai G, Jannasch K, Dullin C, Alves F, Prenzel K, Knoefel WT, Stoecklein NH, Savani RC, Homey B, Fischer JW. Inhibition of oesophageal squamous cell carcinoma progression by in vivo targeting of hyaluronan synthesis. Mol Cancer. 2011; 10:30. https://doi.org/10.1186/1476-4598-10-30. [PubMed].