INTRODUCTION
The development of brain metastases is a growing public health problem affecting more than 100,000 patients in the United States every year [1], including 10–30% of breast cancer patients [2, 3]. This complication is associated with severe morbidity and virtually 100% mortality, as currently there is no treatment strategy with proven efficacy. HER2-positive breast cancer patients are at particularly high risk [4, 5], with around half developing brain metastases during the course of disease [6]. HER2-targeted drug therapies delay the onset of brain metastases in these patients, and improve median survival after a diagnosis of metastatic brain relapse [7–10]. These observations indicate that HER2 plays a critical role in brain relapse. However, the molecular mechanisms underpinning this relationship have not been investigated in detail.
HER2 is an orphan member of the human epidermal growth factor receptor (HER/ERBB) family. It undergoes obligate heterodimerization with HER3, and to a lesser extent HER4 and EGFR [11–14]. The HER3-HER2 dimer is regarded as the major oncogenic unit in HER2-positive breast cancer [11, 15, 16]. Ligand-activated dimers transduce potent survival and proliferation signals through the PI3K-AKT and ERK1/2 pathways [11, 17]. HER3 has two ligands: Heregulin (HRG; also known as neuregulin-1 (NRG1)) and neuregulin-2. HRG is the better studied of the two and is broadly expressed in the brain by neurons, glia and the cerebral endothelium, functioning to promote survival, differentiation, migration and cytoprotection [18–20]. At least 17 HRG isoforms have been described, including secreted, membrane-bound and nuclear ‘back-signaling’ isoforms that are generated through a combination of alternate transcription start sites, splicing and post-translational processing [21]. Importantly, primary breast cancers that over-express HER3 are associated with a significantly higher rate of isolated brain metastases [22], and induction of HER3 is associated with development of brain metastases from both breast and lung cancers [23, 24]. Despite this, the functional relationships between HRG, HER3 and HER2 in breast cancer-brain metastases have not been elucidated.
HRG and HER2 signaling can also induce certain matrix metalloproteinase enzymes (MMPs) [25–28]. MMPs are zinc-dependent endopeptidases that degrade extracellular matrix (ECM) proteins. Their activities facilitate various normal physiologic processes (e.g. wound healing and organ development), and their dysregulation can be associated with pathological processes, including progression-associated changes in the tumor microenvironment. Regulation of MMP expression and activity is complex – they are expressed and stored as zymogens, secreted and activated on-demand by ‘convertases’ including other MMPs, and in vivo, their activities are fine-tuned according to the local balance between MMPs, TIMPs (tissue inhibitors of metalloproteinases) and other physiologic inhibitors [29] (e.g. the metastasis suppressor, RECK [30]). Therefore measurement of MMP expression is not a reliable surrogate for function. MMPs have been strongly implicated in the development of brain metastases from breast cancer [31–35]. For example, expression of MMP-9 is relatively higher in the brain-seeking MDA-MB-231 breast cancer cell line variant compared to parental and bone-homing counterparts [36], and ectopic expression of MMP-2 in MDA-MB-231 cells increased the incidence of brain metastases after intracardiac injection [37].
In order to establish distant brain metastases, disseminated breast cancer cells must initially traverse the blood-brain-barrier: a specialized endothelium that is resistant to diffusion of hydrophilic or large molecules by virtue of endothelial tight-junctions that are unique to the central nervous system. This specialized microvasculature is in close contact with astrocytic foot processes, pericytes and a thick basement membrane, which collectively facilitate the high substrate selectivity that is necessary to protect brain tissue from circulating pathogens and toxins, including chemotherapeutic agents [38, 39]. Extravasation of tumor cells across this barrier is therefore thought to be an active process. Following extravasation, tumor cells must establish growth-promoting interactions with the neural niche [40–43].
This study aimed to investigate molecular mechanisms by which HER3-HER2 signaling may promote the development of brain metastases from breast cancer. In light of the inferred associations between HER3, HER2, MMP activity and brain metastases, and the ubiquitous expression of heregulin in the brain, we hypothesized that heregulin may activate molecular mechanisms conducive to the establishment and growth of HER2-positive breast cancer cells in the brain microenvironment.
RESULTS
Expression of ERBB3 and NRG1 isoforms in breast cancer cell lines
To characterize the expression of heregulin (NRG1 gene) and HER3 (ERBB3 gene) in breast cancer cell line models, and select the most appropriate lines for functional experiments, we investigated the relative baseline mRNA expression levels of ERBB3 and NRG1 (α and β heregulin splice isoforms) in a large panel of breast cancer cell lines by quantitative reverse transcription-PCR (qRT-PCR). The gene expression profile-based molecular subtypes of these cell lines (luminal, luminal-ERBB2-amplified, basal-A and basal-B/claudin-low) were derived from published reports [44–46], marked in Figure 1. This screening experiment revealed an inverse association between ERBB3 and NRG1 expression, with highest levels of ERBB3 in luminal cell lines, and highest levels of NRG1 in claudin-low cell lines, consistent with their mesenchymal-like phenotype [46] (Figure 1). ERBB3/NRG1 expression phenotypes were mixed in basal-A cell lines.
Paracrine activation of HER2-HER3 in luminal breast cancer cell lines
We next investigated the responses of three representative ERBB3-expressing cell lines to treatment with exogenous heregulin (HRG): MDA-MB-361, MCF7 and SKBr3. These three cell lines are luminal-like when stratified based on transcriptomic profile [44–46]. Other key features to note are that MDA-MB-361 and SKBr3 harbor ERBB2 amplification, MDA-MB-361 was isolated from a breast cancer-derived metastatic brain tumor, and SKBr3 cells do not express estrogen receptor (ER-negative) [44]. All three lines are capable of colonizing the brain in animal models ([47, 48] and unpublished observations).
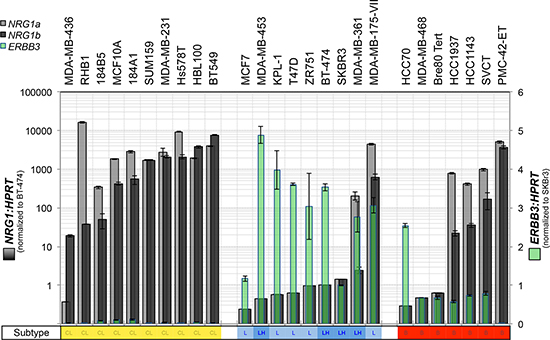
Figure 1: Expression of heregulin and HER3 in claudin-low, luminal and basal A breast cancer cell lines. The breast cancer cell lines indicated were cultured to sub-confluence then total RNA was isolated for quantitative RT-PCR analysis of heregulin (neuregulin-1 α and β splice isoforms: NRG1α and NRG1β) and ERBB3. Data shown are means ± standard deviation. We observed an inverse association between ERBB3 and NRG1 with reciprocal expression in luminal compared to claudin-low (basal B) breast cancer cell lines.
To begin to examine the effects of exogenous HRG, cells were deprived of serum (‘serum-starved’) before HRG treatment, since serum contains many growth factors including HRG itself. Forty-eight hours of HRG treatment resulted in noticeable morphological changes, including stellate features and pseudopodia formation by MCF7 and SKBr3 cells (Figure 2A), consistent with other reports suggesting HRG treatment induces an epithelial-to-mesenchymal phenotypic shift in these cell lines [49, 50]. Morphologic change for MDA-MB-361 was consistent with the other two cell lines but more subtle overall, with cells becoming less cohesive and developing some stellate projections.
We also investigated HER3-HER2 downstream signaling 30 min after HRG treatment. All three cell lines responded to exogenous HRG with phosphorylation of HER3 and its preferred dimerization partner HER2, but not the other HRG receptor HER4 (Figure 2B). There was also HRG-induced phosphorylation of AKT and ERK1/2, important downstream targets of HER2 that regulate tumor cell survival, proliferation and invasion [17]. Though of lesser magnitude than the phosphorylation induction, there was also an increase in total HER3 protein levels. The short time frame of this experiment suggests this may involve post-transcriptional mechanisms, such as protein stabilization or translation efficiency.
In contrast to the HER2/HER3-positive luminal cell lines, three representative claudin-low cell lines (Hs578T, MDA-MB-231 and SUM-159-PT; Figure 1) did not show induction of HER3 expression or phosphorylation following treatment with exogenous HRG (Supplementary Figure 1).
Exogenous HRG treatment induces cell line-dependent proliferation and adhesion of luminal breast cancer cells in vitro
Since AKT and ERK1/2 were potently activated by HRG in three luminal breast cancer cell lines and they are known to induce tumor cell proliferation in other contexts [51], we investigated the effects of HRG on proliferation of HRG-treated versus untreated MDA-MB-361, MCF7 and SKBr3 cells. As shown in Figure 3A, HRG induced a time-dependent proliferative response in MDA-MB-361 and MCF7, but not SKBr3 cells. Others have demonstrated that HER2 promotes proliferation through deregulation of cell cycle checkpoints [52]. We therefore investigated the effects of HRG on cyclin D1 and p27 protein levels by Western blot analysis. As shown in Figure 3B, HRG treatment attenuated p27 and induced cyclin D1 expression in the two proliferative cell lines.
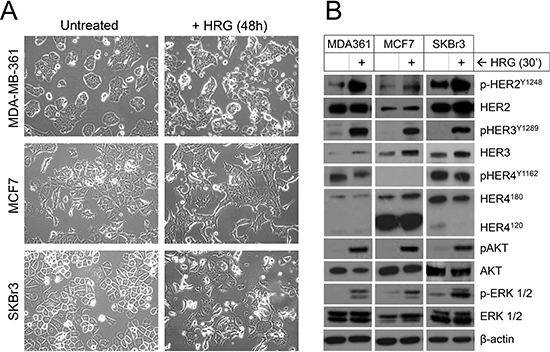
Figure 2: Treatment of luminal HER2+ breast cancer cell lines with exogenous HRG alters cell morphology and activates signaling through HER2, HER3, AKT and ERK. (A) Serum-starved cells were treated with HRG for 48 h then imaged by light microscopy (images acquired at 20x magnification). (B) Serum-starved cells were treated with HRG for 30 min then total and phosphorylated HER, AKT and ERK isoforms were quantified by Western blot. β-actin was used as the loading control.
Tumor cell adhesion to extracellular matrix proteins enhances survival and metastatic potential of circulating tumor cells [53], and adhesion of circulating tumor cells to the brain endothelium is thought to be a critical step preceding endothelial retraction and active extravasation [54, 55]. To investigate whether HER2-HER3 signaling increases the adhesive properties of luminal breast cancer cell lines, we assayed adhesion of HRG-treated cells to collagen I, which is a substrate for a range of cell adhesion molecules. HRG enhanced the adhesion of MDA-MB-361 and SKBr3 cells to collagen I (Figure 4A), concomitant with induction of ICAM-1 (Figure 4B–4C), a β2-integrin receptor associated with enhanced invasion, motility and metastasis in breast cancer [56–58]. Collectively these data show that exogenous HRG promotes proliferation and adhesion of luminal breast cancer cell lines, though these responses could be context-dependent since they were not consistent across the cell lines tested.
Exogenous HRG induces luminal breast cancer cell line invasion and secreted protease activity
AKT signaling induces aggressive breast cancer cell behavior and others have reported that HRG induces motility and invasion through ECM proteins (e.g. [50, 59–61]). Consistent with this, transwell assay experiments showed that the three cell lines migrated toward a HRG chemotactic signal (Figure 5A). Moreover, this response was maintained after coating the transwell inserts with matrigel (Figure 5B). These data show that HRG promotes both migratory and invasive behavior of luminal breast cancer cell lines, which otherwise migrate very poorly in vitro.
Since this invasive activity requires remodeling of ECM proteins, and AKT-associated invasion is marked by secretion of several proteases that are important in invasion and metastasis [62], we next investigated expression of MMP-2, MMP-9, urokinase plasminogen activator receptor (PLAU; uPAR) and its ligand (PLAUR; uPA), and cathepsin B (CSTB) in the HRG-treated cells. Using qRT-PCR analysis we found HRG induced cell line-dependent increases in expression of MMP2, PLAUR and PLAU, and a modest but significant decrease in expression of CSTB (Figure 6A). MMP9 was consistently induced in all three cell lines (Figure 6A). This was also evident at the protein level, with Western blot analysis confirming induction of MMP-9 protein in all three cell lines, and variable changes for the other proteolytic proteins (Figure 6B).
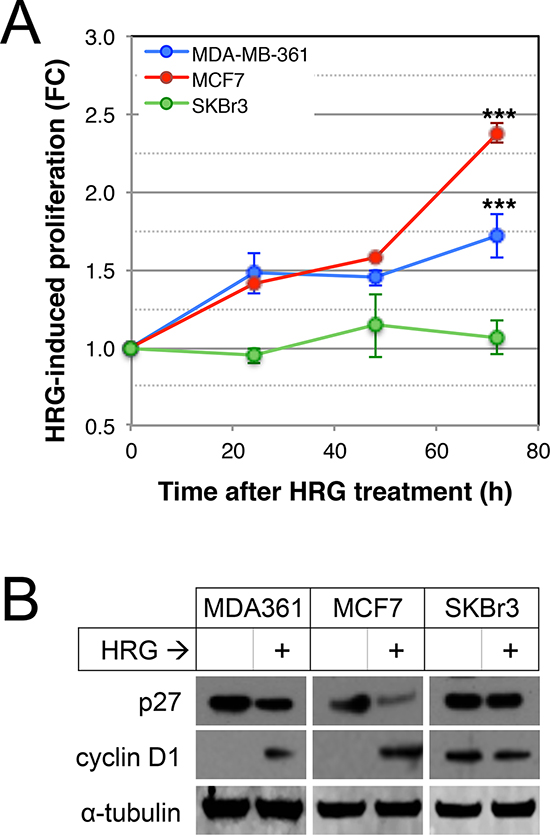
Figure 3: Effects of HRG on proliferation of luminal HER2+ breast cancer cell lines. (A) Serum-starved cells were treated with HRG and proliferation rate was assessed using an MTT assay over the following 72 h. Data shown are means ± standard deviation, normalised to an untreated control group for each cell line at each timepoint. ***p < 0.0001 according to unpaired, 2-tailed student’s t-tests comparing treated vs untreated cells at 72 hours post-treatment. FC, fold-change. (B) Serum-starved cells were treated with HRG for 48 h, then expression of p27 and cyclin D1 proteins was quantified by Western blot (loading control: α-tubulin).
Ultimately we were interested in common HRG-induced changes in the secretion and extracellular activity of ECM proteases, and so we assessed the proteolytic activities of MMP-9 and MMP-2 activity in conditioned media from the HRG-treated cells by gelatin zymography (gelatin is a substrate for both enzymes). This experiment confirmed that HRG-mediated induction of MMP-9 was associated with activation of extracellular MMP-9 activity in cultures of all three cell lines, though the total amount was relatively lower in MDA-MB-361 compared to SKBr3 and MCF7 cells (Figure 6C). MMP-2 activity was induced in MDA-MB-361 and SKBr3 cells.
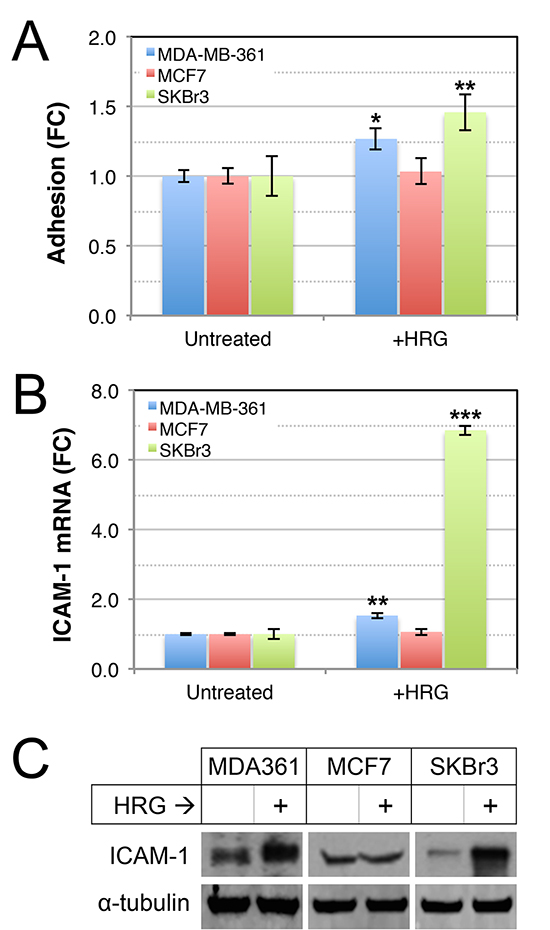
Figure 4: Effects of HRG on adhesive abilities of luminal HER2+ breast cancer cell lines. (A) HRG increases adhesion of MDA-MB-361 and SKBr3 cells to collagen-I. Serum-starved cells were treated with HRG for 48 h, allowed to adhere to collagen-I-coated dishes for 15 min then adhesion potential was determined using optical density measurements of treated versus untreated controls. *p = 0.05–0.01, **p = 0.01–0.001 (2-tailed, unpaired student’s t-tests). HRG increases expression of ICAM-1 RNA (B) and protein (C) in MDA-MB-361 and SKBr3 cells. Cells were treated as above, then total RNA or protein were prepared for quantitative real-time PCR or Western blot of ICAM-1 expression respectively (HPRT1 or α-tubulin loading controls, respectively). **p = 0.001–0.0001; ***p < 0.0001 (2-tailed, unpaired student’s t-tests). FC, fold-change.
Others have previously reported that in both breast tumors and cell lines, there is a negative association between the expression of MMP-9 and one of its natural inhibitors, RECK [30], a key breast cancer metastasis suppressor gene. Furthermore, RECK is repressed in brain metastases compared to primary breast cancers [63]. The KAI1 (CD82) metastasis suppressor has also been implicated in MMP-9 repression [64]. We therefore investigated expression of KAI1 and RECK by qRT-PCR and found that HRG treatment repressed both genes (Figure 6D), suggesting this could be one mechanism by which HRG increases extracellular MMP-9 activity. Interestingly, the two RECK-suppressed cell lines (MDA-MB-361 and SKBr3) are ERBB2-amplified, and RECK is known to functionally oppose oncogenic HER2 signaling by interfering with HER3 dimerization [65].
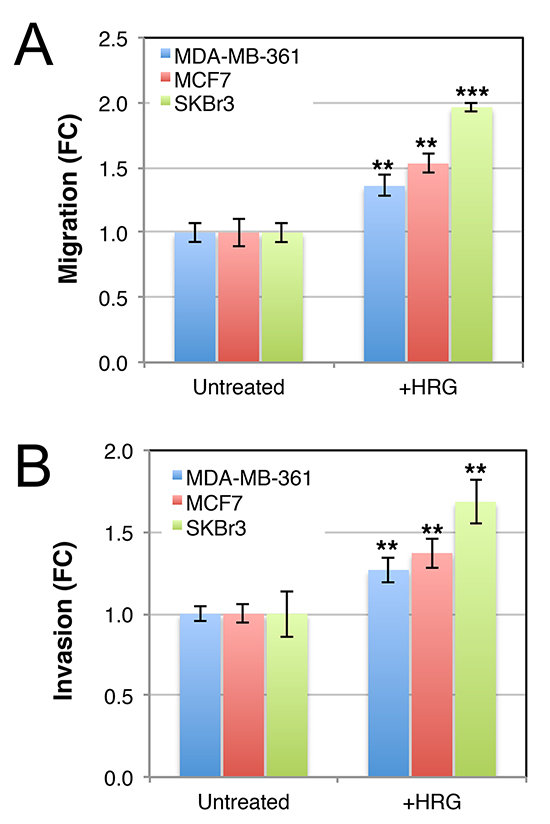
Figure 5: Luminal HER2+ breast cancer cell lines invade across matrigel toward a heregulin chemotactic signal. Serum-starved cells were seeded in 8 μm transwell chambers (A) or 8 μm transwell chambers in which the membranes were pre-coated with matrigel (B). HRG was supplemented in the lower chamber, and media was changed regularly to maintain a concentration gradient. After 48 h, cells on the lower surfaces of the porous membranes were quantified by crystal violet staining. Data shown are means ± standard deviation, normalised to the untreated control group for each cell line. **p = 0.05–0.01, **p = 0.01–0.001 (unpaired, 2-tailed student’s t-tests of HRG-treated cells compared to untreated controls). FC, fold-change.
Finally, we investigated the expression and activity of extracellular cathepsin B, as this has been implicated in mediating invasive behavior of HER2-positive breast cancer cells [66], and activation of MMP-9 and infiltrative tumor cell growth in glioma [67–69]. There was no substantial change in cathepsin B expression following treatment with exogenous HRG (Figure 6E), but there was a significant increase in extracellular cathepsin B activity in all three breast cancer cell lines (Figure 6F). Others have shown that PI3K mediates cathepsin B secretion by lysosomal exocytosis [70], and therefore increased secretion of cathepsin B may be one mechanism by which HRG increases its extracellular activity in breast cancer cell lines.
Exogenous HRG induces transmigration of breast cancer cell lines across a tight barrier of primary human brain microvascular endothelial cells
MMP-2 and MMP-9 have been associated with degradation of endothelial tight junction proteins, permeabilization of the blood-brain-barrier (BBB) and subsequent brain colonization in mouse models of leukemia [71]. Therefore we investigated whether active MMP isoforms in conditioned media from HRG-treated breast cancer cell lines are sufficient to stimulate transmigration across an endothelial barrier. We established an in vitro model of the BBB using primary human brain microvascular endothelial cells (HBMECs) and matrigel to simulate the brain endothelium and basement membrane respectively (Figure 7A). The integrity of this barrier was validated by measuring dextran-FITC diffusion (Figure 7B), and by confirming strong induction of tight junction proteins claudin-5, ZO-1 and occludin in the HBMEC layer (Figure 7C) as described [72].
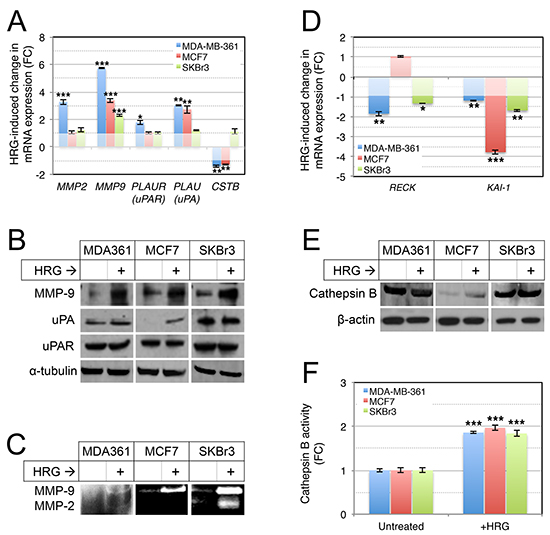
Figure 6: Treatment of luminal breast cancer cell lines with exogenous HRG increases extracellular protease activity. (A, B) HRG increases expression of proteolytic cascade proteins. Serum-starved cells were treated with HRG for 48 h, then total RNA or protein were isolated from the cells for qRT-PCR and Western blot analyses respectively (HPRT1 and α-tubulin were used as normalization and loading controls, respectively). (C) HRG increases secreted MMP-2 and MMP-9 proteolytic activities. Starved cells were treated with HRG as above and then conditioned media was concentrated and analysed for MMP-2 and MMP-9 activity by gelatin zymography (enzymatic activity is proportional to the intensity of the white bands). (D) HRG represses expression of RECK and KAI1 metastasis suppressor genes. qRT-PCR analysis was performed as for (A). HRG treatment does not substantially alter cathepsin B protein expression (E), but increases extracellular cathepsin B proteolytic activity (F). Cathepsin B expression was analyzed by Western blot analysis as for (B), with β-actin as the loading control. Enzyme activity was assayed using a fluorometric enzyme activity assay. *p = 0.01–0.001, **p = 0.001–0.0001, ***p < 0.0001 (unpaired, 2-tailed student’s t-tests comparing treated to untreated control samples). FC, fold-change.
Treatment of co-cultured breast cancer cell lines with exogenous HRG (Figure 7A) caused active migration of all three cell lines across the tight HBMEC layer (Figure 7D). This response was attenuated upon inhibition of HER2, HER3 or MMP activity, assessed by supplementing the upper chambers with saturating doses of humanized monoclonal antibodies against HER2 and HER3 (Herceptin® and EV20 respectively) or the broad-spectrum MMP inhibitor, GM6001. Treatment with GM6001 did not completely abrogate the response, indicating that MMPs may not be the only transmigration mechanism activated by HRG-HER3-HER2 signaling. It is noteworthy that complete inhibition of transmigratory activity was only achieved through combined blockade of HER2 and HER3 (Herceptin® + EV20; Figure 7D, green lines).
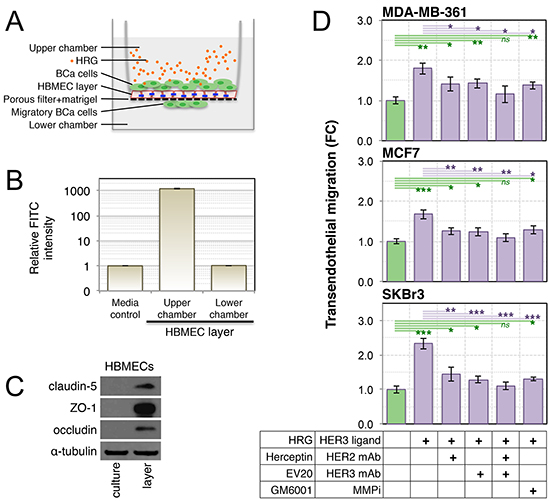
Figure 7: Treatment of luminal breast cancer cell lines with exogenous HRG increases their endothelial transmigration activity. (A) Schematic of the in vitro blood-brain-barrier model used to assay transendothelial migration of HRG-treated breast cancer cell lines (blue lines = tight junctions). Primary human brain microvascular endothelial cells (HBMECs) were seeded into matrigel-coated transwell chambers, then allowed to form a confluent layer. Serum-starved breast cancer cell lines were then seeded over the layer). (B) The functional integrity of the HBMEC layer was validated by measuring dextran-FITC (40 kDa) flux from the upper to lower transwell chambers. (C) HBMEC expression of tight junction proteins in layer-forming culture conditions was confirmed by Western blot analysis. (D) Breast cancer cell line transendothelial migration activity was measured in response to HRG ligand with and without drug treatments as indicated. Data shown are means ± standard deviation (n = 3 from a representative experiment). The statistical significance of differences between treatments and the untreated control was determined using unpaired, 2-tailed student’s t-tests (*p = 0.05–0.01, **p = 0.01–0.001 and ***p < 0.001). BCa, breast cancer; FC, fold-change; mAb, monoclonal antibody; MMPi, matrix metalloproteinase inhibitor.
DISCUSSION
The development of brain metastases is a devastating complication that affects 10–30% of women with breast cancer [2], causing challenging neurological symptoms including headaches, cognitive impairment and seizures [73]. Current treatments can prolong life expectancy and improve quality-of-life, though ultimately they are not curative. Molecular targeted drug therapy is critically under-utilized in the clinic, partly because of deficiencies in our understanding of the molecular mechanisms involved in the seeding and subsequent proliferation of disseminated cells in the brain. Research in this area is now beginning to illuminate some of the mechanisms by which tumor cells exploit and remodel the local microenvironment to facilitate metastatic outgrowth [40–42, 74–76].
Heregulin is critical for the normal development and function of the nervous system. It is expressed by neurons, glia and brain microvascular endothelia, where its functions include promoting glial cell survival and differentiation, neural precursor cell differentiation and migration, and endothelial cell protection from oxidative injury [18–20]. Consistent with the idea that HRG is a brain growth factor exploited by metastatic cells, HER3 is induced and activated in brain metastases compared to matching breast and lung cancers [23, 24] and patients with HER2-positive breast cancer are at high risk of brain relapse [4, 6]. Moreover, primary breast cancers over-expressing HER3 are more likely to relapse as isolated brain metastases than non-HER3-overexpressing tumors [22].
In this study, we found that exposure to HRG stimulated the transendothelial migration of HER2/3-expressing breast cancer cell lines across a tight barrier of primary human brain microvascular endothelia and an associated matrigel layer, and that this was at least partly mediated by MMPs (Figure 7). Specifically, exposure to HRG increased the extracellular activity of MMP-9 in three cell lines, and MMP-2 in two of these lines (Figure 6). Other studies have implicated MMP-2 and -9 in the development of brain metastases [31–34], and the current study now suggests that this could be at least partly due to enhancing vascular permeability. Since HRG is expressed by brain microvascular endothelia [20], these data raise the possibility that HRG-HER3-HER2 signaling is involved in extravasation from the brain microvasculature in vivo, particularly since we also found that HRG increases breast cancer cell adhesion potential (Figure 4). Indeed, MMPs 2 and 9 can mediate vascular leakage in experimental models of cerebral ischemic injury by degrading endothelial tight junction complexes [77–79]. HRG-mediated vascular permeabilization could also be important in established metastases with increasing metabolic demands.
Adjuvant Herceptin® therapy for HER2-positive breast cancer delays the onset of brain metastases [10], and this latency is further extended by the HER2 dimerization blocker Perjeta® [8]. In this context, it is noteworthy that Herceptin® and the humanized HER3 antibody EV20 [80] conferred additive suppression of transmigration in our blood-brain-barrier experiments (Figure 7). There are likely to be multiple mechanisms enabling endothelial transmigration and consequent establishment of micrometastases in vivo (for example, we also found that exposure to exogenous HRG reduces expression of KAI1 and RECK metastasis suppressor genes), however these in vitro experiments may provide some molecular insight into the aforementioned clinical observations.
The cell lines used in this study all migrated and invaded through extracellular matrix proteins towards an HRG chemotactic signal (Figure 5), concomitant with increased activity of extracellular proteases (Figure 6). Consistent with other reports [60, 81], we observed cell proliferation in response to HRG exposure (Figure 3). Collectively these data suggest that HRG-HER3-HER2 signaling could be involved in several aspects of brain metastasis development. In vivo experiments modeling these steps with inhibition of HER2-HER3 dimer function are required in the future.
This study has potential implications in translational oncology, and warrants further investigation into the possibility of targeting the HRG-HER3-HER2 axis for management of brain metastases from HER2-positive breast cancer.
MATERIALS AND METHODS
Breast cancer cell lines and HRG activation assay
The breast cancer cell lines used in this study were obtained from the American Type Culture Collection (ATCC). The panel of cell lines included three molecular subtypes previously defined by expression array profiling and unsupervised cluster analysis and/or surrogate immunohistochemical markers [44–46]: claudin-low (basal B): MDA-MB-436, MDA-MB-231, RHB1, 184B5, MCF10A, 184A1, SUM159, Hs578T, HBL100 and BT549; basal-A: HCC70, MDA-MB-468, Bre80Tert, HCC1937, HCC1143, SVCT, PMC-42-ET; luminal: MCF7, KPL-1, T-47D, ZR751, MDA-MB-175-VII); and luminal/ERBB2-amplified: MDA-MB-453, BT-474, SKBr3 and MDA-MB-361. MDA-MB-361 is the only commercially available breast cancer cell line that was derived from a metastatic brain tumor. All cell lines were authenticated by STR profiling (Cell ID™ system; Promega) and were routinely checked for mycoplasma infection (MycoAlert™; Lonza). Cell cultures were maintained at 37°C in 5% CO2 in a humidified incubator and cultured according to ATCC recommendations.
For heregulin (HRG) activation experiments, cells were routinely seeded at predetermined densities in regular culture medium, then switched from the recommended amount of fetal bovine serum (FBS) to 0.1% FBS (serum-starved conditions) for 24 hours. Cultures were then supplemented with 50 ng/mL HRG-β1 (Sigma) and cultured for either 30 min (signaling analysis) or 48 h (functional experiments including assaying expression of downstream targets of HER2-HER3 signaling).
Analysis of gene expression by quantitative real time reverse transcription-PCR (qRT-PCR)
Trizol (Invitrogen) was used to isolate total RNA from cultured cells. cDNA was prepared using 1 μg of RNA from each sample with the PrimeScript RT reagent kit (Takara Bio Inc.). qPCR was then performed in triplicate on a StepOnePlus instrument using SYBR green (Applied Biosystems) according to standard procedures (see Table 1 for PCR primers). Melt curve analysis was performed to verify amplification of single PCR products. Hypoxanthine phosphoribosyl transferase1 (HPRT1) was amplified as normalizer and fold change in expression of each target mRNA relative to HPRT1 was calculated according to the 2–ΔΔct relative expression formula [82]. HER3 qPCR was performed using the ERBB3 TaqMan® expression assay (Hs00951455_m1; Applied Biosystems).
Western blot analysis
Total protein extracts were prepared in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) containing fresh protease and phosphatase inhibitors (Thermo Scientific) for 30 min at 4°C. Thirty to fifty μg of lysate was resolved by SDS-PAGE, transferred to PVDF membrane (Immobilin P, Millipore) then probed with primary antibodies (Table 2) followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma). Two different HER3 antibodies were used to confirm the increase in HER3 protein levels 30 min after HRG treatment. Blots were probed with α-tubulin and β-actin as loading controls.
Cell proliferation assay
A microculture tetrazolium test (MTT) was performed to determine cell proliferation after treatment of the cells with recombinant HRG. Briefly, cells were plated onto 96-well plates at a density of 4×104/well for 24 h and then starved in 0.1% FBS for 24 h. The cells were then treated with 50 ng/mL of HRG for 24, 48 and 72 h. Untreated cells were used as the control group. 100 μL of MTT (0.5 mg/ml) (Sigma) was added to each well and the cultures were further incubated at 37°C for 2 h. After dissolving the precipitated formazan with 100 μL of dimethyl sulfoxide (DMSO), the optical density was measured at 570 nm.
Cell adhesion assay
Adhesion experiments were conducted as described [14]. Cells were seeded into 6-well plates and after 24 h, washed three times with PBS and starved in 0.1% FBS overnight. The starved cells were treated with 50 ng/mL HRG for 48 h and seeded on collagen I coated 60 mm dishes (Biocoat Cell Environments; Becton Dickinson). After 15 min, cells were washed with cold PBS, stained with 0.5% crystal violet, lysed with 30% acetic acid and the optical density was measured at 590 nm.
Gelatin zymography
Gelatin zymography was carried out as described [83]. Briefly, conditioned media from HRG-treated and untreated cells was collected and centrifuged at high speed for 10 min to pellet cell debris. Protein from the conditioned media was then concentrated as appropriate using a Vacufuge® plus (Eppendorf). Five μg of secreted protein were applied to 10% polyacrylamide gels copolymerized with 1 mg/mL gelatin (Sigma). After electrophoresis, gels were rinsed in 2.5% Triton X-100 (3×30 min) to remove SDS, followed by incubation at 37°C overnight in incubation buffer (0.15 M NaCl, 10 mM CaCl2, 0.02% NaN3 in 50 mM Tris-HCl, pH 7.5). Gels were then stained (0.5% Coomassie Brilliant Blue) and destained with 7% methanol and 5% acetic acid. Areas of enzymatic activity appeared as clear bands over the dark background.
Cell migration and invasion assays
Cell migration was assayed in 24-well, 6.5-mm-internal-diameter transwell plates (8.0 μm pore size; Costar Corp.). Serum-starved cells were placed in the upper chambers, and the lower chamber was filled with media containing 0.1% FBS (control), with or without HRG supplementation (50 ng/mL). Media in both chambers was changed every 6 hours to maintain an HRG gradient. Cells were allowed to migrate for 48 h. After this time, cells on the upper surfaces of the filters were removed by wiping with a cotton swab and migrated cells on the undersides of the filters were fixed with methanol, stained with crystal violet, lysed with 30% acetic acid and the optical density was measured at 590 nm. For invasion, experiments were essentially conducted as above, except that transwell filters were pre-coated with matrigel (1:10 dilution in media; BD Biosciences).
Cathepsin B activity assay
To investigate the effect of HRG on the activity of secreted cathepsin B, conditioned media from HRG-treated and untreated cells was centrifuged at 10,000 rpm for 15 min to remove cell debris, then concentrated appropriately using the Vacufuge® plus (Eppendorf). We used a fluorometric cathepsin B activity assay (Abcam) according to the manufacturer’s instructions.
Blood-brain-barrier transendothelial migration assay
Primary human brain microvascular endothelial cells (HBMECs) and HBMEC culture reagents were purchased from Cell Systems and cells were routinely cultured in CSC Complete Medium on fibronectin (Invitrogen) pre-coated surfaces, according to the manufacturer’s recommendations. Two×104 HBMECs were seeded on matrigel-coated 0.4 μm transwell filters (Costar Corp.) in 200 μL of CSC complete medium. The lower compartment was filled with 600 μL of the same medium. Cells were grown for 4 d to allow complete formation of HBMEC tight junctions (TJs). The impermeability of this in vitro BBB model to small solutes was assayed by measuring dextran-FITC (Invitrogen) flux from the top to bottom chambers. The media was changed to 1% FBS-CSC media plus CultureBoost (Cell Systems) for 24 h. Dextran-FITC (1 mg/mL; Invitrogen) was added to the upper chamber and after 20 min, 50 μL media from upper and lower chambers was removed and fluorescence was measured using a Paradigm™ Detection Platform (Beckman Coulter; 485 nm excitation, 520 nm emission).
Table 1: Primers used for qRT-PCR
Gene |
Accession no. |
Forward primer (5′-3′) |
Reverse primer (5′-3′) |
NRG1α |
NM_013964 |
AAACCAAGAAAAGGCGGAGGAGCT |
GAGGGCGATGCAGATGCCGG |
NRG1β |
NM_013956 |
GCCAGCTTCTACAAGCATCTTGGGA |
GGAGGGCGATGCAGATGCCG |
HPRT1 |
NM_000194 |
TGGACAGGACTGAACGTCTTG |
CCAGCAGGTCAGCAAAGAATTTA |
MMP2 |
NM_004530 |
CTTCCAAGTCTGGAGCGATGT |
TACCGTCAAAGGGGTATCCAT |
MMP9 |
NM_004994 |
GGGACGCAGACATCGTCATC |
TCGTCATCGTCGAAATGGGC |
PLAU (uPA) |
NM_002658 |
TCAAAAACCTGCTATGAGGGGA |
GGGCATGGTACGTTTGCTG |
PLAUR (uPAR) |
NM_002659 |
TGTAAGACCAACGGGGATTGC |
AGCCAGTCCGATAGCTCAGG |
KAI1 |
NM_002231 |
GCCGACAAGAGCAGTTTCATC |
AGGAAAGCAAAGTACAGCCCC |
RECK |
NM_021111 |
TGTGAACTGGCTATTGCCTTG |
GCATAACTGCAACAAACCGAG |
ICAM1 |
NM_000201 |
TTGGGCATAGAGACCCCGTT |
GCACATTGCTCAGTTCATACACC |
CSTB |
NM_147780 |
CTGTCGGATGAGCTGGTCAAC |
TCGGTAAACATAACTCTCTGGGG |
Table 2: Primary antibodies used for western blot analysis
Antigen |
Clone |
Supplier |
HER3 |
2F12 |
Millipore |
HER3 |
C-17 |
Santa Cruz |
HER2 |
Polyclonal |
Millipore |
pHER2Y1248 |
Polyclonal |
Cell Signaling Technology |
pHER3Y1289 |
21D3 |
Cell Signaling Technology |
Akt |
Polyclonal |
Cell Signaling Technology |
pAktS473 |
D9E |
Cell Signaling Technology |
ERK1/2 |
137F5 |
Cell Signaling Technology |
p-ERK1/2T202/Y204 |
197G2 |
Cell Signaling Technology |
Cathepsin B |
G60 |
Cell Signaling Technology |
Cyclin D1 |
H-295 |
Santa Cruz Biotechnology |
ICAM-1 |
H-108 |
Santa Cruz Biotechnology |
p27 |
C-19 |
Santa Cruz Biotechnology |
MMP-9 |
Polyclonal |
Sigma |
uPA |
H-140 |
Sigma |
uPAR |
Polyclonal |
Sigma |
ZO-1 |
ZO1-1A12 |
Invitrogen |
Occludin |
OC-F10 |
Invitrogen |
α-tubulin |
4G1 |
Abcam |
β-actin |
8H10D10 |
Cell Signaling Technology |
For transmigration assays, 2×104 HBMEC cells were seeded into matrigel-coated (BD Biosciences) 24-well transwell inserts (8 μm pores; Costar Corp.). The cells were maintained for 4 d to allow the TJ formation, then media from upper and lower chambers was changed to 1% FBS-CSC media plus CultureBoost for 24 h (as above). Serum-starved breast cancer cell lines (1×105 for MCF7 and SKBr3, or 2×105 for MDA-MB-361) were seeded into the upper chamber in 100 μL of their regular media containing 0.1% FBS, then allowed to attach for 2 h. Cultures were then treated with HRG, (50 ng/mL; Sigma), GM6001 (20 μg/mL; Calbiochem), Herceptin® (20 μg/mL; Experimental Pharmacology Oncology, Berlin) and/or EV20 (20 μg/mL; Mediapharma) for 48 h. The upper surfaces of the transwell filters were then gently wiped clean with a cotton swab to remove non-migrating cells and cells on the undersides were fixed with methanol, stained with crystal violet, lysed with 30% acetic acid and the optical density was measured at 590 nm
Statistical analysis
Data are expressed as mean ± standard deviation (SD). All experiments were performed in triplicate. For statistical analysis, unpaired, two-tailed t-tests were applied. P values of less than 0.05 were considered significant.
ACKNOWLEDGEMENTS
This work was supported by funding from the Australian National Health and Medical Research Council (NHMRC), The University of Queensland scholarships to MM and a National Breast Cancer Foundation (NBCF) fellowship to PTS.
REFERENCES
1. Fox BD, Cheung VJ, Patel AJ, Suki D, Rao G. Epidemiology of metastatic brain tumors. Neurosurg Clin N Am. 2011; 22:1–6, v.
2. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004; 22:2865–2872.
3. Lin NU, Bellon JR, Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004; 22:3608–3617.
4. Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, Anders C, Ewend M, Perou CM. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2011.
5. Pestalozzi BC, Zahrieh D, Price KN, Holmberg SB, Lindtner J, Collins J, Crivellari D, Fey MF, Murray E, Pagani O, Simoncini E, Castiglione-Gertsch M, Gelber RD, Coates AS, Goldhirsch A. International Breast Cancer Study G: Identifying breast cancer patients at risk for Central Nervous System (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG). Ann Oncol. 2006; 17:935–944.
6. Lin NU, Amiri-Kordestani L, Palmieri D, Liewehr DJ, Steeg PS. CNS metastases in breast cancer: old challenge, new frontiers. Clin Cancer Res. 2013; 19:6404–6418.
7. Brufsky AM, Mayer M, Rugo HS, Kaufman PA, Tan-Chiu E, Tripathy D, Tudor IC, Wang LI, Brammer MG, Shing M, Yood MU, Yardley DA. Central nervous system metastases in patients with HER2-positive metastatic breast cancer: incidence, treatment, and survival in patients from registHER. Clin Cancer Res. 2011; 17:4834–4843.
8. Swain SM, Baselga J, Miles D, Im YH, Quah C, Lee LF, Cortes J. Incidence of central nervous system metastases in patients with HER2-positive metastatic breast cancer treated with pertuzumab, trastuzumab, and docetaxel: results from the randomized phase III study CLEOPATRA. Ann Oncol. 2014; 25:1116–1121.
9. Bartsch R, Berghoff A, Pluschnig U, Bago-Horvath Z, Dubsky P, Rottenfusser A, DeVries C, Rudas M, Fitzal F, Dieckmann K, Mader RM, Gnant M, Zielinski CC, Steger GG. Impact of anti-HER2 therapy on overall survival in HER2-overexpressing breast cancer patients with brain metastases. Br J Cancer. 2012; 106:25–31.
10. Park YH, Park MJ, Ji SH, Yi SY, Lim DH, Nam DH, Lee JI, Park W, Choi DH, Huh SJ, Ahn JS, Kang WK, Park K, Im YH. Trastuzumab treatment improves brain metastasis outcomes through control and durable prolongation of systemic extracranial disease in HER2-overexpressing breast cancer patients. Br J Cancer. 2009; 100:894–900.
11. Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008; 68:5878–5887.
12. Campiglio M, Ali S, Knyazev PG, Ullrich A. Characteristics of EGFR family-mediated HRG signals in human ovarian cancer. J Cell Biochem. 1999; 73:522–532.
13. Zhang Y, Opresko L, Shankaran H, Chrisler WB, Wiley HS, Resat H. HER/ErbB receptor interactions and signaling patterns in human mammary epithelial cells. BMC Cell Biol. 2009; 10:78.
14. Ueno Y, Sakurai H, Tsunoda S, Choo MK, Matsuo M, Koizumi K, Saiki I. Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int J Cancer. 2008; 123:340–347.
15. Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, Manning HC, Chang J, Arteaga CL. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. PNAS. 2011; 108:5021–5026.
16. Vaught DB, Stanford JC, Young C, Hicks DJ, Wheeler F, Rinehart C, Sanchez V, Koland J, Muller WJ, Arteaga CL, Cook RS. HER3 is required for HER2-induced preneoplastic changes to the breast epithelium and tumor formation. Cancer Res. 2012; 72:2672–2682.
17. Eccles SA. The role of c-erbB-2/HER2/neu in breast cancer progression and metastasis. J Mammary Gland Biol Neoplasia. 2001; 6:393–406.
18. Bernstein HG, Lendeckel U, Bertram I, Bukowska A, Kanakis D, Dobrowolny H, Stauch R, Krell D, Mawrin C, Budinger E, Keilhoff G, Bogerts B. Localization of neuregulin-1alpha (heregulin-alpha) and one of its receptors, ErbB-4 tyrosine kinase, in developing and adult human brain. Brain Res Bull. 2006; 69:546–559.
19. Pinkas-Kramarski R, Eilam R, Spiegler O, Lavi S, Liu N, Chang D, Wen D, Schwartz M, Yarden Y. Brain neurons and glial cells express Neu differentiation factor/heregulin: a survival factor for astrocytes. PNAS. 1994; 91:9387–9391.
20. Lok J, Sardi SP, Guo S, Besancon E, Ha DM, Rosell A, Kim WJ, Corfas G, Lo EH. Neuregulin-1 signaling in brain endothelial cells. J Cereb Blood Flow Metab. 2009; 29:39–43.
21. Britsch S. The neuregulin-I/ErbB signaling system in development and disease. Adv Anat Embryol Cell Biol. 2007; 190:1–65.
22. Berghoff AS, Bartsch R, Preusser M, Ricken G, Steger GG, Bago-Horvath Z, Rudas M, Streubel B, Dubsky P, Gnant M, Fitzal F, Zielinski CC, Birner P. Co-overexpression of HER2/HER3 is a predictor of impaired survival in breast cancer patients. Breast. 2014; 23:637–643.
23. Da Silva L, Simpson PT, Smart CE, Cocciardi S, Waddell N, Lane A, Morrison BJ, Vargas A, Healey S, Beesley J, Pakkiri P, Parry S, Kurniawan N, Reid L, Keith P, Faria P, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010; 12:R46.
24. Sun M, Behrens C, Feng L, Ozburn N, Tang X, Yin G, Komaki R, Varella-Garcia M, Hong WK, Aldape KD, Wistuba II. HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clin Cancer Res. 2009; 15:4829–4837.
25. Yu D, Hamada J, Zhang H, Nicolson GL, Hung MC. Mechanisms of c-erbB2/neu oncogene-induced metastasis and repression of metastatic properties by adenovirus 5 E1A gene products. Oncogene. 1992; 7:2263–2270.
26. Asrani K, Keri RA, Galisteo R, Brown SA, Morgan SJ, Ghosh A, Tran NL, Winkles JA. The HER2- and heregulin beta1 (HRG)-inducible TNFR superfamily member Fn14 promotes HRG-driven breast cancer cell migration, invasion, and MMP9 expression. Mol Cancer Res. 2013; 11:393–404.
27. Yao J, Xiong S, Klos K, Nguyen N, Grijalva R, Li P, Yu D. Multiple signaling pathways involved in activation of matrix metalloproteinase-9 (MMP-9) by heregulin-beta1 in human breast cancer cells. Oncogene. 2001; 20:8066–8074.
28. Xu FJ, Stack S, Boyer C, O’Briant K, Whitaker R, Mills GB, Yu YH, Bast RC, Jr. Heregulin and agonistic anti-p185(c-erbB2) antibodies inhibit proliferation but increase invasiveness of breast cancer cells that overexpress p185(c-erbB2): increased invasiveness may contribute to poor prognosis. Clin Cancer Res. 1997; 3:1629–1634.
29. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010; 141:52–67.
30. Figueira RC, Gomes LR, Neto JS, Silva FC, Silva ID, Sogayar MC. Correlation between MMPs and their inhibitors in breast cancer tumor tissue specimens and in cell lines with different metastatic potential. BMC Cancer. 2009; 9:20.
31. Mendes O, Kim HT, Stoica G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin Exp Metastasis. 2005; 22:237–246.
32. Mendes O, Kim HT, Lungu G, Stoica G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin Exp Metastasis. 2007; 24:341–351.
33. Wang L, Cossette SM, Rarick KR, Gershan J, Dwinell MB, Harder DR, Ramchran R. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS One. 2013; 8:e80933.
34. Lee KY, Kim YJ, Yoo H, Lee SH, Park JB, Kim HJ. Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier. Anticancer Res. 2011; 31:4307–4313.
35. Liu H, Kato Y, Erzinger SA, Kiriakova GM, Qian Y, Palmieri D, Steeg PS, Price JE. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer. 2012; 12:583.
36. Stark AM, Anuszkiewicz B, Mentlein R, Yoneda T, Mehdorn HM, Held-Feindt J. Differential expression of matrix metalloproteinases in brain- and bone-seeking clones of metastatic MDA-MB-231 breast cancer cells. J Neurooncol. 2007; 81:39–48.
37. Tester AM, Waltham M, Oh SJ, Bae SN, Bills MM, Walker EC, Kern FG, Stetler-Stevenson WG, Lippman ME, Thompson EW. Pro-matrix metalloproteinase-2 transfection increases orthotopic primary growth and experimental metastasis of MDA-MB-231 human breast cancer cells in nude mice. Cancer Res. 2004; 64:652–658.
38. Pardridge WM. Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv. 2003; 3:90–105, 151.
39. Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK. The biology of brain metastases-translation to new therapies. Nat Rev Clin Oncol. 2011; 8:344–356.
40. Fidler IJ, Balasubramanian K, Lin Q, Kim SW, Kim SJ. The brain microenvironment and cancer metastasis. Mol Cells. 2010; 30:93–98.
41. Neman J, Choy C, Kowolik CM, Anderson A, Duenas VJ, Waliany S, Chen BT, Chen MY, Jandial R. Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin Exp Metastasis. 2013; 30:753–768.
42. Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, Li H, Hambrecht AC, Roberts E, Jandial R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. PNAS. 2014; 111:984–989.
43. Termini J, Neman J, Jandial R. Role of the neural niche in brain metastatic cancer. Cancer Res. 2014; 74:4011–4015.
44. Hollestelle A, Nagel JHA, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, Riaz M, Koopman DG, ten Hagen TLM, Leeuw BHCGM, Zwarthoff EC, Teunisse A, et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat. 2009; 121:53–64.
45. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe J-P, Tong F. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006; 10:515–527.
46. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast cancer research: BCR. 2010; 12:R68.
47. Zhang RD, Fidler IJ, Price JE. Relative malignant potential of human breast carcinoma cell lines established from pleural effusions and a brain metastasis. Invasion Metastasis. 1991; 11:204–215.
48. Bergman I, Barmada MA, Griffin JA, Slamon DJ. Treatment of meningeal breast cancer xenografts in the rat using an anti-p185/HER2 antibody. Clin Cancer Res. 2001; 7:2050–2056.
49. Hijazi MM, Thompson EW, Tang C, Coopman P, Torri JA, Yang D, Mueller SC, Lupu R. Heregulin regulates the actin cytoskeleton and promotes invasive properties in breast cancer cell lines. Int J Oncol. 2000; 17:629–641.
50. Kim J, Jeong H, Lee Y, Kim C, Kim H, Kim A. HRG-beta1-driven ErbB3 signaling induces epithelial-mesenchymal transition in breast cancer cells. BMC Cancer. 2013; 13:383.
51. Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, Singh S, Arribas J, Rosen N, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011; 30:2547–2557.
52. Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001; 61:6583–6591.
53. Zetter BR. Adhesion molecules in tumor metastasis. Semin Cancer Biol. 1993; 4:219–229.
54. Soto MS, Serres S, Anthony DC, Sibson NR. Functional role of endothelial adhesion molecules in the early stages of brain metastasis. Neuro Oncol. 2014; 16:540–551.
55. Arshad F, Wang L, Sy C, Avraham S, Avraham HK. Blood-brain barrier integrity and breast cancer metastasis to the brain. Pathology research international. 2010; 2011:920509.
56. Roland CL, Harken AH, Sarr MG, Barnett CC, Jr. ICAM-1 expression determines malignant potential of cancer. Surgery. 2007; 141:705–707.
57. Regidor PA, Callies R, Regidor M, Schindler AE. Expression of the cell adhesion molecules ICAM-1 and VCAM-1 in the cytosol of breast cancer tissue, benign breast tissue and corresponding sera. Eur J Gynaecol Oncol. 1998; 19:377–383.
58. Rosette C, Roth RB, Oeth P, Braun A, Kammerer S, Ekblom J, Denissenko MF. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis. 2005; 26:943–950.
59. Sollome JJ, Thavathiru E, Camenisch TD, Vaillancourt RR. HER2/HER3 regulates extracellular acidification and cell migration through MTK1 (MEKK4). Cell Signal. 2014; 26:70–82.
60. Ruan SQ, Wang ZH, Wang SW, Fu ZX, Xu KL, Li DB, Zhang SZ. Heregulin-beta1-induced GPR30 upregulation promotes the migration and invasion potential of SkBr3 breast cancer cells via ErbB2/ErbB3-MAPK/ERK pathway. Biochem Biophys Res Commun. 2012; 420:385–390.
61. Lazaro G, Smith C, Goddard L, Jordan N, McClelland R, Barrett-Lee P, Nicholson RI, Hiscox S. Targeting focal adhesion kinase in ER+/HER2+ breast cancer improves trastuzumab response. Endocr Relat Cancer. 2013; 20:691–704.
62. Shukla S, Maclennan GT, Hartman DJ, Fu P, Resnick MI, Gupta S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int J Cancer. 2007; 121:1424–1432.
63. Wikman H, Westphal L, Schmid F, Pollari S, Kropidlowski J, Sielaff-Frimpong B, Glatzel M, Matschke J, Westphal M, Iljin K, Huhtala H, Terracciano L, Kallioniemi A, Sauter G, Muller V, Witzel I, et al. Loss of CADM1 expression is associated with poor prognosis and brain metastasis in breast cancer patients. Oncotarget. 2014; 5:3076–3087.
64. Jee BK, Park KM, Surendran S, Lee WK, Han CW, Kim YS, Lim Y. KAI1/CD82 suppresses tumor invasion by MMP9 inactivation via TIMP1 up-regulation in the H1299 human lung carcinoma cell line. Biochem Biophys Res Commun. 2006; 342:655–661.
65. Hong KJ, Hsu MC, Hou MF, Hung WC. The tumor suppressor RECK interferes with HER-2/Neu dimerization and attenuates its oncogenic signaling. FEBS Lett. 2011; 585:591–595.
66. Rafn B, Nielsen CF, Andersen SH, Szyniarowski P, Corcelle-Termeau E, Valo E, Fehrenbacher N, Olsen CJ, Daugaard M, Egebjerg C, Bottzauw T, Kohonen P, Nylandsted J, Hautaniemi S, Moreira J, Jaattela M, et al. ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol Cell. 2012; 45:764–776.
67. Rao Malla R, Gopinath S, Alapati K, Gorantla B, Gondi CS, Rao JS. Knockdown of cathepsin B and uPAR inhibits CD151 and alpha3beta1 integrin-mediated cell adhesion and invasion in glioma. Mol Carcinog. 2013; 52:777–790.
68. Sivaparvathi M, Sawaya R, Wang SW, Rayford A, Yamamoto M, Liotta LA, Nicolson GL, Rao JS. Overexpression and localization of cathepsin B during the progression of human gliomas. Clin Exp Metastasis. 1995; 13:49–56.
69. Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003; 3:489–501.
70. Steffan JJ, Snider JL, Skalli O, Welbourne T, Cardelli JA. Na+/H+ exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic. 2009; 10:737–753.
71. Feng S, Cen J, Huang Y, Shen H, Yao L, Wang Y, Chen Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS One. 2011; 6:e20599.
72. Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 2010; 6:e1000874.
73. Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. J Neurooncol. 2005; 75:5–14.
74. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, Massague J. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014; 156:1002–1016.
75. Gril B, Palmieri D, Qian Y, Anwar T, Liewehr DJ, Steinberg SM, Andreu Z, Masana D, Fernandez P, Steeg PS, Vidal-Vanaclocha F. Pazopanib inhibits the activation of PDGFRbeta-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am J Pathol. 2013; 182:2368–2379.
76. Chuang HN, van Rossum D, Sieger D, Siam L, Klemm F, Bleckmann A, Bayerlova M, Farhat K, Scheffel J, Schulz M, Dehghani F, Stadelmann C, Hanisch UK, Binder C, Pukrop T. Carcinoma cells misuse the host tissue damage response to invade the brain. Glia. 2013; 61:1331–1346.
77. Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010; 30:837–848.
78. Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000; 20:1681–1689.
79. Seo JH, Guo S, Lok J, Navaratna D, Whalen MJ, Kim KW, Lo EH. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr Pharm Des. 2012; 18:3645–3648.
80. Sala G, Rapposelli IG, Ghasemi R, Piccolo E, Traini S, Capone E, Rossi C, Pelliccia A, Di Risio A, D’Egidio M, Tinari N, Muraro R, Iacobelli S. EV20, a Novel Anti-ErbB-3 Humanized Antibody, Promotes ErbB-3 Down-Regulation and Inhibits Tumor Growth In Vivo. Transl Oncol. 2013; 6:676–684.
81. Zhang Y, Akinmade D, Hamburger AW. Inhibition of heregulin mediated MCF-7 breast cancer cell growth by the ErbB3 binding protein EBP1. Cancer Lett. 2008; 265:298–306.
82. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3:1101–1108.
83. Momeny M, Malehmir M, Zakidizaji M, Ghasemi R, Ghadimi H, Shokrgozar MA, Emami AH, Nafissi S, Ghavamzadeh A, Ghaffari SH. Silibinin inhibits invasive properties of human glioblastoma U87MG cells through suppression of cathepsin B and nuclear factor kappa B-mediated induction of matrix metalloproteinase 9. Anticancer Drugs. 2010; 21:252–260.