
Cancers, or malignant tumors, are defined and diagnosed as tumors whose cells migrate into normal tissues, while benign tumors as those whose cells fail to do so [1]. Why the cells of some tumors migrate and the cells of others do not is not entirely clear, as can be judged by the near absence of drugs that target cancer cell migration [2, 3], by expert opinions that metastasis, the process of migrating to distant organs, is an “almost intractable facet of cancer medicine” [4] in which “attempts to specifically target metastatic pathways have met with near-universal failures” [5], and by the fact that surviving a disseminated cancer is still less likely, and by far, than surviving a round of Russian roulette [5–7], a comparison that is relevant because a cancer patient’s survival is also predicted statistically, that is to say, as a matter of luck.
This situation is not due to the want of effort, as attested by at least 3.3 million articles published on cancer (searching PubMed on March 26, 2023 with ‘cancer [MeSH Major Topic]’ returned 3,374,062 results) since a prominent researcher concluded a century ago that “despite an immense accumulation of data, the solution of the tumor problem waits upon fresh findings” [8]. That this conclusion is still valid reminds us that since it was made some intractable medical problems have been solved not by continuing to accumulate data, but by revisiting or rediscovering neglected observations and models [9–14].
One of the models that are still neglected proposes that tumor cells become migratory not by accumulating genomic aberrations, as a century-old view which dominates cancer research and drug development posits [15–21], but by acquiring this ability from normal migratory cells, like a company that merges with another business to acquire a technology needed to expand into new markets (Figure 1).
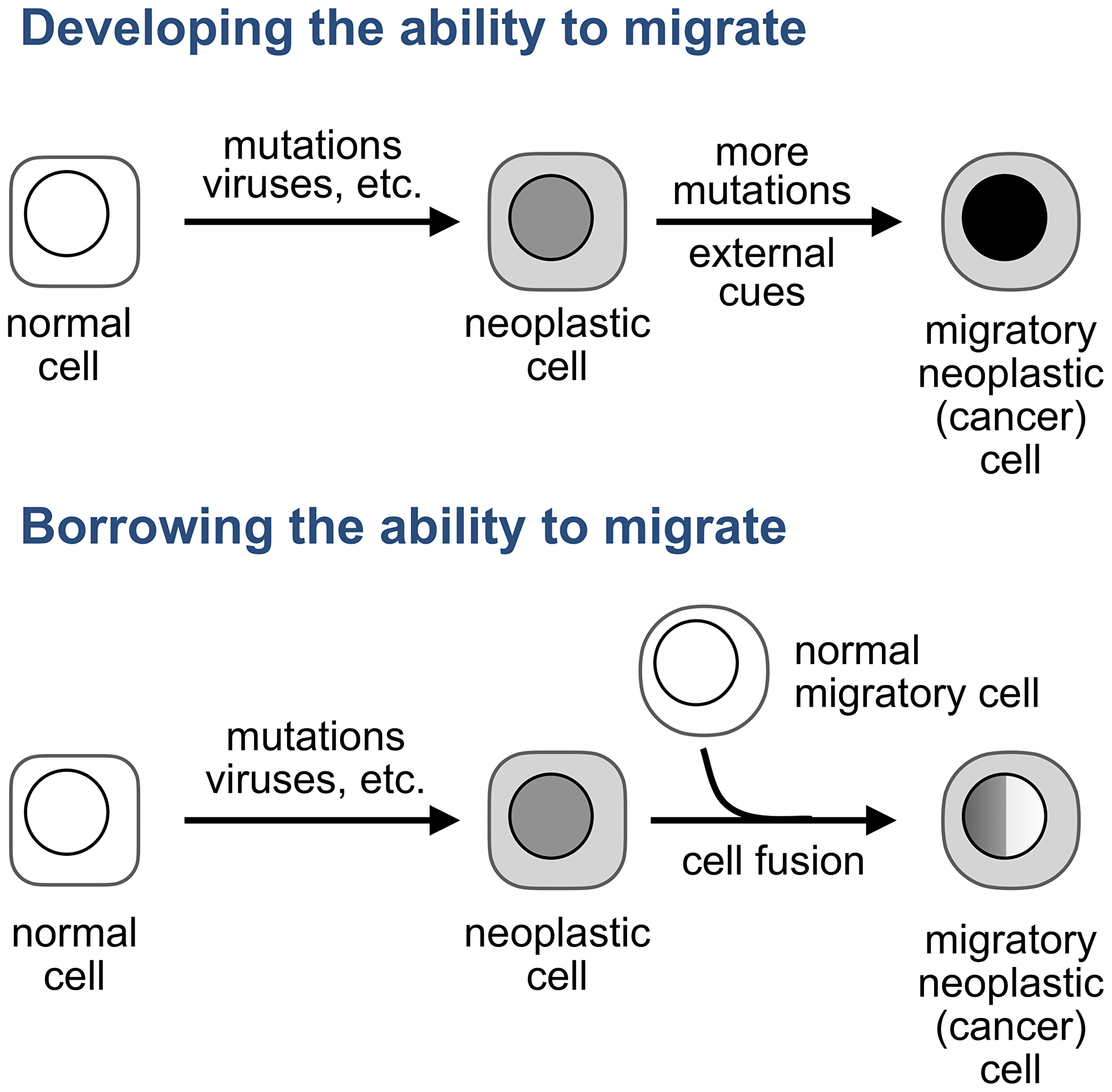
Figure 1: How do tumor cells become migratory? A prevailing view (top) is that tumor cells develop the ability to migrate from within: by acquiring mutations, in response to external cues, or by a combination of these stimuli. An alternative explanations (bottom) is that the ability to migrate is acquired from normal migratory cells. The original hypothesis posited that this acquisition results from fusion between a normal or neoplastic sedentary cell and a normal migratory cell, such as a leukocyte. We will discuss later other modalities of transferring properties from one cell to another.
The original hypothesis, also a century old [22, 23], proposed that the ability of cancer cells to migrate is acquired through cell fusion, a process that combines two or more cells into one by merging their plasma membranes and then combining entire cell contents [24]. The idea was that if a normal cell, or a cell of a benign tumor, fuses to a leucocyte, a migratory cell of the immune system, the resulting hybrids, like cancer cells, would proliferate as one parent and migrate as the other. Also like cancer cells, these hybrids would be diverse because some would “have more the wanderlust of the leukocyte, others more the ability of the somatic cell to function in a sedentary manner,” with additional heterogeneity resulting from the unequal distribution of chromosomes following cell fusion [23].
This hypothesis had laid fallow for six decades, until a serendipitous discovery revealed that human cancers grafted into laboratory animals can become metastatic and evade the immune response by forming hybrids with the cells of the host [25, 26]. Since then, the ability of cell fusion to enable metastases in a variety of animal systems has been confirmed by many studies (reviewed in: [27–30]) and complemented by findings that cell fusion can also produce dormant tumor cells, change their drug sensitivity, suppress the ability to form tumors, which led to the concept of tumor suppressors [31], induce genomic and phenotypic instability [32], affect tumor cell evolution [33, 34], change cell metabolism, produce circulating tumor cells, affect immune response, and, in synergy with oncogenes, produce invasive tumors that are similar to human cancers (reviewed in [27, 29, 35–42])
Are these observations relevant to human cancers? This question prompted a search for cell hybrids in human tumors.
How to find a hybrid?
To find a hybrid in a laboratory animal, the animal is made chimeric, that is, composed of two or more genetically distinct cell populations. For example, to make two populations distinct and their hybrids easier to detect, one population is commonly modified to carry a gene encoding a green fluorescent protein, while the other a gene encoding a red protein. Hybrids are then identified as cells that carry both genes.
Chimeric animals are required for two reasons. First, unlike other features that may be specific to parental cells, nuclear genomes cannot be changed by cell fusion beyond recognition, are replicated during each cell cycle, and, consequently, can be identified unambiguously even after numerous cell divisions, which is why genomic analysis is also used in forensics to identify humans and their genealogical relationships. Second, most cells of a laboratory mouse or a human have the same genetic background because they are the progeny of one cell, the fertilized egg. As a result, cell hybrids remain “invisible” to genomic approaches unless the analyzed organism is chimeric.
Hence, the search for hybrids in human cancers turned to patients who are chimeric because prior to a cancer diagnosis these individuals received an organ transplant and, as a result, are made of two genetically distinct cell populations: one has the genome of the recipient, the other the genome of the donor. Analyzing patients who received an organ from the donor of the opposite sex, say a female who received a bone marrow transplant from a male (Figure 2), is particularly informative because the origin of individual cells (donor or recipient) in a tissue can be revealed by visualizing sex chromosomes and whether these cells are normal or neoplastic determined by histopathology.
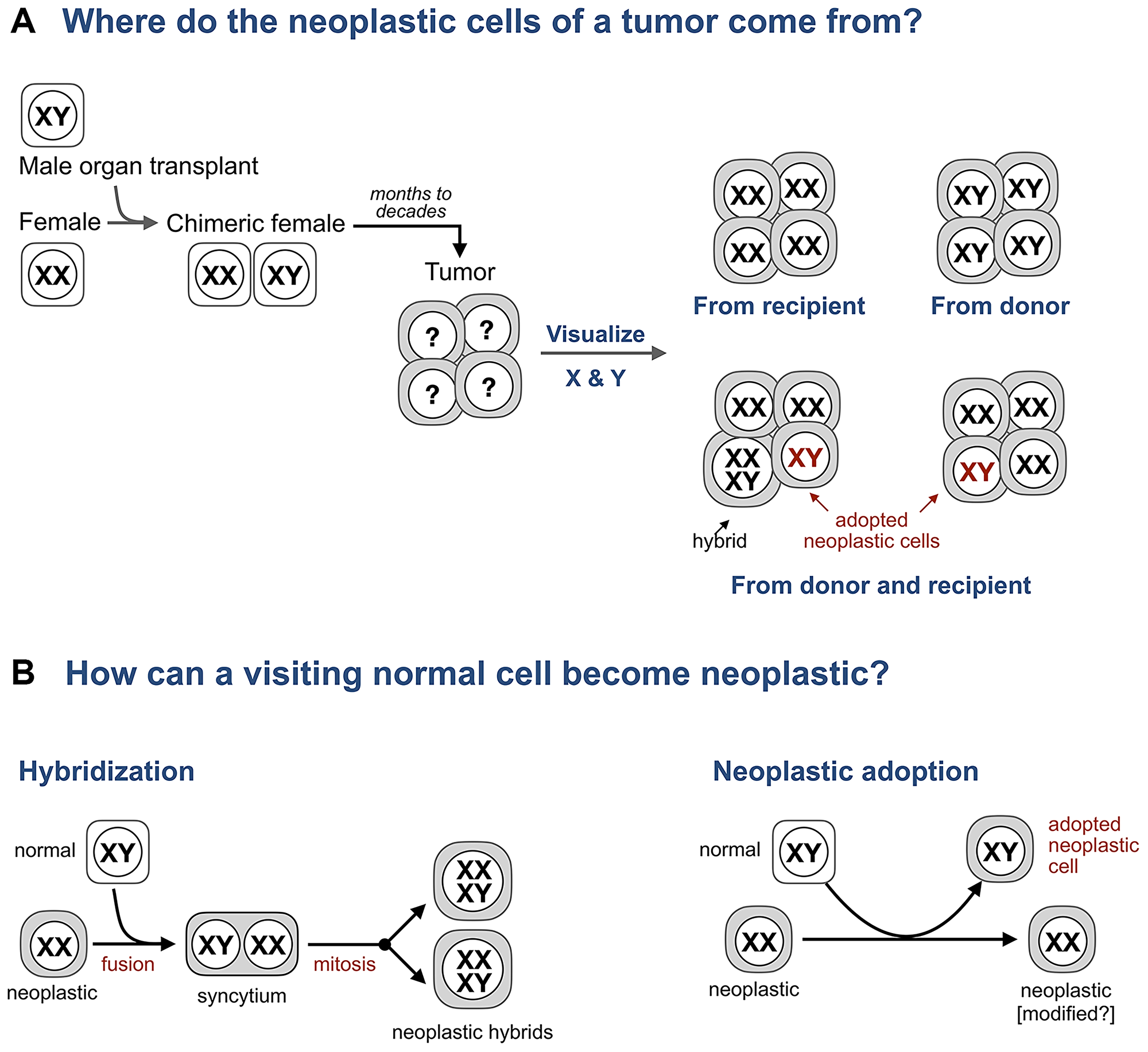
Figure 2: Adopted neoplastic cells in tumors from sex-mismatched transplant patients. (A) A typical experiment to test if tumor cells form hybrids with bone marrow derived cells in tumors from transplant patients. A female who received a bone marrow transplant from a male subsequently develops a tumor. Whether the cells of this tumor come from the donor or the recipient is revealed by visualizing sex chromosomes (XY in males, XX in females, and XXXY in predicted hybrids) and whether these cells are neoplastic or normal is determined by histopathology. (B) Expected intermediates for hybridization and neoplastic adoption.
These so called sex-mismatch patients are rare – for example, only four were found among 12,000 transplant patients by one study [43] – and so have been studies that have searched for cell hybrids in human tumors. This rarity, and the potential to reveal what had been invisible, both literally and figuratively, make the obtained results precious.
Adopted neoplastic cells
The analysis of tissues from sex-mismatch patients has revealed two groups of tumors (Figure 2).
In the first group, which included basal cell carcinomas, skin squamous cell carcinomas, and oral squamous cell carcinomas, all of the analyzed neoplastic cells in each tumor contained the sex chromosomes of either the donor or the recipient [44–46].
This result is consistent with the current view that all neoplastic cells in a tumor are a clone of a single cell, the cell of origin [21]. Indeed, this model predicts that in a transplant patient the cell of origin can come either from the recipient, in which case all neoplastic cells of the resulting tumor should have the genome of the recipient, or from the donor, in which case all neoplastic cells should have the genome of the donor.
However, in the second group of tumors, which included lung adenocarcinoma, laryngeal squamous cell carcinoma, glioblastoma, Kaposi sarcoma, colon adenomas, esophageal carcinoma, basal cell carcinoma, squamous cell carcinoma, and pancreatic ductal carcinoma, 1% to 40%, or “a proportion” [47], of neoplastic cells identified by pathologists as “histologically malignant,” “as carcinoma cells,” “invasive,” “consistent with neoplastic colonic adenoma cells” contained the sex chromosomes of the donor, with the rest containing only the sex chromosomes of the recipient [43–45, 48–51]. Hence, each of these tumors was made of two genetically distinct populations of neoplastic cells – one derived from the recipient, another from the donor – and thus could not be a clone of one cell.
Several additional observations support this conclusion. First, not all tumors analyzed by the same approaches in the same laboratory had detectable neoplastic donor cells [45–47, 50], meaning that these cells are a feature of some tumors rather than an artifact of the methods used. Second, neoplastic donor cells were also detected among tumor cells found in the blood (circulating tumor cells, or CTC) [51], which rules out potential artifacts associated with analyzing solid tissues. Third, some of the studies verified the donor origin of neoplastic cells using multiple approaches, such as tracing a rare mutation, analyzing mitochondrial DNA, genotyping microdissected tumor cells, staining for HLA antigens, and evaluating the ploidy of donor-derived neoplastic cells by visualizing non-sex chromosomes [43, 47, 50].
The presence of donor-derived neoplastic cells could be explained, as the model which prompted the search for the hybrids had predicted, if some normal donor cells fused to the neoplastic cells of the recipient, yielding hybrids in which the neoplastic phenotype becomes dominant. However, genomic evidence for hybridization has been found only for a fraction of neoplastic donor cells and only in some tumors [48, 49, 52]. No such evidence was found [43, 45, 50] or available [44, 51] for other tumors, implying that at least some of the donor-derived neoplastic cells are not hybrids.
If they are not hybrids, then what are they?
The lack of evidence for their hybrid origin, the observation that these cells are a minority among neoplastic cells, and the finding that these cells are detected in tumors diagnosed as early as two months after an organ transplant [45] can be explained if some normal donor cells migrate into a tumor and acquire the phenotype of tumor cells without acquiring their genome, as has indeed been suggested [43, 45, 50, 53]. I will refer to such cells as adopted neoplastic cells [53], irrespective of how they become adopted, because these cells are not the genetic offspring of resident neoplastic cells.
How are these cells adopted? Who are the “adoptive parents”? What types of cells can be adopted? What is the fate of adopted cells? What kind of tumors can adopt cells? Are these cells restricted to transplant patients? To see if these questions are worth discussing, let us first consider how the presence of adopted cells would affect tumor development.
What would the existence of adopted neoplastic cells imply?
Adopted neoplastic cells not only look as resident neoplastic cells to expert pathologists, which is by itself remarkable because how tumor cells look is used to diagnose tumors and to predict their clinical course [1], but at least some of them may have properties required to expand or seed tumors. Indeed, some donor-derived neoplastic cells were found in clusters or nests [43, 44, 48, 49], as would be expected if these cells proliferate. Whether solitary adopted cells also proliferate is unclear because migratory cells can wander away from each other instead of forming a cluster. That at least some of the adopted cells are migratory, perhaps because they retain some features of their normal precursors, follows from their presence among circulating tumor cells [30, 51, 52], an observation that also implies that adopted cells can retain their neoplastic phenotype for some time after leaving the tumor.
The ability of adopted neoplastic cells to proliferate, migrate, and retain their phenotype prompts a hypothesis that even if the majority of the adopted cells vanish, as adopting a neoplastic phenotype might trigger cell suicide, cell cycle arrest, or an attack by the immune system, some hopeful monsters, to use an evolutionary term [54] particularly fitting cancer cells, could survive, multiply, and evolve.
If so, the existence of adopted neoplastic cells would have consequences that might explain some outstanding observations.
Tumor evolution can be genetically discontinuous
A model taken by cancer research and oncology as a fact, despite contradicting evidence [55, 56], posits that all neoplastic cells of a tumor are the genetic progeny of a single cell, the cell of origin, which becomes neoplastic by acquiring genomic and epigenetic aberrations [18, 20, 21] (Figure 3). This model implies that new neoplastic cells in a tumor are produced only by the division of existing neoplastic cells, or, in other words, that tumor evolution is genetically continuous.
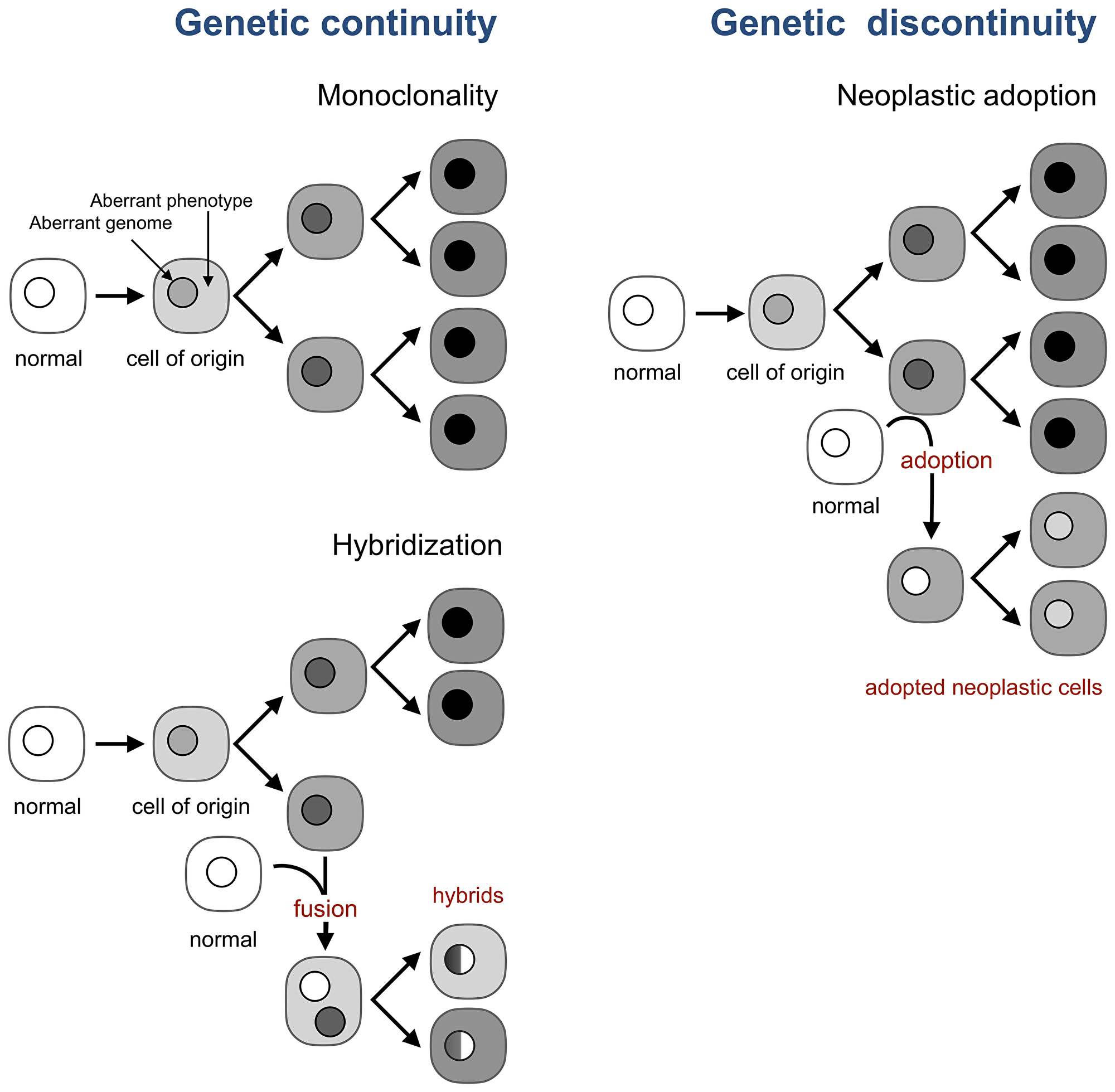
Figure 3: Neoplastic adoption enables genetic discontinuity in tumor development. A current view (top left) is that all neoplastic cells in a tumor are the progeny of the cell of origin, which implies genetic continuity of tumor development. This continuity holds even if tumor cells form hybrids with normal cells (bottom left). This continuity breaks, however, when a normal cell acquires the neoplastic phenotype of resident neoplastic cells without acquiring their genome (right).
For example, the notion of genetic continuity underlies the effort to identify and target genomic aberrations that cause the cell of origin to become cancerous and thus are expected to be inherited by all neoplastic cells in a tumor. Hence, the hope is that targeting these aberrations would kill all tumor cells with one arrow. However, if normal cells can acquire a neoplastic phenotype without acquiring the tumor genome and thus its aberrations, targeting these aberrations is bound to fail, as has indeed been the case for reasons that are only partially understood [57–59].
This unfortunate implication of genetic discontinuity may not require many adopted cells, as a single cancer cell can seed a tumor [60], cancers can relapse in patients with undetectable residual disease [61], and metastases can emerge, apparently from single disseminated cells, years or decades after the primary tumor has been excised [62–64]. To put these facts in perspective, a tumor in which 1% of neoplastic cells are adopted could have 107 of hopeful monsters per each 1 cm3 of the tumor [61].
Genetically discontinuous evolution can also help to explain some observations related to metastasis.
“Lost” aberrations
All metastases are thought to be genetically continuous with a primary tumor [4]. Yet, some genomic aberrations found in a primary tumor, including deletions, can be absent in its metastases [4, 62, 65–67].
How are these aberrations lost?
The parallel model of metastasis [62] explains this paradox by positing that neoplastic cells that seed metastases leave the primary tumor early in its development, before the “lost” aberrations appear (Figure 4). The tumor and the “seeds” then evolve in parallel, with the potential of convergent genomic evolution characteristic to the cells of the same cell type [68]. This model can explain why single breast cancer cells found in bone marrow have few or no chromosomal aberrations which are abundant in primary tumors [62, 69], and why metastases can be present with no detectable primary tumor [70].
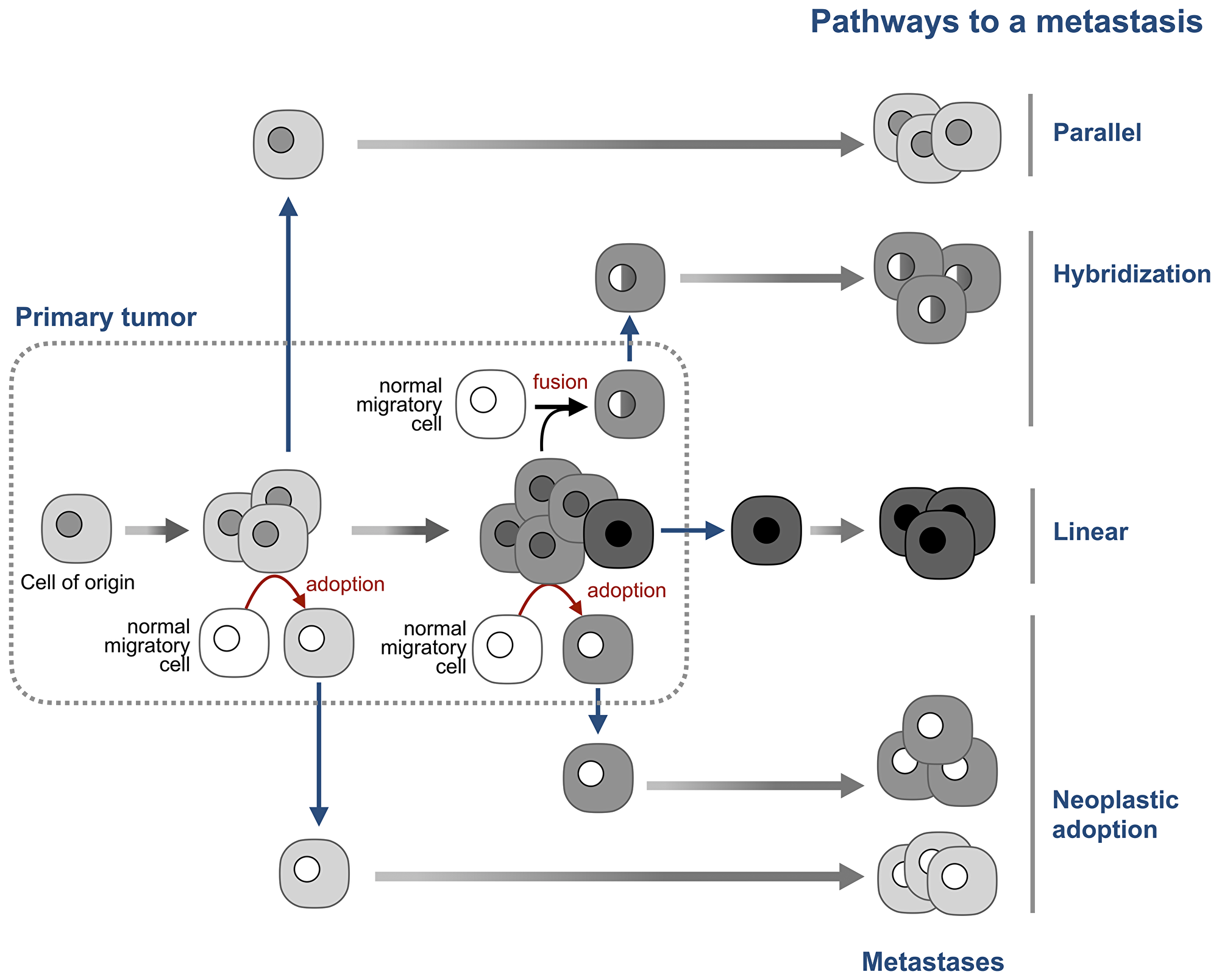
Figure 4: Neoplastic adoption as a pathway of metastasis. The prevailing linear model, posits that tumor cells become able to seed new tumors by accumulating additional mutations late in tumor development. The parallel model posits that tumor cells disseminate early in tumor development and then continue to evolve in parallel with the primary tumor. The hybridization model argues that metastases are formed by hybrids between tumor cells and normal migratory cells. The neoplastic adoption model suggests that metastases can be seeded by normal migratory cells that acquire a neoplastic phenotype of tumor cells without acquiring their genome. Both hybridization and neoplastic adoption models suggest that metastatic cells can be formed at any stage of tumor development. Note that these four models are not mutually exclusive.
Adopted neoplastic cells would also seed metastases that “lost” the aberrations of the primary tumor, but for a different reason – because adopted cells do not inherit the tumor genome. This difference also means that adopted cells can seed metastases with “lost” aberrations at any time of tumor development, not only at its early stages, as the parallel model suggests.
A leap
The failure to find mutations that cause metastasis prompted a search for a “discrete step in tumor evolution that may be independent of specific oncogene pathways or mutations and instead co-opts cellular traits that mitigate immunologic, genotoxic and therapeutic stressors accreted during tumorigenesis.“ [5]. In evolutionary biology such steps are known as saltational (from the Latin for leap), as they suddenly (on the evolutionary time scale) enable “profound phenotypic novelties or species” [54].
For example, the metastasis seed preselection model proposes that neoplastic cells become “bone marrow-like” under the influence of bone marrow-derived cells, which are abundant in tumors, and thus can prosper and proliferate once they reach the bone marrow, a common site for metastasis [71, 72]. The existence of adopted cells derived from bone marrow transplants [43, 45, 51] implies that the influence can flow in the opposite direction as well, to make some bone marrow-derived cells neoplastic.
Several properties of neoplastic adoption make it a suitable candidate for the sought after “leap”. First, this phenotypic switch is a discrete step and it happens suddenly on the time scale of tumor development. Second, adopted cells do not inherit the burden of genomic aberrations, and the consequent aberrant antigens targeted by the immune system [73], because they do not inherit the tumor genome. Finally, retaining some properties of normal migratory cells [51] enables safe passage to distant organs and helps to settle there.
Puzzling circulating tumor cells
Circulating tumor cells (CTC), discovered a century and a half ago [74], are neoplastic cells present in the blood of patients with solid tumors and are thought to include the precursors of metastases [75].
Paradoxically, a fraction or, in some cases, most of CTC released by some non-hematological tumors, such as melanoma, breast, ovarian, and pancreatic cancers, were found to carry CD45, a protein whose expression is normally restricted to bone-marrow derived cells [30, 51, 76–78]. Hence, these cells (CTC-CD45) were viewed as a persistent artifact until an appeal to give them a closer look [77] revealed that they indeed exist [79] and that their concentration inversely correlates with the survival of patients with pancreatic cancer [30, 51].
The presence of a lymphocyte protein on the cells of non-hematological tumors prompted a hypothesis that CTC-CD45 are hybrids between tumor cells and leukocytes [79]. As a result, these cells have been reported as macrophage-tumor cell fusions [80], circulating hybrid cells [30], or simply hybrids [78]. However, genomic evidence for the hybrid origin of these cells is still unavailable, leaving open other explanations: that CD45 is present due to aberrant gene expression, which is common in cancer, or that cancer cells acquire this protein through trogocytosis [81], a process that enables the intercellular exchange of membrane proteins [82], or by a similar phenomenon termed vampirization [83].
The existence of adopted cells suggests another potential explanations: that some CTC-CD45 are bone marrow derived cells that adopted a neoplastic phenotype. This explanation is consistent with a report that only a fraction of CTC-CD45 recovered from a pancreatic cancer patient have a mutation characteristic to this cancer [52]. Given that only one of 10,000 CTC is estimated to seed a metastasis [74], adopted cells may be a candidate for this subpopulation.
What kind of cells can be adopted?
The fact that various normal cells are commonly present in tumors means that only some of these cells can be adopted. What type of cells could they be?
Adoptable cells should be migratory to enter a tumor, able to adopt the phenotype of surrounding cells, and capable of residing in more than one organ, as adopted cells were detected in patients transplanted with all transplanted organs that were analyzed: bone marrow [43, 45, 49, 51], mobilized peripheral blood stem cells [45], and kidney [44, 47, 50].
This profile matches that of mesenchymal stem cells (MSCs), a group of migratory cell types that are found in bone marrow and some other organs, are capable of assuming a variety of phenotypes, and are prone to home to damaged tissues and tumors [84–87]. Since MSCs transplantation, including genetically modified and allogeneic cells, has been used in more than 1,000 clinical trials [87], the treated patients can be used to test whether MSCs contribute to the neoplastic population of tumor cells.
While MSCs are a suitable candidate for a cell type susceptible to adoption, the spectrum of cell types that can be adopted will need to be determined. These studies can be informed by considering potential mechanisms of adoption.
How can a normal cell adopt a neoplastic phenotype?
The lack of genetic evidence for hybridization prompted a hypothesis that cells derived from bone marrow can adopt the phenotype of resident tumor cells by “subjection to locally released growth factors and cell-cell contact,” a mechanism named developmental mimicry [45] (Figure 5, left panel). However, this hypothesis poses a conundrum: How can a normal cell mimic a neoplastic phenotype by responding to external signals if, as the prevailing view of cancer posits, this phenotype results from genomic aberrations which a normal cell lacks by definition? This puzzle suggests two, not mutually exclusive, solutions.
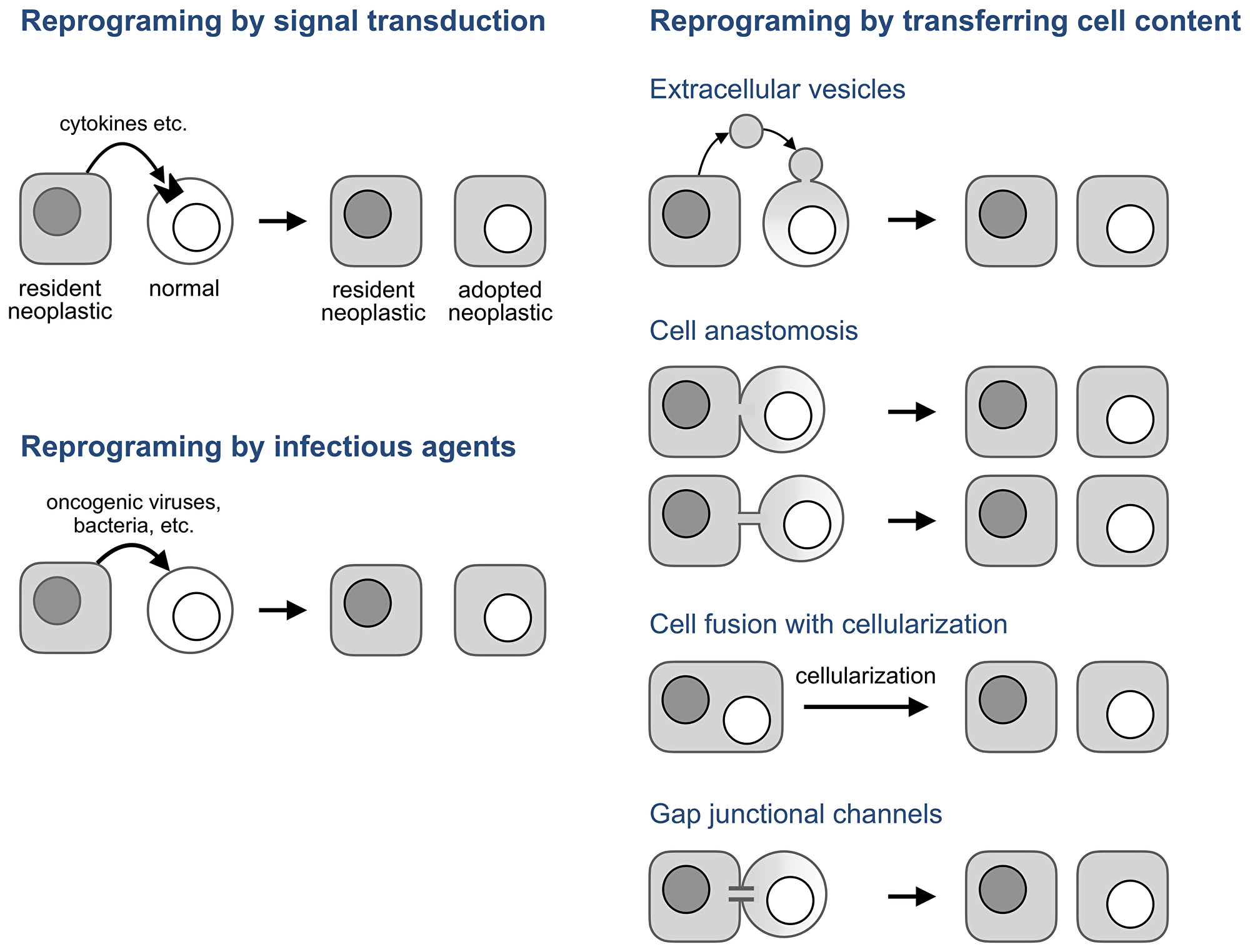
Figure 5: Potential mechanisms of neoplastic adoption.
One, that at least some neoplastic phenotypes are caused not by genomic aberrations. For example, they may be caused by aberrations in molecular and electric processes that organize cells into a tissue and determine their phenotypes [88–90]. If so, then some normal cells entering a tumor could be made neoplastic by the same forces and mechanisms that created resident neoplastic cells in the first place.
Another explanation is that a neoplastic phenotype, whatever its origin, can be transmitted from a tumor cell to a normal cell, or, in other words, that a cancer cell can make a normal cell cancerous. This century-old concept [91] was rediscovered four decades ago as horizontal oncogenesis to explain how a human adenocarcinoma transplanted into a mouse could induce a mouse sarcoma, a tumor which arises from a different cell type, in the adjacent connective tissue [92, 93]. Two mechanisms of transmission were initially considered: an oncogenic infectious agent and fusion between the transplanted and host cells that results in neoplastic hybrids. As we have discussed, such hybrids have indeed been documented, but so have been grafted human cancers that induced mouse tumors of the same or different type without any evidence for hybridization [94, 95].
In the absence of an infectious agent, these observations and neoplastic adoption can be explained if transcription factors and other molecules that determine a neoplastic phenotype are transmitted between cells without forming cell hybrids. Several mechanisms can do that (Figure 5, right panel).
Extracellular vesicles are membrane-enclosed cell fragments of various origin, content, and size that are released by cells and can deliver their content to a target cell by fusing to its plasma membrane [96–98]. This transfer has been implicated in various aspects of cancer development [98–100] and in the transfer of neoplastic properties to normal cells in particular.
For example, vesicles isolated from human colorectal cancers induced “tumor-like morphological changes and marked growth rate increase” in human mesenchymal stromal cells isolated from normal colon [101]. Likewise, vesicles derived from human prostate cancer cell lines enabled normal adipose stem cells isolated from prostate cancer patients to form neoplastic lesions “that were grossly and histologically comparable with those developed by [prostate cancer] cells” [102], while vesicles from a breast cancer cell line enabled an immortalized breast epithelial cell line to form tumors [103]. These findings are consistent with earlier results obtained by using cytoplasts, cell fragments made by enucleating cells and similar in size to some naturally occurring extracellular vesicles [96]. Fusing cytoplasts from tumorigenic cells with normal human lymphocytes produced immortalized cell lines which “exhibited morphological diversity ranging from adherent cells to free floating round cells” [104], while fusing cytoplasts from a tumorigenic breast epithelial line to its non-tumorigenic predecessor yielded tumorigenic cells [105]. In essence, extracellular vesicles and cytoplasts produced adopted neoplastic cells in the dish.
However, the oncogenic effects of extracellular vesicles have been questioned, in part because the resulting phenotypes were transient in an experimental system [106] and their persistence “would violate several tenets of the existing cancer progression paradigm” [107], as if some of these tenets were not contradicted by facts, logic, and clinical outcomes [108–113]. Hence, “the possibility that horizontal transfer of oncogenic material [by vesicles] can lead to tumor formation is the subject of considerable debate” [99]. The evidence for the presence of adopted neoplastic cells in human cancers suggests that this debate is no longer only academic, especially because extracellular vesicles are not the only mechanism for transferring cellular components nor is it most efficient.
While extracellular vesicles transfer cell content in small packets, cell anastomosis (Figure 5) bridges cells directly through pores (anastomoses) made by merging the plasma membranes of adjacent cells [114–118]. These pores, which range from 100 nm to a few microns [115], can bridge adjacent cell bodies, in which case the connection has been called partial [115] or transient [119] cell fusion, or form anastomosis tubes by anastomosing a protrusion of one cell with the body of another, or two protrusions to each other.
Protrusions that transmit “dyestuff, mitochondria and granules from one cell to another” [120] had been reported a century ago [120, 121] and were rediscovered more recently [122, 123] as a family of intercellular tubes with diverse properties, functions, and names that enable cells change the properties of other cells by transferring cellular components [122–127].
Unlike cell fusion, which combines the entirety of two cells into one morphologically and functionally distinct unit, a syncytium, which can then produce hybrids, cell anastomosis enables the transient, localized, and regulated sharing and exchange of cellular components, including whole nuclei [128–130], while preserving the morphology of the bridged cells. As a result, anastomosis can be overlooked even in experimental systems by approaches that focus on detecting cell hybrids, as no hybrids or syncytia are formed. Anastomoses can be visualized by electron microscopy [114–118] and their presence detected by methods that register the consequences of transient cell bridging, such as DNA modifications made by the Cre/loxP system [119, 131] or mitochondria transfer. For example the finding that a transmissible canine cancer has acquired mitochondrial but not nuclear genomes from several of its hosts [132] can be explained by anastomosis.
Cell anastomosis is a candidate for processes that enable neoplastic adoption because mesenchymal stromal cells are prone to anastomose with other cells [133, 134] and because anastomoses, along with other intercellular bridges, tie neoplastic and normal cells of human cancers into a network, a process whose significance has been increasingly appreciated [125, 129, 130, 135–139]. Adopted neoplastic cells might emerge as a part of this process.
Cell fusion may also be involved in neoplastic adoption if syncytia it forms undergo cellularization, an enigmatic but well documented process by which multinucleated cells split into mononuclear cells without entering mitosis. Cellularization is common in protists [140], is a required part of Drosophila development [141], is involved in tissue regeneration in the newt [142], and also happens in mouse osteoclasts [143], myotubes [144, 145], and, under the name of neosis, in transformed multinucleated mouse cells [146]. Some small molecules that induce cellularization in the newt also do so in mouse cells [142] implying that the mechanisms of cellularization are conserved among species.
While extracellular vesicles, anastomosis, and cell fusion use membrane fusion to transfer cellular components, gap junctional channels enable this transfer by piercing the membranes of adjacent cells. These transmembrane protein complexes can transmit metabolites smaller than 1.5 kDa, of which a cell has about forty thousand [147], regulatory RNAs, and some proteins [148–151]. The role of this transfer in cancer has been studied for half a century to implicate it in nearly all aspects of this disease [147]. However, these channels might contribute to neoplastic adoption not only by transferring molecules but also by functioning as “biological transistors” [152, 153] which enable bioelectrical signaling between non-neuronal cells, a process implicated in cancer [90, 154–156].
Overall, several mechanisms, including infectious agents, “classic” signaling between autonomous cells, tissue organization fields, and intercellular component transfer can potentially explain how a normal cell can acquire the neoplastic phenotype of surrounding cells. I find component transfer intellectually attractive because transferring activities and structures that determine a phenotype can readily explain how this phenotype, normal or abnormal, can be imposed on another cell. This mechanism can also explain how the imposed and suppressed phenotypes can blend at various ratios, and how this blending can produce emergent properties [32, 157].
What components need to be transferred to induce a neoplastic phenotype?
This question is difficult to answer definitively because how a cell becomes neoplastic and how its phenotype is maintained is still a matter of debate. However, the bridging mechanisms that we have discussed (Figure 5) can transfer practically any component mentioned in this debate, from oncogenes to oncometabolites [158, 159].
For example, the activity responsible for the ability of cytoplasts from tumorigenic cells to immortalize human lymphocytes [104] was identified as two short species of endogenous cytoplasmic DNA [160]. Cytoplasmic DNA includes retrotransposons, linear and circular chromatin fragments of various size, mitochondrial DNA, and micronuclei [161], all of which are transferrable by anastomosis [162] or by extracellular vesicles [163–165].
Micronuclei are of particular interest because they encapsulate chromosomes and their fragments and can rearrange them by a process called chromothripsis, which breaks chromatin into fragments and then stitches them in apparently random order [166]. Besides other consequences, this process can yield circular extrachromosomal DNA (ecDNA) [167], which was found in nearly half of human cancers, “almost never found in normal cells,” and affects tumor evolution and drug resistance presumably by harboring amplified oncogenes [168–170].
Transferring tumor mitochondria to a normal cell by anastomoses or extracellular vesicles would transmit mutations in mitochondria DNA that have been considered oncogenic [171], and also has the potential to make the target cell neoplastic by reprograming its metabolism [172], with the concomitant production of oncometabolites, which are metabolites that deregulate gene expression if present at an increased concentration [158, 159]. Given their size, oncometabolites can also be transferred by gap junctional channels.
Likewise, merging plasma membranes by anastomosis or transferring membrane fragments by extracellular vesicles enables the migration of membrane-associated molecules, including growth factor and cytokine receptors implicated in causing and maintaining cancer phenotypes [58].
Overall, multiple components that are known to be transferred between cells can contribute to neoplastic transformation. Learning which of these components enable neoplastic adoption and whether this process involves component transfer at all may be helped by knowing the reasons for neoplastic adoption.
Why are normal cells converted into neoplastic?
One possibility is that neoplastic adoption illustrate the notion that no good deed goes unpunished (Figure 6).
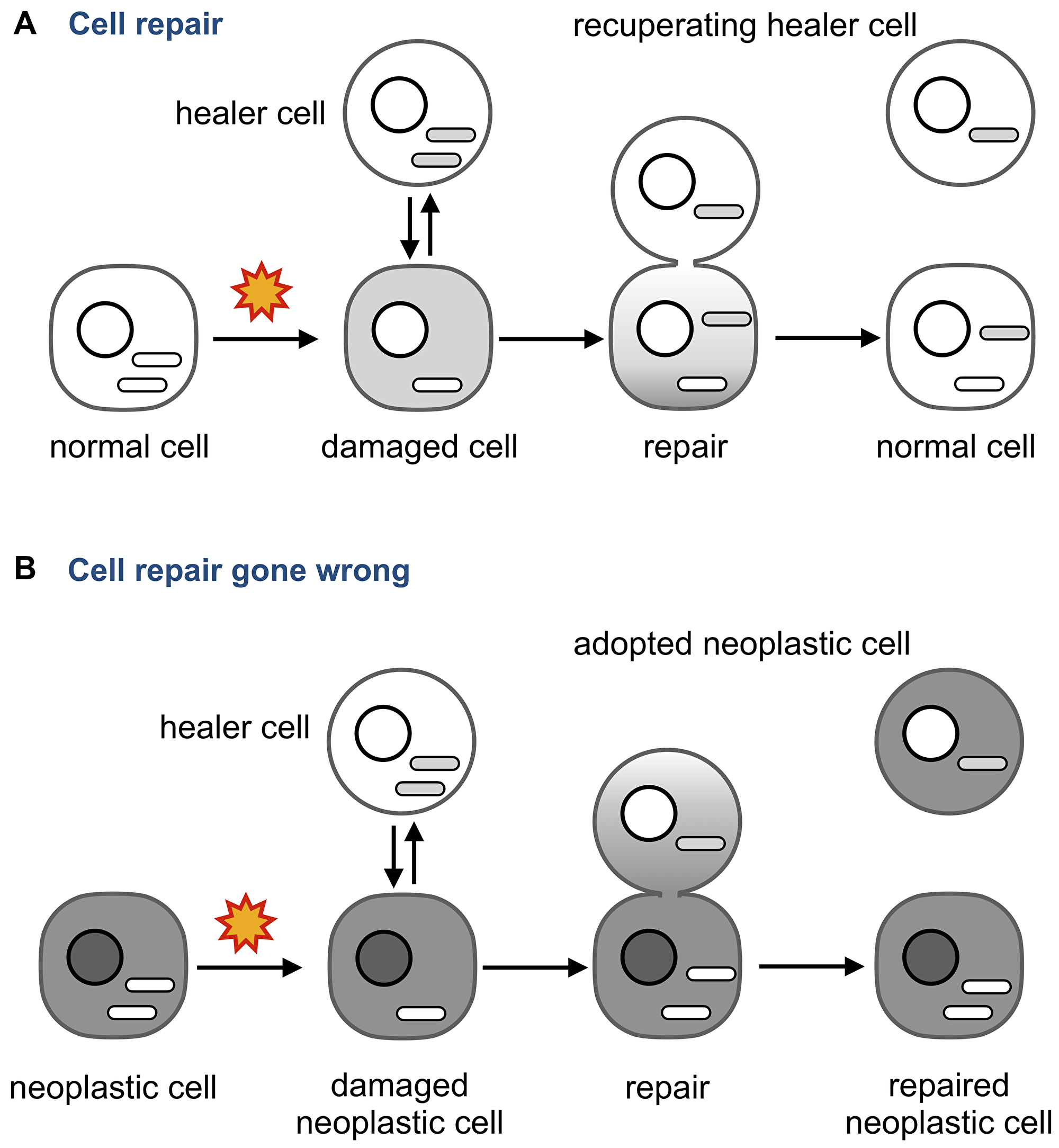
Figure 6: Neoplastic adoption as a side effect of cell repair. (A) An injured cell whose mitochondria and other cytoplasmic components are damaged can receive them from an intact “healer” cell. The transfer is unidirectional to make sure the damaged components are not transferred from the “patient”. (B) If the injured cell is neoplastic, the transfer may become bidirectional because multiple functions of the neoplastic cells are deregulated. As a result, regulatory molecules that maintain a neoplastic phenotype are transferred to the healer and change its phenotype into neoplastic. Note that the repaired neoplastic cell may also change its properties by acquiring the content of the healer.
Tissue injury, such as irradiation or chronic inflammation, prompts circulating bone marrow-derived cells to fuse to damaged cells [173, 174] or to repair them by delivering molecules and organelles through anastomoses [123, 124, 134, 175–177]. These observations pointed to the existence of a cell repair mechanism that heals injured cells by providing intact components, and mitochondria in particular [123]. This mechanism has been already explored as a therapeutic approach using MSCs as “healer” cells, even though the underlying molecular mechanisms and possible side effects are yet to be understood [87, 134, 178].
For example, what would happen if the damaged cell is neoplastic? Can the “healer” cell contract the neoplastic “disease” through the same anastomosis tubes that deliver the “cure,” perhaps because some abnormalities of the neoplastic cell affect how the two cells are bridged, what components are transferred, and in which direction? For example, cytoskeleton, which is deregulated in cancer [179], is involved in forming intercellular bridges [180], determines what these bridges transmit [181], and regulates the size of pores formed by the fusion of plasma membranes [182].
The possibility of contracting a neoplastic phenotype as a side effect of repairing a neoplastic cell can explain why adopted cells were detected not in all tumors, as a cell would be adopted nor merely because it wanders among neoplastic cells but because it attempts to repair them. If so, the incidence of neoplastic adoption in a tumor would depend on the extent of genomic and other aberrations, hypoxia, inflammation, infection, or other internal and (micro) environmental factors that can result in cell damage.
The model that neoplastic adoption is a side effect of cell repair, and the observation that adopted cells are present in colon adenomas [45], which are benign tumors that can progress to cancers, suggest that non-cancerous neoplastic lesions can progress to cancerous by adopting migratory cells. This hypothesis can explain why cancers can arise without a detectable precursor lesion, as happens with the majority of melanomas [183] and some types of lung cancer [184]. For example, this phenomenon would be expected if some cells are adopted by a microscopic neoplastic lesion, which are by far more abundant than cancers [185, 186], and then evolve on their own, locally or elsewhere, even if the adopting lesion vanishes.
This model would also be consistent with the exponential increase of cancer incidence with aging, a process associated with accumulating cellular damage of various kind [187], and the fact that chronic inflammation, which is associated with the recruitment of bone marrow derived cells to abnormal tissues, increases the incidence of cancer [188, 189].
Finally, the model that neoplastic adoption is a consequence of repair implies that a treatment that damages cancer cells without killing them can create adopted neoplastic cells. These cells may take revenge even if the “adopting parents” eventually die, reminding us, to rephrase a quote, that if you strike at an emperor, you must kill him [190, 191].
Another possibility is that neoplastic adoption is not a side effect of cell repair but results from the propensity of neoplastic cells to bridge neoplastic and normal cells into the networks using anastomosis tubes and gap junctional channels, as has been documented in glioblastoma [129, 130, 139, 192, 193].
Both the side effect model and the network model imply that neoplastic adoption may affects the “adoptive parents,” for example if the unidirectional transfer of components from the “healer” to the “patient” turns into intercellular exchange. If so, then neoplastic adoption not only would produce adopted neoplastic cells but would also modify resident neoplastic cells. For example, resident cells can become migratory or “invisible” to the immune system. If so, neoplastic adoption would further increase the diversity of cell types in a tumor and thus its ability to preempt our assassination attempts when they are still on the drawing boards of pharmaceutical companies.
Are adopted neoplastic cells present in non-transplant patients and how can these cells be detected?
Adopted cells have been revealed by analyzing tumors from transplant patients. Because these individuals have a medical history that differs from that of most cancer patients, it is reasonable to ask if adopted cells are also present in the tumors of non-transplant patients and how these cells can be detected.
A hint that neoplastic adoption is not limited to transplant patients comes from the studies of fetomaternal microchimerism, a condition in which cells exchanged between a woman and her child during pregnancy persist in their bodies, sometimes for decades [194–196]. If the fetus is a male, fetal cells can be identified in the mother’s tissues by visualizing the Y chromosome, the same approach that was used to look for cell hybrids in transplant patients.
This approach has revealed that some fetal cells have properties of migratory progenitors with multilineage differentiation capacity [197, 198]. Like mesenchymal stromal cells, these progenitors home to damaged organs and tumors, become morphologically “undistinguishable from the surrounding cells in terms of size and nuclear shape“ [199], and begin to express antigens specific to the host tissues, whether normal or neoplastic [196, 199–202].
Yet, fetal cells that have the features of surrounding tumor cells have not been considered neoplastic because these cells were not found in clusters “even if they expressed antigens that can be found within the adenocarcinoma“ [199]. However, some fetal cells in cervical cancers were found in clusters [200], and one can argue that some proliferating cells would not be expected to form clusters if these cells are migratory. Hence, a suggestion that fetal cells can “adapt a malignant phenotype and potentially fuel tumorigenesis” [196].
The similarities between fetal progenitors and mesenchymal stromal cells in their ability to home to damaged tissues and adopt the phenotype of the resident cells suggests that fetal cells could be “tracers” of adult cell populations that have similar properties, including the ability to adopt a neoplastic phenotype.
The uncertainty about whether fetal cells can be neoplastic, as well as about some of their other properties, stems from their rarity – a few to a hundred are found per million of maternal cells analyzed [199] – although this incidence may be underestimated [196]. However, given the increasing sophistication of automatic tissue analysis [203] this rarity may be compensated by the abundance of microchimeric cancer patients, as the majority of women have fetal cells even if their pregnancy was incomplete or went unnoticed [198, 204].
Pregnancy provides another, yet to be explored, opportunity – to detect adopted neoplastic cells and cell hybrids by analyzing gestational tumors. Gestational tumors, which include moles and gestational choriocarcinoma, arise from the cells of the conceptus and thus are genetically distinct from the cells of the mother [205]. Hence, adopted cells, as well as hybrids between neoplastic and normal cells, can be detected by the approaches applied to transplant patients [53], especially because some gestational tumors have only a male genome, which facilitates the analysis. Another advantage of analyzing gestational tumors is the ability to trace normal cells derived from all organs of the patient rather than only from a transplanted organ.
To look beyond chimeric tumors, the concept of neoplastic adoption predicts that some histologically neoplastic cells of genomically aberrant tumors should have a normal genome, and that the offspring of these cells evolves in parallel with the resident cells of the tumor. This prediction can be tested by comparing the genomes and histopathology of single cells from non-chimeric tumors. Given precedents with cell types, such as CTC-CD45, that were neglected because they were not supposed to exist [79], it might well be that adopted cells have been put aside as outliers that are too unusual to consider while searching for something anticipated.
Finally, as experiments with humans can go only that far, and fortunately so, testing the hypotheses we have discussed will require experimental systems, such as human tumor explants which have been explored to reveal intercellular bridging [129, 130] and chimeric animals designed to monitor cell fate, cell fusion, and component transfer [206]. Keeping in mind that this transfer can be mediated by more than one mechanism and that intercellular signaling that does not involve component transfer may also be involved can help to use these systems to their full potential. The information obtained from existing and new experimental systems may suggest new approaches for detecting adopted cells in human tumors and how to use these cells for clinical needs.
Meanwhile, I hope that considering and testing the concept of adopted neoplastic cells will prove to be useful in explaining puzzling observations related to neoplasia and would lead to ideas, discoveries, and technologies beneficial to future cancer patients.
ACKNOWLEDGMENTS
I am grateful to Andy Koff, George Parris, Marta Gyselova, Andriy Marusyk, Frank Rösl, Jan Brábek, Karel Smetana, Hunter Pitney, Jyoti Raychaudhuri, and Egor Lazebnik for discussion, comments, advice, and encouragement.
CONFLICTS OF INTEREST
Author has no conflicts of interest to declare.
References
1. Rubin R, Strayer DS, Rubin E, McDonald JM. Rubin’s Pathology: Clinicopathologic Foundations of Medicine. Lippincott Williams & Wilkins; 2008. https://books.google.com/books?id=kD9VZ267wDEC.
2. Rosel D, Fernandes M, Sanz-Moreno V, Brábek J. Migrastatics: Redirecting R&D in Solid Cancer Towards Metastasis? Trends Cancer. 2019; 5:755–56. https://doi.org/10.1016/j.trecan.2019.10.011. [PubMed].
3. Solomon J, Raškova M, Rösel D, Brábek J, Gil-Henn H. Are We Ready for Migrastatics? Cells. 2021; 10:1845. https://doi.org/10.3390/cells10081845. [PubMed].
4. Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016; 352:169–75. https://doi.org/10.1126/science.aaf2784. [PubMed].
5. Esposito M, Ganesan S, Kang Y. Emerging strategies for treating metastasis. Nat Cancer. 2021; 2:258–70. https://doi.org/10.1038/s43018-021-00181-0. [PubMed].
6. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022; 72:7–33. https://doi.org/10.3322/caac.21708. [PubMed].
7. Russian roulette. Wikipedia. 2022. https://en.wikipedia.org/wiki/Russian_roulette.
8. Rous P. An Avian Tumor in Its Relation to the Tumor Problem. Proc Am Philos Soc. American Philosophical Society. 1912; 51:201–5. https://www.jstor.org/stable/984137.
9. Kidd JG, Rous P. Cancers deriving from the virus papillomas of wild rabbits under natural conditions. J Exp Med. 1940; 71:469–94. https://doi.org/10.1084/jem.71.4.469. [PubMed].
10. zur Hausen H, Gissmann L, Steiner W, Dippold W, Dreger I. Human papilloma viruses and cancer. Bibl Haematol. 1975; 569–71. https://doi.org/10.1159/000399220. [PubMed].
11. Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer. 1988; 61:1942–56. https://doi.org/10.1002/1097-0142(19880515)61:10%3c1942::aid-cncr2820611003%3e3.0.co;2-j. [PubMed].
12. Foster W, Raoult A. Early descriptions of antibiosis. J R Coll Gen Pract. 1974; 24:889–94. [PubMed].
13. Kidd M, Modlin IM. A century of Helicobacter pylori: paradigms lost-paradigms regained. Digestion. 1998; 59:1–15. https://doi.org/10.1159/000007461. [PubMed].
14. Rigas B, Feretis C, Papavassiliou ED. John Lykoudis: an unappreciated discoverer of the cause and treatment of peptic ulcer disease. Lancet. 1999; 354:1634–35. https://doi.org/10.1016/S0140-6736(99)06034-1. [PubMed].
15. Bignold LP, Coghlan BLD, Jersmann HPA. David Paul von Hansemann: Contributions to Oncology: Context, Comments and Translations. 2007th edition. Boston: Birkhäuser Basel. 2006; 395. https://doi.org/10.1007/978-3-7643-7769-4.
16. Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008 (Suppl 1); 121:1–84. https://doi.org/10.1242/jcs.025742. [PubMed].
17. Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953; 7:68–72. https://doi.org/10.1038/bjc.1953.8. [PubMed].
18. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976; 194:23–28. https://doi.org/10.1126/science.959840. [PubMed].
19. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004; 10:789–99. https://doi.org/10.1038/nm1087. [PubMed].
20. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009; 458:719–24. https://doi.org/10.1038/nature07943. [PubMed].
21. Visvader JE. Cells of origin in cancer. Nature. 2011; 469:314–22. https://doi.org/10.1038/nature09781. [PubMed].
22. Aichel O. Eine neue Hypothese über Ursachen und Wesen bösartiger Geschwülste. Santiago de Chile: Universo. 1908; 42. http://archive.org/details/b3061210x.
23. Aichel O. Über zellverschmelzung mit qualitativ abnormer chromosomenverteilung als ursache der geschwulstbildung. Leipzig: Verlag Von Wilhelm Engelmann. 1911; 115. https://books.google.com/books?id=8Sk4AQAAMAAJ.
24. Brukman NG, Uygur B, Podbilewicz B, Chernomordik LV. How cells fuse. J Cell Biol. 2019; 218:1436–51. https://doi.org/10.1083/jcb.201901017. [PubMed].
25. Goldenberg DM, Müller E, Witte S. In vivo proliferation of heterotransplanted human cancer cells. Eur J Cancer (1965). 1967; 3:315–19. https://doi.org/10.1016/0014-2964(67)90013-8. [PubMed].
26. Goldenberg DM, Pavia RA, Tsao MC. In vivo hybridisation of human tumour and normal hamster cells. Nature. 1974; 250:649–51. https://doi.org/10.1038/250649a0. [PubMed].
27. Duelli D, Lazebnik Y. Cell fusion: a hidden enemy? Cancer Cell. 2003; 3:445–48. https://doi.org/10.1016/s1535-6108(03)00114-4. [PubMed].
28. Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008; 8:377–86. https://doi.org/10.1038/nrc2371. [PubMed].
29. Parris GE. Historical perspective of cell-cell fusion in cancer initiation and progression. Crit Rev Oncog. 2013; 18:1–18. https://doi.org/10.1615/critrevoncog.v18.i1-2.20. [PubMed].
30. Sutton TL, Patel RK, Anderson AN, Bowden SG, Whalen R, Giske NR, Wong MH. Circulating Cells with Macrophage-like Characteristics in Cancer: The Importance of Circulating Neoplastic-Immune Hybrid Cells in Cancer. Cancers (Basel). 2022; 14:3871. https://doi.org/10.3390/cancers14163871. [PubMed].
31. Harris H. How tumour suppressor genes were discovered. FASEB J. 1993; 7:978–79. https://doi.org/10.1096/fasebj.7.10.8344496. [PubMed].
32. Koulakov AA, Lazebnik Y. The problem of colliding networks and its relation to cell fusion and cancer. Biophys J. 2012; 103:2011–20. https://doi.org/10.1016/j.bpj.2012.08.062. [PubMed].
33. Parris GE. The role of viruses in cell fusion and its importance to evolution, invasion and metastasis of cancer clones. Med Hypotheses. 2005; 64:1011–14. https://doi.org/10.1016/j.mehy.2004.11.012. [PubMed].
34. Miroshnychenko D, Baratchart E, Ferrall-Fairbanks MC, Velde RV, Laurie MA, Bui MM, Tan AC, Altrock PM, Basanta D, Marusyk A. Spontaneous cell fusions as a mechanism of parasexual recombination in tumour cell populations. Nat Ecol Evol. 2021; 5:379–91. https://doi.org/10.1038/s41559-020-01367-y. [PubMed].
35. Duelli D, Lazebnik Y. Cell-to-cell fusion as a link between viruses and cancer. Nat Rev Cancer. 2007; 7:968–76. https://doi.org/10.1038/nrc2272. [PubMed].
36. Shabo I, Svanvik J, Lindström A, Lechertier T, Trabulo S, Hulit J, Sparey T, Pawelek J. Roles of cell fusion, hybridization and polyploid cell formation in cancer metastasis. World J Clin Oncol. 2020; 11:121–35. https://doi.org/10.5306/wjco.v11.i3.121. [PubMed].
37. Lu X, Kang Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009; 69:8536–39. https://doi.org/10.1158/0008-5472.CAN-09-2159. [PubMed].
38. Mirzayans R, Murray D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int J Mol Sci. 2020; 21:1308. https://doi.org/10.3390/ijms21041308. [PubMed].
39. Lartigue L, Merle C, Lagarde P, Delespaul L, Lesluyes T, Le Guellec S, Pérot G, Leroy L, Coindre JM, Chibon F. Genome remodeling upon mesenchymal tumor cell fusion contributes to tumor progression and metastatic spread. Oncogene. 2020; 39:4198–211. https://doi.org/10.1038/s41388-020-1276-6. [PubMed].
40. Hass R, von der Ohe J, Dittmar T. Hybrid Formation and Fusion of Cancer Cells In Vitro and In Vivo. Cancers (Basel). 2021; 13:4496. https://doi.org/10.3390/cancers13174496. [PubMed].
41. Zhou Y, Cheng JT, Feng ZX, Wang YY, Zhang Y, Cai WQ, Han ZW, Wang XW, Xiang Y, Yang HY, Liu BR, Peng XC, Cui SZ, Xin HW. Could gastrointestinal tumor-initiating cells originate from cell-cell fusion in vivo? World J Gastrointest Oncol. 2021; 13:92–108. https://doi.org/10.4251/wjgo.v13.i2.92. [PubMed].
42. Zhang H, Ma H, Yang X, Fan L, Tian S, Niu R, Yan M, Zheng M, Zhang S. Cell Fusion-Related Proteins and Signaling Pathways, and Their Roles in the Development and Progression of Cancer. Front Cell Dev Biol. 2021; 9:809668. https://doi.org/10.3389/fcell.2021.809668. [PubMed].
43. Avital I, Moreira AL, Klimstra DS, Leversha M, Papadopoulos EB, Brennan M, Downey RJ. Donor-derived human bone marrow cells contribute to solid organ cancers developing after bone marrow transplantation. Stem Cells. 2007; 25:2903–9. https://doi.org/10.1634/stemcells.2007-0409. [PubMed].
44. Aractingi S, Kanitakis J, Euvrard S, Le Danff C, Peguillet I, Khosrotehrani K, Lantz O, Carosella ED. Skin carcinoma arising from donor cells in a kidney transplant recipient. Cancer Res. 2005; 65:1755–60. https://doi.org/10.1158/0008-5472.CAN-04-2783. [PubMed].
45. Cogle CR, Theise ND, Fu D, Ucar D, Lee S, Guthrie SM, Lonergan J, Rybka W, Krause DS, Scott EW. Bone marrow contributes to epithelial cancers in mice and humans as developmental mimicry. Stem Cells. 2007; 25:1881–87. https://doi.org/10.1634/stemcells.2007-0163. [PubMed].
46. Janin A, Murata H, Leboeuf C, Cayuela JM, Gluckman E, Legrès L, Desveaux A, Varna M, Ratajczak P, Soulier J, de Thé H, Bertheau P, Socié G. Donor-derived oral squamous cell carcinoma after allogeneic bone marrow transplantation. Blood. 2009; 113:1834–40. https://doi.org/10.1182/blood-2008-07-171702. [PubMed].
47. Barozzi P, Luppi M, Facchetti F, Mecucci C, Alù M, Sarid R, Rasini V, Ravazzini L, Rossi E, Festa S, Crescenzi B, Wolf DG, Schulz TF, Torelli G. Post-transplant Kaposi sarcoma originates from the seeding of donor-derived progenitors. Nat Med. 2003; 9:554–61. https://doi.org/10.1038/nm862. [PubMed].
48. Yilmaz Y, Lazova R, Qumsiyeh M, Cooper D, Pawelek J. Donor Y chromosome in renal carcinoma cells of a female BMT recipient: visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transplant. 2005; 35:1021–24. https://doi.org/10.1038/sj.bmt.1704939. [PubMed].
49. Hutchinson L, Stenstrom B, Chen D, Piperdi B, Levey S, Lyle S, Wang TC, Houghton J. Human Barrett’s adenocarcinoma of the esophagus, associated myofibroblasts, and endothelium can arise from bone marrow-derived cells after allogeneic stem cell transplant. Stem Cells Dev. 2011; 20:11–17. https://doi.org/10.1089/scd.2010.0139. [PubMed].
50. Verneuil L, Varna M, Leboeuf C, Plassa LF, Elbouchtaoui M, Loisel-Ferreira I, Bouhidel F, Peraldi MN, Lebbé C, Ratajczak P, Janin A. Donor-derived keratinocytes in actinic keratosis and squamous cell carcinoma in patients with kidney transplant. J Invest Dermatol. 2013; 133:1108–11. https://doi.org/10.1038/jid.2012.422. [PubMed].
51. Gast CE, Silk AD, Zarour L, Riegler L, Burkhart JG, Gustafson KT, Parappilly MS, Roh-Johnson M, Goodman JR, Olson B, Schmidt M, Swain JR, Davies PS, et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci Adv. 2018; 4:eaat7828. https://doi.org/10.1126/sciadv.aat7828. [PubMed].
52. Dietz MS, Sutton TL, Walker BS, Gast CE, Zarour L, Sengupta SK, Swain JR, Eng J, Parappilly M, Limbach K, Sattler A, Burlingame E, Chin Y, et al. Relevance of circulating hybrid cells as a non-invasive biomarker for myriad solid tumors. Sci Rep. 2021; 11:13630. https://doi.org/10.1038/s41598-021-93053-7. [PubMed].
53. Lazebnik Y. Gestational tumors as a model to probe reticulate evolution in human neoplasia. Oncotarget. 2019; 10:259–62. https://doi.org/10.18632/oncotarget.26510. [PubMed].
54. Theissen G. Saltational evolution: hopeful monsters are here to stay. Theory Biosci. 2009; 128:43–51. https://doi.org/10.1007/s12064-009-0058-z. [PubMed].
55. Parsons BL. Many different tumor types have polyclonal tumor origin: evidence and implications. Mutat Res. 2008; 659:232–47. https://doi.org/10.1016/j.mrrev.2008.05.004. [PubMed].
56. Parsons BL. Multiclonal tumor origin: Evidence and implications. Mutat Res Rev Mutat Res. 2018; 777:1–18. https://doi.org/10.1016/j.mrrev.2018.05.001. [PubMed].
57. Cesi G, Philippidou D, Kozar I, Kim YJ, Bernardin F, Van Niel G, Wienecke-Baldacchino A, Felten P, Letellier E, Dengler S, Nashan D, Haan C, Kreis S. A new ALK isoform transported by extracellular vesicles confers drug resistance to melanoma cells. Mol Cancer. 2018; 17:145. https://doi.org/10.1186/s12943-018-0886-x. [PubMed].
58. Shaurova T, Zhang L, Goodrich DW, Hershberger PA. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front Genet. 2020; 11:281. https://doi.org/10.3389/fgene.2020.00281. [PubMed].
59. Marusyk A, Janiszewska M, Polyak K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell. 2020; 37:471–84. https://doi.org/10.1016/j.ccell.2020.03.007. [PubMed].
60. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008; 456:593–98. https://doi.org/10.1038/nature07567. [PubMed].
61. Luskin MR, Murakami MA, Manalis SR, Weinstock DM. Targeting minimal residual disease: a path to cure? Nat Rev Cancer. 2018; 18:255–63. https://doi.org/10.1038/nrc.2017.125. [PubMed].
62. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009; 9:302–12. https://doi.org/10.1038/nrc2627. [PubMed].
63. Schumacher S, Bartenhagen C, Hoffmann M, Will D, Fischer JC, Baldus SE, Vay C, Fluegen G, Dizdar L, Vallböhmer D, Klein CA, Knoefel WT, Stoecklein NH, Möhlendick B. Disseminated tumour cells with highly aberrant genomes are linked to poor prognosis in operable oesophageal adenocarcinoma. Br J Cancer. 2017; 117:725–33. https://doi.org/10.1038/bjc.2017.233. [PubMed].
64. Cackowski FC, Wang Y, Decker JT, Sifuentes C, Weindorf S, Jung Y, Wang Y, Decker AM, Yumoto K, Szerlip N, Buttitta L, Pienta KJ, Morgan TM, Taichman RS. Detection and isolation of disseminated tumor cells in bone marrow of patients with clinically localized prostate cancer. Prostate. 2019; 79:1715–27. https://doi.org/10.1002/pros.23896. [PubMed].
65. Gow CH, Chang YL, Hsu YC, Tsai MF, Wu CT, Yu CJ, Yang CH, Lee YC, Yang PC, Shih JY. Comparison of epidermal growth factor receptor mutations between primary and corresponding metastatic tumors in tyrosine kinase inhibitor-naive non-small-cell lung cancer. Ann Oncol. 2009; 20:696–702. https://doi.org/10.1093/annonc/mdn679. [PubMed].
66. Zhao ZM, Zhao B, Bai Y, Iamarino A, Gaffney SG, Schlessinger J, Lifton RP, Rimm DL, Townsend JP. Early and multiple origins of metastatic lineages within primary tumors. Proc Natl Acad Sci U S A. 2016; 113:2140–45. https://doi.org/10.1073/pnas.1525677113. [PubMed].
67. Kim KM, Lee SH, Kim SM, Kim NY, Gwak HS, Shin SH, Kwon JW, Yoo H. Discordance of Epidermal Growth Factor Receptor Mutation between Brain Metastasis and Primary Non-Small Cell Lung Cancer. Brain Tumor Res Treat. 2019; 7:137–40. https://doi.org/10.14791/btrt.2019.7.e44. [PubMed].
68. Patkar S, Heselmeyer-Haddad K, Auslander N, Hirsch D, Camps J, Bronder D, Brown M, Chen WD, Lokanga R, Wangsa D, Wangsa D, Hu Y, Lischka A, et al. Hard wiring of normal tissue-specific chromosome-wide gene expression levels is an additional factor driving cancer type-specific aneuploidies. Genome Med. 2021; 13:93. https://doi.org/10.1186/s13073-021-00905-y. [PubMed].
69. Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003; 100:7737–42. https://doi.org/10.1073/pnas.1331931100. [PubMed].
70. Economopoulou P, Mountzios G, Pavlidis N, Pentheroudakis G. Cancer of Unknown Primary origin in the genomic era: Elucidating the dark box of cancer. Cancer Treat Rev. 2015; 41:598–604. https://doi.org/10.1016/j.ctrv.2015.05.010. [PubMed].
71. Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, Massagué J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013; 154:1060–73. https://doi.org/10.1016/j.cell.2013.07.036. [PubMed].
72. Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell. 2013; 24:410–21. https://doi.org/10.1016/j.ccr.2013.09.007. [PubMed].
73. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018; 359:1350–55. https://doi.org/10.1126/science.aar4060. [PubMed].
74. Vidlarova M, Rehulkova A, Stejskal P, Prokopova A, Slavik H, Hajduch M, Srovnal J. Recent Advances in Methods for Circulating Tumor Cell Detection. Int J Mol Sci. 2023; 24:3902. https://doi.org/10.3390/ijms24043902. [PubMed].
75. Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol. 2017; 11:40–61. https://doi.org/10.1002/1878-0261.12022. [PubMed].
76. Clawson GA, Kimchi E, Patrick SD, Xin P, Harouaka R, Zheng S, Berg A, Schell T, Staveley-O’Carroll KF, Neves RI, Mosca PJ, Thiboutot D. Circulating tumor cells in melanoma patients. PLoS One. 2012; 7:e41052. https://doi.org/10.1371/journal.pone.0041052. [PubMed].
77. Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One. 2012; 7:e33788. https://doi.org/10.1371/journal.pone.0033788. [PubMed].
78. Ruano APC, Gadelha Guimarães AP, Braun AC, Flores BCT, Tariki MS, Abdallah EA, Torres JA, Nunes DN, Tirapelli B, de Lima VCC, Fanelli MF, Colombo PE, da Costa AAB, et al. Fusion Cell Markers in Circulating Tumor Cells from Patients with High-Grade Ovarian Serous Carcinoma. Int J Mol Sci. 2022; 23:14687. https://doi.org/10.3390/ijms232314687. [PubMed].
79. Clawson GA. Cancer. Fusion for moving. Science. 2013; 342:699–700. https://doi.org/10.1126/science.1244270. [PubMed].
80. Clawson GA, Matters GL, Xin P, McGovern C, Wafula E, dePamphilis C, Meckley M, Wong J, Stewart L, D’Jamoos C, Altman N, Imamura Kawasawa Y, Du Z, et al. “Stealth dissemination” of macrophage-tumor cell fusions cultured from blood of patients with pancreatic ductal adenocarcinoma. PLoS One. 2017; 12:e0184451. https://doi.org/10.1371/journal.pone.0184451. [PubMed].
81. Yamanaka N, Wong CJ, Gertsenstein M, Casper RF, Nagy A, Rogers IM. Bone marrow transplantation results in human donor blood cells acquiring and displaying mouse recipient class I MHC and CD45 antigens on their surface. PLoS One. 2009; 4:e8489. https://doi.org/10.1371/journal.pone.0008489. [PubMed].
82. Miyake K, Karasuyama H. The Role of Trogocytosis in the Modulation of Immune Cell Functions. Cells. 2021; 10:1255. https://doi.org/10.3390/cells10051255. [PubMed].
83. Portela M, Venkataramani V, Fahey-Lozano N, Seco E, Losada-Perez M, Winkler F, Casas-Tintó S. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019; 17:e3000545. https://doi.org/10.1371/journal.pbio.3000545. [PubMed].
84. Charbord P. Bone marrow mesenchymal stem cells: historical overview and concepts. Hum Gene Ther. 2010; 21:1045–56. https://doi.org/10.1089/hum.2010.115. [PubMed].
85. Viswanathan S, Shi Y, Galipeau J, Krampera M, Leblanc K, Martin I, Nolta J, Phinney DG, Sensebe L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy. 2019; 21:1019–24. https://doi.org/10.1016/j.jcyt.2019.08.002. [PubMed].
86. Hass R. Role of MSC in the Tumor Microenvironment. Cancers (Basel). 2020; 12:2107. https://doi.org/10.3390/cancers12082107. [PubMed].
87. García-Bernal D, García-Arranz M, Yáñez RM, Hervás-Salcedo R, Cortés A, Fernández-García M, Hernando-Rodríguez M, Quintana-Bustamante Ó, Bueren JA, García-Olmo D, Moraleda JM, Segovia JC, Zapata AG. The Current Status of Mesenchymal Stromal Cells: Controversies, Unresolved Issues and Some Promising Solutions to Improve Their Therapeutic Efficacy. Front Cell Dev Biol. 2021; 9:650664. https://doi.org/10.3389/fcell.2021.650664. [PubMed].
88. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953; 6:963–68. https://doi.org/10.1002/1097-0142(195309)6:5%3c963::aid-cncr2820060515%3e3.0.co;2-q. [PubMed].
89. Sonnenschein C, Soto AM. Carcinogenesis explained within the context of a theory of organisms. Prog Biophys Mol Biol. 2016; 122:70–76. https://doi.org/10.1016/j.pbiomolbio.2016.07.004. [PubMed].
90. Levin M. Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell. 2021; 184:1971–89. https://doi.org/10.1016/j.cell.2021.02.034. [PubMed].
91. Goldenberg DM, Pavia RA. Horizontal transmission of malignant conditions rediscovered. N Engl J Med. 1981; 305:283–84. https://doi.org/10.1056/nejm198107303050513. [PubMed].
92. Goldenberg DM, Pavia RA. Malignant potential of murine stromal cells after transplantation of human tumors into nude mice. Science. 1981; 212:65–67. https://doi.org/10.1126/science.7209521. [PubMed].
93. Goldenberg DM, Pavia RA. In vivo horizontal oncogenesis by a human tumor in nude mice. Proc Natl Acad Sci U S A. 1982; 79:2389–92. https://doi.org/10.1073/pnas.79.7.2389. [PubMed].
94. Pathak S, Nemeth MA, Multani AS. Human tumor xenografts in nude mice are not always of human origin: a warning signal. Cancer. 1998; 83:1891–93. https://doi.org/10.1002/(sici)1097-0142(19981101)83:9<1891::aid-cncr3>3.0.co;2-u. [PubMed].
95. Caretti V, Sewing ACP, Lagerweij T, Schellen P, Bugiani M, Jansen MHA, van Vuurden DG, Navis AC, Horsman I, Vandertop WP, Noske DP, Wesseling P, Kaspers GJL, et al. Human pontine glioma cells can induce murine tumors. Acta Neuropathol. 2014; 127:897–909. https://doi.org/10.1007/s00401-014-1272-4. [PubMed].
96. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, Krummel MF. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016; 531:513–17. https://doi.org/10.1038/nature16985. [PubMed].
97. Mathieu M, Martin-Jaular L, Lavieu G, Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019; 21:9–17. https://doi.org/10.1038/s41556-018-0250-9. [PubMed].
98. Gurung S, Perocheau D, Touramanidou L, Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal. 2021; 19:47. https://doi.org/10.1186/s12964-021-00730-1. [PubMed].
99. Daßler-Plenker J, Küttner V, Egeblad M. Communication in tiny packages: Exosomes as means of tumor-stroma communication. Biochim Biophys Acta Rev Cancer. 2020; 1873:188340. https://doi.org/10.1016/j.bbcan.2020.188340. [PubMed].
100. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-Mediated Metastasis: Communication from a Distance. Dev Cell. 2019; 49:347–60. https://doi.org/10.1016/j.devcel.2019.04.011. [PubMed].
101. Lugini L, Valtieri M, Federici C, Cecchetti S, Meschini S, Condello M, Signore M, Fais S. Exosomes from human colorectal cancer induce a tumor-like behavior in colonic mesenchymal stromal cells. Oncotarget. 2016; 7:50086–98. https://doi.org/10.18632/oncotarget.10574. [PubMed].
102. Abd Elmageed ZY, Yang Y, Thomas R, Ranjan M, Mondal D, Moroz K, Fang Z, Rezk BM, Moparty K, Sikka SC, Sartor O, Abdel-Mageed AB. Neoplastic reprogramming of patient-derived adipose stem cells by prostate cancer cell-associated exosomes. Stem Cells. 2014; 32:983–97. https://doi.org/10.1002/stem.1619. [PubMed].
103. Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, Lucci A, Ivan C, Calin GA, Kalluri R. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014; 26:707–21. https://doi.org/10.1016/j.ccell.2014.09.005. [PubMed].
104. Abken H, Jungfer H, Albert WH, Willecke K. Immortalization of human lymphocytes by fusion with cytoplasts of transformed mouse L cells. J Cell Biol. 1986; 103:795–805. https://doi.org/10.1083/jcb.103.3.795. [PubMed].
105. Israel BA, Schaeffer WI. Cytoplasmic mediation of malignancy. In Vitro Cell Dev Biol. 1988; 24:487–90. https://doi.org/10.1007/BF02628504. [PubMed].
106. Lee TH, Chennakrishnaiah S, Meehan B, Montermini L, Garnier D, D’Asti E, Hou W, Magnus N, Gayden T, Jabado N, Eppert K, Majewska L, Rak J. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget. 2016; 7:51991–2002. https://doi.org/10.18632/oncotarget.10627. [PubMed].
107. Choi D, Lee TH, Spinelli C, Chennakrishnaiah S, D’Asti E, Rak J. Extracellular vesicle communication pathways as regulatory targets of oncogenic transformation. Semin Cell Dev Biol. 2017; 67:11–22. https://doi.org/10.1016/j.semcdb.2017.01.003. [PubMed].
108. Rous P. Surmise and fact on the nature of cancer. Nature. 1959; 183:1357–61. https://doi.org/10.1038/1831357a0. [PubMed].
109. Harris H. A long view of fashions in cancer research. Bioessays. 2005; 27:833–38. https://doi.org/10.1002/bies.20263. [PubMed].
110. Kato S, Lippman SM, Flaherty KT, Kurzrock R. The Conundrum of Genetic “Drivers” in Benign Conditions. J Natl Cancer Inst. 2016; 108:djw036. https://doi.org/10.1093/jnci/djw036. [PubMed].
111. Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, Fitzgerald RC, Handford PA, Campbell PJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018; 362:911–17. https://doi.org/10.1126/science.aau3879. [PubMed].
112. Naxerova K. Mutation fingerprints encode cellular histories. Nature. 2021; 597:334–36. https://doi.org/10.1038/d41586-021-02269-0. [PubMed].
113. Baker SG. The case for a cancer paradox initiative. Carcinogenesis. 2021; 42:1023–25. https://doi.org/10.1093/carcin/bgab052. [PubMed].
114. Peracchia C. Direct communication between axons and sheath glial cells in crayfish. Nature. 1981; 290:597–98. https://doi.org/10.1038/290597a0. [PubMed].
115. Driesen RB, Dispersyn GD, Verheyen FK, van den Eijnde SM, Hofstra L, Thoné F, Dijkstra P, Debie W, Borgers M, Ramaekers FCS. Partial cell fusion: a newly recognized type of communication between dedifferentiating cardiomyocytes and fibroblasts. Cardiovasc Res. 2005; 68:37–46. https://doi.org/10.1016/j.cardiores.2005.05.020. [PubMed].
116. Acquistapace A, Bru T, Lesault PF, Figeac F, Coudert AE, le Coz O, Christov C, Baudin X, Auber F, Yiou R, Dubois-Randé JL, Rodriguez AM. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells. 2011; 29:812–24. https://doi.org/10.1002/stem.632. [PubMed].
117. Hardtke-Wolenski M, Kraus L, Schmetz C, Trautewig B, Noyan F, Vondran FWR, Bektas H, Klempnauer J, Jaeckel E, Lieke T. Exchange of cytosolic content between T cells and tumor cells activates CD4 T cells and impedes cancer growth. PLoS One. 2013; 8:e78558. https://doi.org/10.1371/journal.pone.0078558. [PubMed].
118. Eugenin E, Camporesi E, Peracchia C. Direct Cell-Cell Communication via Membrane Pores, Gap Junction Channels, and Tunneling Nanotubes: Medical Relevance of Mitochondrial Exchange. Int J Mol Sci. 2022; 23:6133. https://doi.org/10.3390/ijms23116133. [PubMed].
119. Sawamiphak S, Kontarakis Z, Filosa A, Reischauer S, Stainier DYR. Transient cardiomyocyte fusion regulates cardiac development in zebrafish. Nat Commun. 2017; 8:1525. https://doi.org/10.1038/s41467-017-01555-8. [PubMed].
120. Vaubel E. The form and function of synovial cells in tissue cultures: I. Morphology of the cells under varying conditions. J Exp Med. 1933; 58:63–83. https://doi.org/10.1084/jem.58.1.63. [PubMed].
121. Fischer A. Tissue culture: studies in experimental morphology and general physiology of tissue cells in vitro. London: Heinemann. 1925; 332. https://doi.org/10.5962/bhl.title.6566.
122. Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004; 303:1007–10. https://doi.org/10.1126/science.1093133. [PubMed].
123. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A. 2006; 103:1283–88. https://doi.org/10.1073/pnas.0510511103. [PubMed].
124. Wang X, Gerdes HH. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015; 22:1181–91. https://doi.org/10.1038/cdd.2014.211. [PubMed].
125. Pinto G, Brou C, Zurzolo C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer. 2020; 6:874–88. https://doi.org/10.1016/j.trecan.2020.04.012. [PubMed].
126. Tiwari V, Koganti R, Russell G, Sharma A, Shukla D. Role of Tunneling Nanotubes in Viral Infection, Neurodegenerative Disease, and Cancer. Front Immunol. 2021; 12:680891. https://doi.org/10.3389/fimmu.2021.680891. [PubMed].
127. Zampieri LX, Silva-Almeida C, Rondeau JD, Sonveaux P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int J Mol Sci. 2021; 22:3245. https://doi.org/10.3390/ijms22063245. [PubMed].
128. Pennanen P, Alanne MH, Fazeli E, Deguchi T, Näreoja T, Peltonen S, Peltonen J. Diversity of actin architecture in human osteoclasts: network of curved and branched actin supporting cell shape and intercellular micrometer-level tubes. Mol Cell Biochem. 2017; 432:131–39. https://doi.org/10.1007/s11010-017-3004-2. [PubMed].
129. Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, Huang L, Ratliff M, Karimian Jazi K, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015; 528:93–98. https://doi.org/10.1038/nature16071. [PubMed].
130. Osswald M, Solecki G, Wick W, Winkler F. A malignant cellular network in gliomas: potential clinical implications. Neuro Oncol. 2016; 18:479–85. https://doi.org/10.1093/neuonc/now014. [PubMed].
131. Searles SC, Santosa EK, Bui JD. Cell-cell fusion as a mechanism of DNA exchange in cancer. Oncotarget. 2018; 9:6156–73. https://doi.org/10.18632/oncotarget.23715. [PubMed].
132. Strakova A, Murchison EP. The cancer which survived: insights from the genome of an 11000 year-old cancer. Curr Opin Genet Dev. 2015; 30:49–55. https://doi.org/10.1016/j.gde.2015.03.005. [PubMed].
133. Matula Z, Mikala G, Lukácsi S, Matkó J, Kovács T, Monostori É, Uher F, Vályi-Nagy I. Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers (Basel). 2021; 13:3461. https://doi.org/10.3390/cancers13143461. [PubMed].
134. Gomzikova MO, James V, Rizvanov AA. Mitochondria Donation by Mesenchymal Stem Cells: Current Understanding and Mitochondria Transplantation Strategies. Front Cell Dev Biol. 2021; 9:653322. https://doi.org/10.3389/fcell.2021.653322. [PubMed].
135. Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K, Moore MAS. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One. 2012; 7:e33093. https://doi.org/10.1371/journal.pone.0033093. [PubMed].
136. Polak R, de Rooij B, Pieters R, den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood. 2015; 126:2404–14. https://doi.org/10.1182/blood-2015-03-634238. [PubMed].
137. Venkatesh VS, Lou E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep (Hoboken). 2019; 2:e1185. https://doi.org/10.1002/cnr2.1185. [PubMed].
138. Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, Zhou Z, Auger MJ, Bowles KM, Rushworth SA. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019; 79:2285–97. https://doi.org/10.1158/0008-5472.CAN-18-0773. [PubMed].
139. Venkataramani V, Schneider M, Giordano FA, Kuner T, Wick W, Herrlinger U, Winkler F. Disconnecting multicellular networks in brain tumours. Nat Rev Cancer. 2022; 22:481–91. https://doi.org/10.1038/s41568-022-00475-0. [PubMed].
140. Demin S, Berdieva M, Goodkov A. Cell-cell fusions and cell-in-cell phenomena in healthy cells and cancer: Lessons from protists and invertebrates. Semin Cancer Biol. 2022; 81:96–105. https://doi.org/10.1016/j.semcancer.2021.03.005. [PubMed].
141. Sokac AM, Biel N, De Renzis S. Membrane-actin interactions in morphogenesis: Lessons learned from Drosophila cellularization. Semin Cell Dev Biol. 2023; 133:107–22. https://doi.org/10.1016/j.semcdb.2022.03.028. [PubMed].
142. Um J, Jung DW, Williams DR. Lessons from the swamp: developing small molecules that confer salamander muscle cellularization in mammals. Clin Transl Med. 2017; 6:13. https://doi.org/10.1186/s40169-017-0143-8. [PubMed].
143. Jansen IDC, Vermeer JAF, Bloemen V, Stap J, Everts V. Osteoclast fusion and fission. Calcif Tissue Int. 2012; 90:515–22. https://doi.org/10.1007/s00223-012-9600-y. [PubMed].
144. Odelberg SJ, Kollhoff A, Keating MT. Dedifferentiation of mammalian myotubes induced by msx1. Cell. 2000; 103:1099–109. https://doi.org/10.1016/s0092-8674(00)00212-9. [PubMed].
145. McGann CJ, Odelberg SJ, Keating MT. Mammalian myotube dedifferentiation induced by newt regeneration extract. Proc Natl Acad Sci U S A. 2001; 98:13699–704. https://doi.org/10.1073/pnas.221297398. [PubMed].
146. Sundaram M, Guernsey DL, Rajaraman MM, Rajaraman R. Neosis: a novel type of cell division in cancer. Cancer Biol Ther. 2004; 3:207–18. https://doi.org/10.4161/cbt.3.2.663. [PubMed].
147. Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer. 2017; 17:74. https://doi.org/10.1038/nrc.2016.142. [PubMed].
148. Cieniewicz AM, Woodruff RI. Passage through vertebrate gap junctions of 17/18kDa molecules is primarily dependent upon molecular configuration. Tissue Cell. 2010; 42:47–52. https://doi.org/10.1016/j.tice.2009.07.003. [PubMed].
149. Lemcke H, David R. Potential mechanisms of microRNA mobility. Traffic. 2018; 19:910–17. https://doi.org/10.1111/tra.12606. [PubMed].
150. Laird DW, Lampe PD. Therapeutic strategies targeting connexins. Nat Rev Drug Discov. 2018; 17:905–21. https://doi.org/10.1038/nrd.2018.138. [PubMed].
151. Peterson NG, Stormo BM, Schoenfelder KP, King JS, Lee RR, Fox DT. Cytoplasmic sharing through apical membrane remodeling. Elife. 2020; 9:e58107. https://doi.org/10.7554/eLife.58107. [PubMed].
152. Palacios-Prado N, Bukauskas FF. Heterotypic gap junction channels as voltage-sensitive valves for intercellular signaling. Proc Natl Acad Sci U S A. 2009; 106:14855–60. https://doi.org/10.1073/pnas.0901923106. [PubMed].
153. Levin M. The Computational Boundary of a “Self”: Developmental Bioelectricity Drives Multicellularity and Scale-Free Cognition. Front Psychol. 2019; 10:2688. https://doi.org/10.3389/fpsyg.2019.02688. [PubMed].
154. Morokuma J, Blackiston D, Adams DS, Seebohm G, Trimmer B, Levin M. Modulation of potassium channel function confers a hyperproliferative invasive phenotype on embryonic stem cells. Proc Natl Acad Sci U S A. 2008; 105:16608–13. https://doi.org/10.1073/pnas.0808328105. [PubMed].
155. Blackiston D, Adams DS, Lemire JM, Lobikin M, Levin M. Transmembrane potential of GlyCl-expressing instructor cells induces a neoplastic-like conversion of melanocytes via a serotonergic pathway. Dis Model Mech. 2011; 4:67–85. https://doi.org/10.1242/dmm.005561. [PubMed].
156. Payne SL, Levin M, Oudin MJ. Bioelectric Control of Metastasis in Solid Tumors. Bioelectricity. 2019; 1:114–30. https://doi.org/10.1089/bioe.2019.0013. [PubMed].
157. Lazebnik Y. The shock of being united and symphiliosis. Another lesson from plants? Cell Cycle. 2014; 13:2323–29. https://doi.org/10.4161/cc.29704. [PubMed].
158. Nowicki S, Gottlieb E. Oncometabolites: tailoring our genes. FEBS J. 2015; 282:2796–805. https://doi.org/10.1111/febs.13295. [PubMed].
159. Yong C, Stewart GD, Frezza C. Oncometabolites in renal cancer. Nat Rev Nephrol. 2020; 16:156–72. https://doi.org/10.1038/s41581-019-0210-z. [PubMed].
160. Abken H, Hegger R, Bützler C, Willecke K. Short DNA sequences from the cytoplasm of mouse tumor cells induce immortalization of human lymphocytes in vitro. Proc Natl Acad Sci U S A. 1993; 90:6518–22. https://doi.org/10.1073/pnas.90.14.6518. [PubMed].
161. Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, Adams PD. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell. 2021; 184:5506–26. https://doi.org/10.1016/j.cell.2021.09.034. [PubMed].
162. Driscoll J, Gondaliya P, Patel T. Tunneling Nanotube-Mediated Communication: A Mechanism of Intercellular Nucleic Acid Transfer. Int J Mol Sci. 2022; 23:5487. https://doi.org/10.3390/ijms23105487. [PubMed].
163. Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. 2011; 2:180. https://doi.org/10.1038/ncomms1180. [PubMed].
164. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, Takasugi M, Watanabe S, Kanemaki MT, Obuse C, Hara E. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. 2017; 8:15287. https://doi.org/10.1038/ncomms15287. [PubMed].
165. Clancy JW, Sheehan CS, Boomgarden AC, D’Souza-Schorey C. Recruitment of DNA to tumor-derived microvesicles. Cell Rep. 2022; 38:110443. https://doi.org/10.1016/j.celrep.2022.110443. [PubMed].
166. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011; 144:27–40. https://doi.org/10.1016/j.cell.2010.11.055. [PubMed].
167. Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, Fang R, Castillon GA, Yu M, Li JSZ, Sun Y, Ellisman MH, Ren B, et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature. 2021; 591:137–41. https://doi.org/10.1038/s41586-020-03064-z. [PubMed].
168. Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, Kornblum HI, Taylor MD, Kaushal S, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017; 543:122–25. https://doi.org/10.1038/nature21356. [PubMed].
169. Verhaak RGW, Bafna V, Mischel PS. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer. 2019; 19:283–88. https://doi.org/10.1038/s41568-019-0128-6. [PubMed].
170. Noer JB, Hørsdal OK, Xiang X, Luo Y, Regenberg B. Extrachromosomal circular DNA in cancer: history, current knowledge, and methods. Trends Genet. 2022; 38:766–81. https://doi.org/10.1016/j.tig.2022.02.007. [PubMed].
171. Kim M, Mahmood M, Reznik E, Gammage PA. Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer. 2022; 8:1046–59. https://doi.org/10.1016/j.trecan.2022.08.001. [PubMed].
172. Seyfried TN. Cancer as a mitochondrial metabolic disease. Front Cell Dev Biol. 2015; 3:43. https://doi.org/10.3389/fcell.2015.00043. [PubMed].
173. Nygren JM, Liuba K, Breitbach M, Stott S, Thorén L, Roell W, Geisen C, Sasse P, Kirik D, Björklund A, Nerlov C, Fleischmann BK, Jovinge S, Jacobsen SE. Myeloid and lymphoid contribution to non-haematopoietic lineages through irradiation-induced heterotypic cell fusion. Nat Cell Biol. 2008; 10:584–92. https://doi.org/10.1038/ncb1721. [PubMed].
174. Johansson CB, Youssef S, Koleckar K, Holbrook C, Doyonnas R, Corbel SY, Steinman L, Rossi FMV, Blau HM. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat Cell Biol. 2008; 10:575–83. https://doi.org/10.1038/ncb1720. [PubMed].
175. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012; 18:759–65. https://doi.org/10.1038/nm.2736. [PubMed].
176. Naphade S, Sharma J, Gaide Chevronnay HP, Shook MA, Yeagy BA, Rocca CJ, Ur SN, Lau AJ, Courtoy PJ, Cherqui S. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells. 2015; 33:301–9. https://doi.org/10.1002/stem.1835. [PubMed].
177. Kalargyrou AA, Basche M, Hare A, West EL, Smith AJ, Ali RR, Pearson RA. Nanotube-like processes facilitate material transfer between photoreceptors. EMBO Rep. 2021; 22:e53732. https://doi.org/10.15252/embr.202153732. [PubMed].
178. Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, Gao J. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther. 2021; 6:65. https://doi.org/10.1038/s41392-020-00440-z. [PubMed].
179. Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharmacol. 2014; 171:5507–23. https://doi.org/10.1111/bph.12704. [PubMed].
180. Ljubojevic N, Henderson JM, Zurzolo C. The Ways of Actin: Why Tunneling Nanotubes Are Unique Cell Protrusions. Trends Cell Biol. 2021; 31:130–42. https://doi.org/10.1016/j.tcb.2020.11.008. [PubMed].
181. Latario CJ, Schoenfeld LW, Howarth CL, Pickrell LE, Begum F, Fischer DA, Grbovic-Huezo O, Leach SD, Sanchez Y, Smith KD, Higgs HN. Tumor microtubes connect pancreatic cancer cells in an Arp2/3 complex-dependent manner. Mol Biol Cell. 2020; 31:1259–72. https://doi.org/10.1091/mbc.E19-11-0605. [PubMed].
182. Chen A, Leikina E, Melikov K, Podbilewicz B, Kozlov MM, Chernomordik LV. Fusion-pore expansion during syncytium formation is restricted by an actin network. J Cell Sci. 2008; 121:3619–28. https://doi.org/10.1242/jcs.032169. [PubMed].
183. Marks R, Dorevitch AP, Mason G. Do all melanomas come from “moles”? A study of the histological association between melanocytic naevi and melanoma. Australas J Dermatol. 1990; 31:77–80. https://doi.org/10.1111/j.1440-0960.1990.tb00656.x. [PubMed].
184. Klebe S, Henderson DW. Facts and fiction: premalignant lesions of lung tissues. Pathology. 2013; 45:305–15. https://doi.org/10.1097/PAT.0b013e32835f45fd. [PubMed].
185. Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011; 17:320–29. https://doi.org/10.1038/nm.2328. [PubMed].
186. Zlotta AR, Egawa S, Pushkar D, Govorov A, Kimura T, Kido M, Takahashi H, Kuk C, Kovylina M, Aldaoud N, Fleshner N, Finelli A, Klotz L, et al. Prevalence of prostate cancer on autopsy: cross-sectional study on unscreened Caucasian and Asian men. J Natl Cancer Inst. 2013; 105:1050–58. https://doi.org/10.1093/jnci/djt151. [PubMed].
187. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75:685–705. https://doi.org/10.1146/annurev-physiol-030212-183653. [PubMed].
188. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009; 30:1073–81. https://doi.org/10.1093/carcin/bgp127. [PubMed].
189. Schmitt M, Greten FR. The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol. 2021; 21:653–67. https://doi.org/10.1038/s41577-021-00534-x. [PubMed].
190. What Emerson Said. The New York Times. 1989. https://www.nytimes.com/1989/10/15/books/l-what-emerson-said-124389.html.
191. Mukherjee S. The Emperor of All Maladies: A Biography of Cancer. Reprint edition. New York: Scribner. 2011; 608. https://doi.org/10.5005/jp-journals-10028-1025.
192. Milkina E, Ponomarenko A, Korneyko M, Lyakhova I, Zayats Y, Zaitsev S, Mischenko P, Eliseikina M, Khotimchenko Y, Shevchenko V, Sharma H, Bryukhovetskiy I. Interaction of hematopoietic CD34+ CD45+ stem cells and cancer cells stimulated by TGF-β1 in a model of glioblastoma in vitro. Oncol Rep. 2018; 40:2595–607. https://doi.org/10.3892/or.2018.6671. [PubMed].
193. Filippova N, Nabors LB. ELAVL1 Role in Cell Fusion and Tunneling Membrane Nanotube Formations with Implication to Treat Glioma Heterogeneity. Cancers (Basel). 2020; 12:3069. https://doi.org/10.3390/cancers12103069. [PubMed].
194. Fjeldstad HE, Johnsen GM, Staff AC. Fetal microchimerism and implications for maternal health. Obstet Med. 2020; 13:112–19. https://doi.org/10.1177/1753495X19884484. [PubMed].
195. Cómitre-Mariano B, Martínez-García M, García-Gálvez B, Paternina-Die M, Desco M, Carmona S, Gómez-Gaviro MV. Feto-maternal microchimerism: Memories from pregnancy. iScience. 2022; 25:103664. https://doi.org/10.1016/j.isci.2021.103664. [PubMed].
196. Sedov E, McCarthy J, Koren E, Fuchs Y. Fetomaternal microchimerism in tissue repair and tumor development. Dev Cell. 2022; 57:1442–52. https://doi.org/10.1016/j.devcel.2022.05.018. [PubMed].
197. Khosrotehrani K, Johnson KL, Cha DH, Salomon RN, Bianchi DW. Transfer of fetal cells with multilineage potential to maternal tissue. JAMA. 2004; 292:75–80. https://doi.org/10.1001/jama.292.1.75. [PubMed].
198. Bianchi DW, Khosrotehrani K, Way SS, MacKenzie TC, Bajema I, O’Donoghue K. Forever Connected: The Lifelong Biological Consequences of Fetomaternal and Maternofetal Microchimerism. Clin Chem. 2021; 67:351–62. https://doi.org/10.1093/clinchem/hvaa304. [PubMed].
199. Dubernard G, Aractingi S, Oster M, Rouzier R, Mathieu MC, Uzan S, Khosrotehrani K. Breast cancer stroma frequently recruits fetal derived cells during pregnancy. Breast Cancer Res. 2008; 10:R14. https://doi.org/10.1186/bcr1860. [PubMed].
200. Cha D, Khosrotehrani K, Kim Y, Stroh H, Bianchi DW, Johnson KL. Cervical cancer and microchimerism. Obstet Gynecol. 2003; 102:774–81. https://doi.org/10.1016/s0029-7844(03)00615-x. [PubMed].
201. Cirello V, Recalcati MP, Muzza M, Rossi S, Perrino M, Vicentini L, Beck-Peccoz P, Finelli P, Fugazzola L. Fetal cell microchimerism in papillary thyroid cancer: a possible role in tumor damage and tissue repair. Cancer Res. 2008; 68:8482–88. https://doi.org/10.1158/0008-5472.CAN-08-0672. [PubMed].
202. Nguyen Huu S, Oster M, Avril MF, Boitier F, Mortier L, Richard MA, Kerob D, Maubec E, Souteyrand P, Moguelet P, Khosrotehrani K, Aractingi S. Fetal microchimeric cells participate in tumour angiogenesis in melanomas occurring during pregnancy. Am J Pathol. 2009; 174:630–37. https://doi.org/10.2353/ajpath.2009.080566. [PubMed].
203. Erickson A, He M, Berglund E, Marklund M, Mirzazadeh R, Schultz N, Kvastad L, Andersson A, Bergenstråhle L, Bergenstråhle J, Larsson L, Alonso Galicia L, Shamikh A, et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature. 2022; 608:360–67. https://doi.org/10.1038/s41586-022-05023-2. [PubMed].
204. Cirello V, Fugazzola L. Novel insights into the link between fetal cell microchimerism and maternal cancers. J Cancer Res Clin Oncol. 2016; 142:1697–704. https://doi.org/10.1007/s00432-015-2110-3. [PubMed].
205. Hoffner L, Surti U. The genetics of gestational trophoblastic disease: a rare complication of pregnancy. Cancer Genet. 2012; 205:63–77. https://doi.org/10.1016/j.cancergen.2012.01.004. [PubMed].
206. Marti Gutierrez N, Mikhalchenko A, Ma H, Koski A, Li Y, Van Dyken C, Tippner-Hedges R, Yoon D, Liang D, Hayama T, Battaglia D, Kang E, Lee Y, et al. Horizontal mtDNA transfer between cells is common during mouse development. iScience. 2022; 25:103901. https://doi.org/10.1016/j.isci.2022.103901. [PubMed].