
Introduction
HNSCC ranks sixth amongst cancers diagnosed worldwide [1]. Despite recent advancements in treatment modalities, 50–60% of human papillomavirus (HPV) negative advanced stage HNSCC patients develop locoregional recurrence after definitive treatment [1, 2]. TP53 is the most frequently mutated gene in ≥80% of HPV-negative tumors [3–5]. This is unlike p53 wild-type (wt) tumors seen more commonly in HPV-positive HNSCC which have excellent survival rates that now require de-escalation of treatment. In addition to losing tumor-suppressive functions, mutant p53 proteins acquire additional biological functions with transforming abilities that promote tumorigenesis [6, 7]. Mutant p53 causes sustained activation of the mTOR pathway, thereby contributing to cancer pathogenesis [8, 9]. Our prior work has shown that overexpression of eIF4E in surgical margins predicts recurrence and that overexpression of eIF4E is functionally active through activation of the Akt/mTOR pathway [10, 11]. Results from a window of opportunity clinical trial concluded that the mTOR pathway was a potential therapeutic target for HNSCC [12]. In our recently reported phase-2 multi-institutional adjuvant trial with everolimus in stage IV HNSCC patients at high risk for recurrence, treatment with everolimus demonstrated significant improvement in 2-year progression-free survival (PFS) in patients with TP53 mutations compared to the placebo group [13]. Our results are even more exciting as this subset of patients with TP53 mutations has the highest risk of recurrence and therefore, the greatest need for adjuvant therapy [14, 15]. Hence, this study focuses on p53 mutated tumors as it is this group of patients that requires adjuvant therapy to improve survival. HNSCC cells exhibit a significant upregulation in lymphatic endothelial cell invasiveness and proliferation [16, 17]. The role of mTOR inhibitors (mTORi) as potent growth inhibitory and antiangiogenic/anti-lymphangiogenic agents in HNSCC is well established [18]. Moreover, mTORi significantly suppressed baseline invasiveness of endothelial and HNSCC tumor cells [19]. However, the underlying molecular mechanisms for mutant p53 protein-mediated activation of the mTOR pathway which drive the oncologic processes in HNSCC are yet to be elucidated. Previous studies have shown that mutant p53 causes stabilization of HIF-1α to upregulate its transcriptional activity, promoting a variety of oncologic processes, including angiogenesis and lymphangiogenesis [20–22]. Moreover, mutant p53-mediated activation of HIF-1α is both transcriptionally and translationally regulated through mTOR [23, 24]. HIF-1α target genes, such as VEGF-A and VEGF-C, are involved in tumor angiogenesis and lymphangiogenesis [25–27], leading to lymph node metastasis and recurrence [28–30]. Accordingly, we sought to investigate the mechanism for everolimus-induced inhibition of TP53 HNSCC.
Results
Everolimus inhibits the growth of TP53 mutant HNSCC both in vitro and in vivo
To evaluate the effect of everolimus on the cell growth of TP53 mutant HNSCC, we utilized FaDu, SCC114, and HN31 cell lines. Consistent with our previous study, everolimus significantly reduced cell viability at 10 nm and 100 nm concentrations (Figure 1A). Moreover, we also evaluated the effect of everolimus on TP53 mutant HNSCC tumor growth in vivo in a tumor mouse xenograft model. Treatment with everolimus (5 mg/kg, oral gavage daily) for 23 days significantly inhibited the growth of both HN31 (Figure 1B) and FaDu xenografts suggesting the inhibitory effect is not cell line specific (Figure 1C).
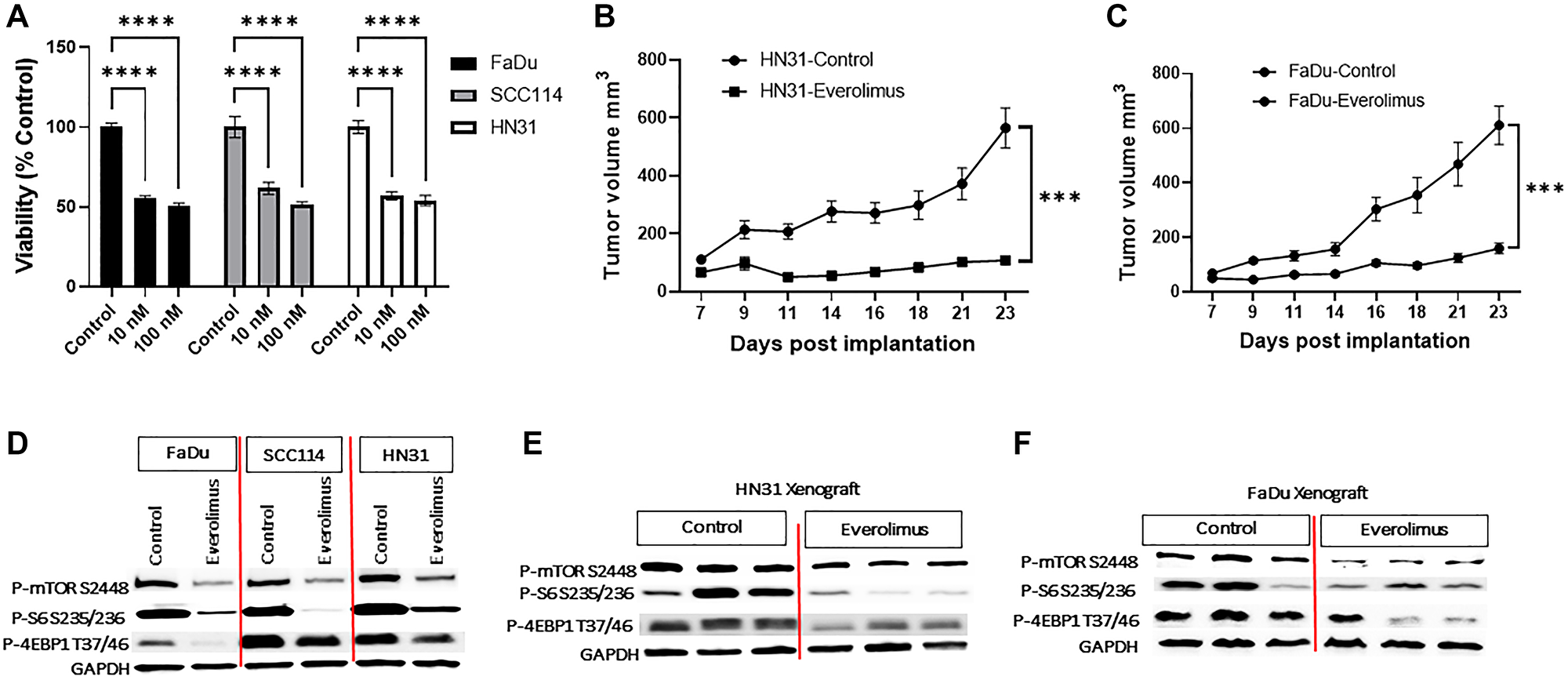
Figure 1: Everolimus downregulates mTORC1 pathway and inhibits growth of TP53 mutant HNSCC cell lines and xenografts. (A) The effect of everolimus on cell viability was measured using an MTS assay after six days of treatment. Cell viability of everolimus treated cells was measured as a percent of control (untreated cells). ****P < 0.00005 vs. control, ANOVA. Data represent the mean +/− SEM of three independent experiments, each experiment comprising samples in triplicate. (B) The growth curve of HN31 xenograft showing everolimus significantly reduces tumor volume. ***P < 0.0005 vs. control, Student’s t-test. Data represent mean +/− SEM, N = 20 mice in each group. (C) The growth curve of FaDu xenograft showing that everolimus significantly reduces tumor volume. ***P < 0.0005 vs. control, Student’s t-test. Data represent mean +/− SEM, N = 20 mice in each group. (D) Representative western blot for mTOR pathway proteins, prepared from cells treated with 100 nm everolimus for 24 hours. (E and F) Representative western blot for mTOR pathway proteins in HN31 and FaDu xenografts. The levels of P-mTOR, P-S6 and P-4EBP1 were reduced in everolimus treated cell lines and xenografts. The western blot experiment was repeated three times.
Everolimus downregulates mTORC1 pathway in TP53 mutant HNSCC
We next evaluated the effect of everolimus on the oncogenic mTOR signaling pathway. Treatment of FaDu, SCC114, and HN31 cells with everolimus significantly downregulated P-mTOR S2448, P-S6 S235/236, and P-4EBP1 T37/46 in these cell lines (Figure 1D). To further translate our in vitro studies in vivo, we investigated the effect of everolimus on the mTORC1 pathway in HN31 and FaDu tumor cell xenografts. Consistent with the effect of everolimus in vitro, oral administration of everolimus was associated with a significant downregulation of P-mTOR S2448, P-S6 S235/236, and P-4EBP1 T37/46 in HN31 and FaDu xenografts (Figure 1E and 1F).
Everolimus inhibits STAT3 phosphorylation and downregulates HIF-1α, VEGF-A and VEGF-C in TP53 mutant HNSCC
As the activity of the mTORC1 pathway is upstream of HIF-1α, we investigated whether everolimus can reduce the level of HIF-1α and its targets VEGF-A and VEGF-C both in cell lines and xenografts [31]. Western blot analysis demonstrated that everolimus reduced P-STAT3 Y705 and P-STAT3 S727 not only in cell lines in vitro, but also in tumor-cell xenografts (Figure 2A–2E). The levels of HIF-1α, VEGF-A and VEGF-C were also decreased by everolimus treatment in cell lines (Figure 2A) as well as in xenografts (Figure 2B–2E). Moreover, mRNA levels of HIF-1α, VEGF-A and VEGF-C were significantly reduced by everolimus treatment both in vitro (Figure 3A–3C) and in vivo (Figure 3D and 3E). Secretion of VEGF-A was also reduced when cells were treated with everolimus (Figure 3F), as measured by ELISA.
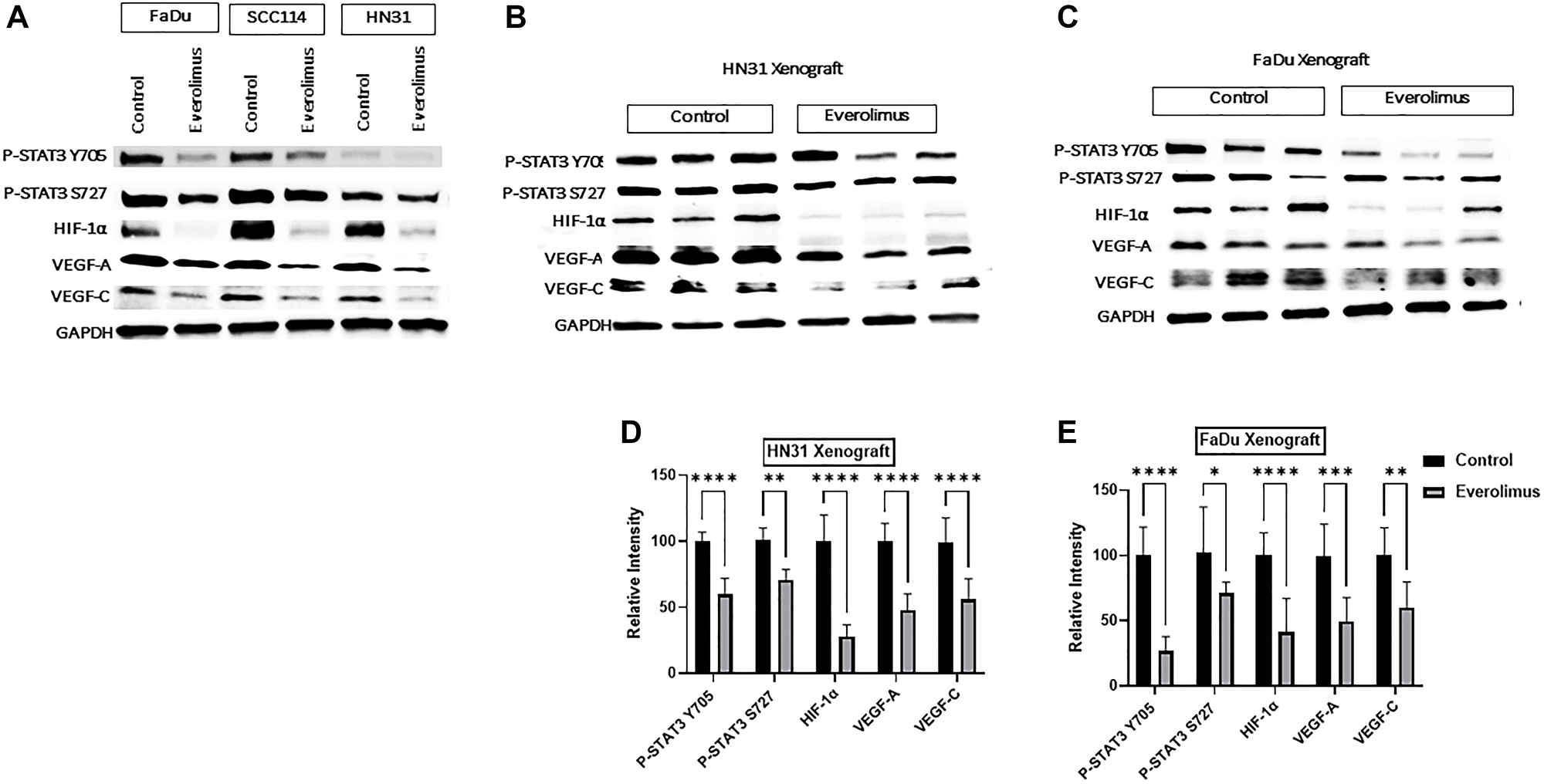
Figure 2: Everolimus inhibits STAT3 phosphorylation and downregulates HIF-1α, VEGF-A and VEGF-C in TP53 mutant HNSCC cell lines and xenografts. (A) Representative western blot of P-STAT3, HIF-1α, VEGF-A and VEGF-C in FaDu, SCC114, and HN31 cell lines. Proteins were prepared from cells treated with 100 nm everolimus for 24 hours. (B and C) Representative western blot for P-STAT3, HIF-1α, VEGF-A, and VEGF-C in HN31 and FaDu xenograft. Proteins were isolated from the xenograft after mice were treated with 5 mg/kg of everolimus daily for 21 days. The western blot experiment was repeated three times. Everolimus also downregulated HIF-1α and its target genes VEGF-A and VEGF-C in FaDu, SCC114, and HN31 cell lines. This downregulation was also seen in both xenograft models (B–E). Moreover, the mRNA levels of HIF-1α, VEGF-A and VEGF-C were significantly reduced by everolimus treatment both in vitro (Figure 3A–3C) and in vivo (Figure 3D–3E). Secretion of VEGF-A was also reduced when cells were treated with everolimus (Figure 3F), as measured by ELISA.
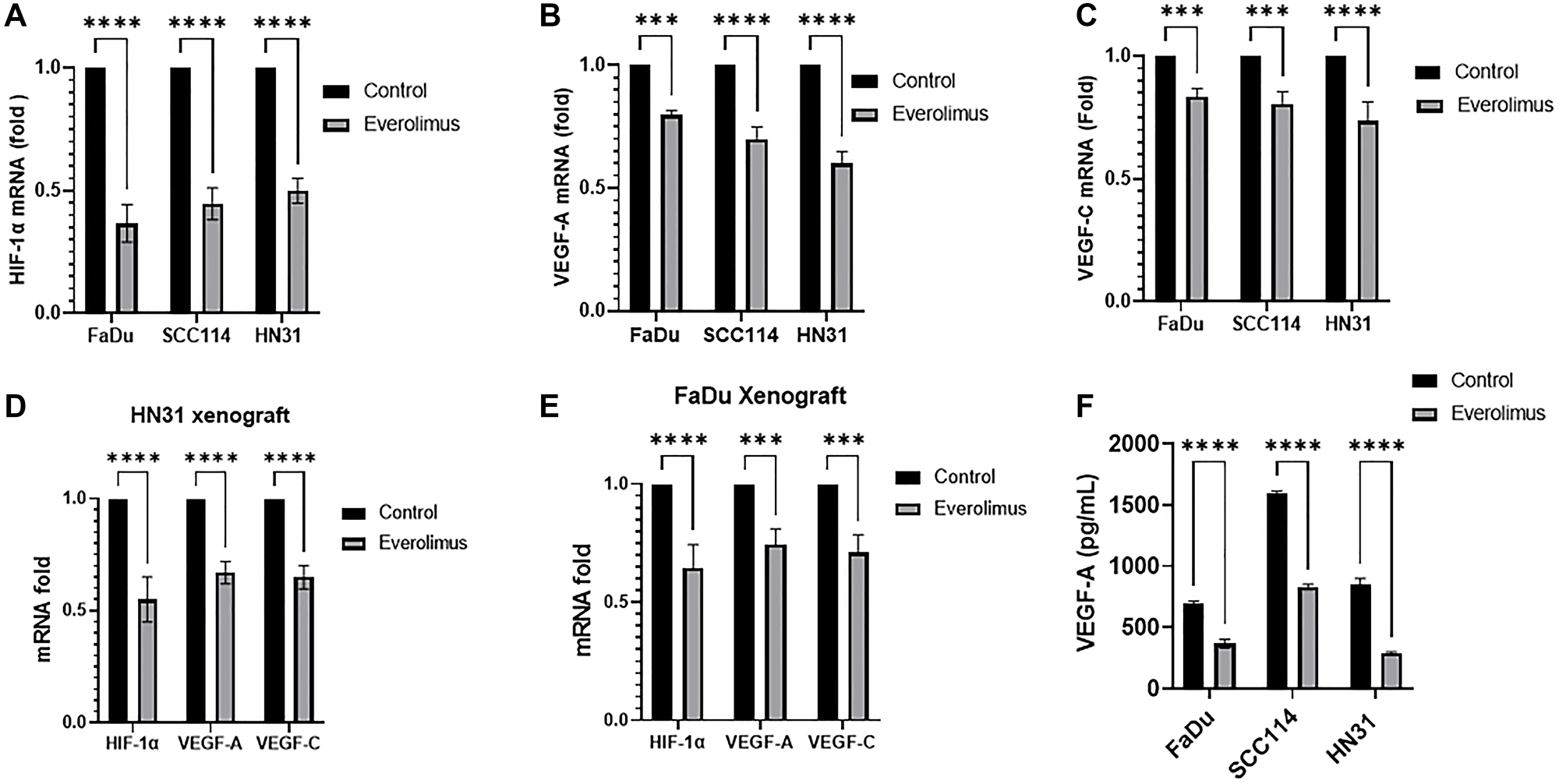
Figure 3: Everolimus reduces mRNA levels of HIF-1α, VEGF-A and VEGF-C in TP53 mutant HNSCC cell lines and xenografts. qRT-PCR was employed to measure the relative amount of mRNA. (A–C) mRNA fold change for HIF-1α, VEGF-A and VEGF-C in FaDu, SCC114, and HN31 cell lines, fold changes are shown relative to control. **P < 0.005; ***P < 0.0005; ****P < 0.00005 vs. control, ANOVA. Data represent the mean +/− SEM of three independent experiments, each comprising samples in triplicate. (D and E) mRNA fold change for HIF-1α, VEGF-A and VEGF-C in HN31 and FaDu xenograft. ***P < 0.0005; ****P < 0.00005 vs. control, ANOVA. Data represent the mean +/− SEM of three independent experiments comprising n = 3 mice. (F) Everolimus reduces the secretion of VEGF-A in cell culture medium. The level of VEGF-A in cell culture medium was determined using ELISA. ****P < 0.00005 vs. control, ANOVA. Data represent the mean +/− SEM of three independent experiments comprising triplicate samples in each experiment.
Everolimus inhibits tumor angiogenesis and lymphangiogenesis in mouse xenograft model
We investigated the effect of everolimus on tumor angiogenesis and lymphangiogenesis. CD31 immunohistochemistry demonstrated that everolimus significantly reduced microvessel density in HN31 and FaDu xenografts (Figure 4A). To identify tumor-associated lymphatic vessels, tumors were immunostained with mouse-specific LYVE-1. Consistent with the inhibitory effect of everolimus on angiogenesis, lymphatic vessel density was significantly reduced by everolimus treatment in FaDu and HN31 xenografts (Figure 4B).
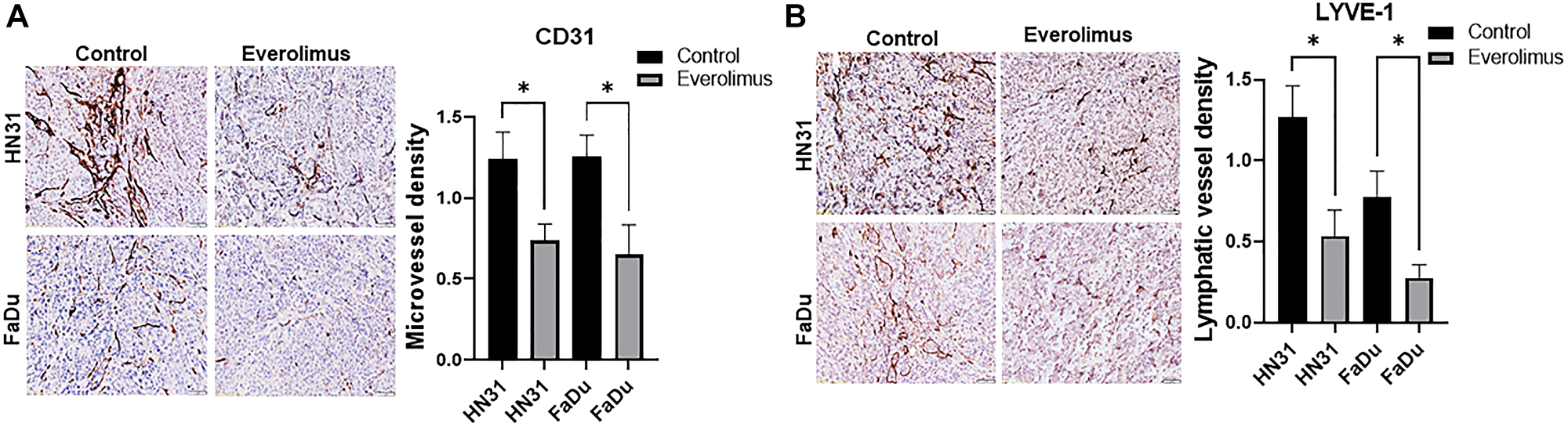
Figure 4: Everolimus reduces microvessel density (MVD) and lymphatic vessel density (LVD) in TP53 mutant HNSCC xenografts. (A) Representative image of CD31 immunostaining and quantification of MVD in HN31 and FaDU xenografts. (B) Representative image of LYVE-1 immunostaining and quantification of LVD in HN31 and FaDU xenografts. Prior to tumor harvesting, mice were treated with 5 mg/kg of everolimus daily for 21 days. *P < 0.05 vs. control, ANOVA. Data represent the mean +/− SEM, N = 20 mice in each group.
The effect of everolimus on human microvascular endothelial (HMEC-1) cells
Angiogenesis involves multiple processes of neovascularization that includes endothelial cell proliferation, migration, and formation of lumen morphogenesis on matrigel. Everolimus significantly inhibited cell proliferation of HMEC-1 when cells were treated with 100 nm for 72 hours (Supplementary Figure 1A). Next, we evaluated in vitro tube formation in cells treated with 100 nm everolimus for 16 hours, where everolimus significantly reduced tube morphology and network formation of HMEC-1 cells (Supplementary Figure 1B). The evaluation of everolimus on endothelial cell migration, where cells were treated with 100 nm everolimus for 8 hours, showed a significant reduction in the number of migrated cells (Supplementary Figure 1C). Moreover, P-mTOR S2448, P-S6 S235/236, P-4EBP1 T37/46 and P-STAT3 were downregulated in both HMEC-1 (Figure 5A) and HMEC-1A (Figure 5D) cell lines. Consistent with the HNSCC cell lines, everolimus reduced levels of HIF-1α, VEGF-A and VEGF-C in both HMEC-1 (Figure 5B) and HMEC-1A (Figure 5E) cells. Finally, the mRNA levels of HIF-1α, VEGF-A and VEGF-C were significantly reduced by everolimus treatment in both HMEC-1 (Figure 5C) and HMEC-1A (Figure 5F).
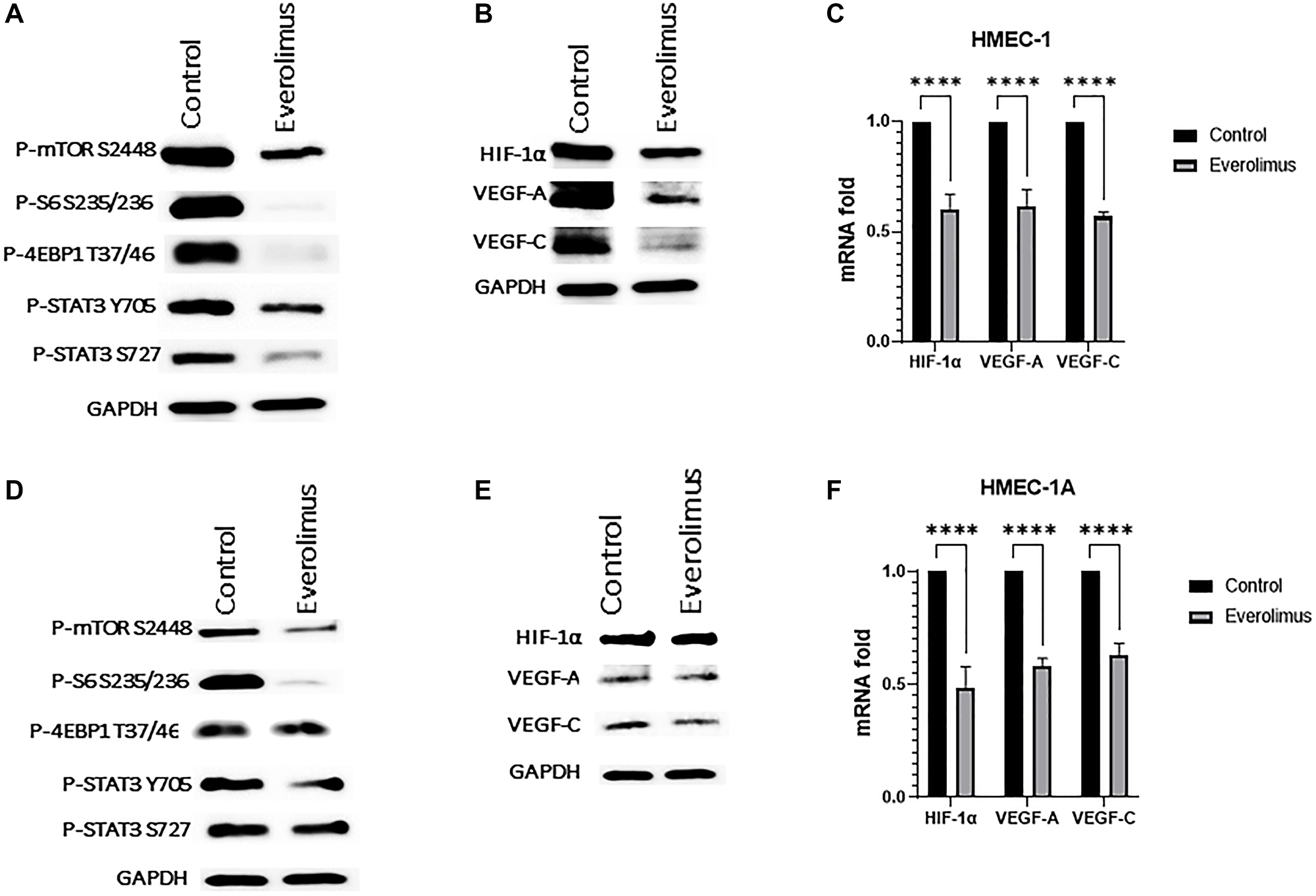
Figure 5: Everolimus inhibits mTORC1/STAT3 pathway and downregulates HIF-1α, VEGF-A and VEGF-C in HMEC-1 and HMEC-1A cells. (A) Proteins were prepared from HMEC-1 treated with 100 nm of everolimus for 24 hours. Representative western blot showing inhibition of mTORC1 pathway proteins and STAT3 phosphorylation. (B) Representative western blot of HIF-1α, VEGF-A and VEGF-C in HMEC-1 showing downregulation by everolimus treatment. (C) Quantification of qRT-PCR data showing mRNA fold change for HIF-1α, VEGF-A and VEGF-C in HMEC-1. ****P < 0.00005 vs. control, ANOVA. Data represent the mean +/− SEM of three independent experiments, each comprising samples in triplicate. mRNA levels of all were significantly reduced by everolimus treatment. (D) Proteins were prepared from HMEC-1A cells treated with 100 nm of everolimus for 24 hours. Representative western blot showing inhibition of mTORC1 and STAT3 phosphorylation in HMEC-1A. (E) Representative western blot of HIF-1α, VEGF-A and VEGF-C in HMEC-1A showing downregulation by everolimus treatment. (F) Quantification of qRT-PCR data showing mRNA fold change for HIF-1α, VEGF-A and VEGF-C in HMEC-1A. ****P < 0.00005 vs. control, ANOVA. Data represent the mean +/− SEM of three independent experiments, each comprising samples in triplicate. mRNA levels of all were significantly reduced by everolimus treatment.
DISCUSSION
TP53 mutations are associated with treatment resistance and shorter survival [32, 33]. Therefore, patients with TP53 mutations often exhibit persistent disease or MRD and potentially could benefit from adjuvant therapy. A multi-institutional phase-2 clinical trial subset analysis determined that HNSCC patients with TP53 mutations benefited when everolimus was administered as adjuvant therapy [13]. Everolimus inhibits STAT3/HIF-1α/VEGF pathways in wt TP53 cell lines as well (data not shown). However, our goal was not to compare p53 mutated tumors to p53 wt tumors, as patients with p53 wt respond so well and do not require adjuvant therapy. Most trials are aimed at de-escalation of treatment for p53 wt tumor patients to decrease side effects. However, we sought specifically to determine the mechanism of why patients with p53 mutated tumors benefited from mTORi.
We employed TP53 mutant cell lines and tumor-cell xenografts to investigate the underlying mechanisms of the anti-tumorigenic effects of everolimus. To recapitulate the clinical trial design, we utilized the minimum residual disease (MRD) model using HN31 and FaDu xenografts [34]. MRD is closely associated with disease persistence/recurrence. Therefore, a better understanding of the underlying mechanism and targets related to the MRD mouse model will be essential to effectively preventing the progression of upper aerodigestive tract cancers. Our prior published studies established mTOR inhibitors as possible adjuvant therapy for microscopic residual disease in HNSCC [34]. However, the underlying mechanism for mTORi growth inhibitory effects in MRD model is largely unknown. Treatment with everolimus significantly inhibited the growth of TP53 mutant cell lines and attenuated the growth kinetics of tumor-cell xenografts. In accordance with our previous studies [35], everolimus inhibited mTORC1 activity by downregulating P-mTOR S2448, P-S6 S235/236, and 4EBP1 T70. Moreover, both in cell lines and xenografts, P-STAT3, HIF-1α, VEGF-A and VEGF-C were downregulated with everolimus treatment. Interestingly, some studies suggest that mTORi do not affect the stability of HIF-1α and act independently of the Von Hippel-Lindau (VHL) protein [36, 37]. Instead, mTORi inhibits the translation of HIF-1α through downregulation of P-4EBP1 and represses transcription of HIF-1α through reduction of P-STAT3 [24]. Our study showed similar results in TP53 mutant HNSCC cell lines and xenografts. Everolimus downregulated HIF-1α, accompanied by the decrease in P-4EBP1 and P-STAT3. Therefore, in line with other studies we postulate that everolimus-mediated downregulation of HIF-1α involves inhibition of its translation (through downregulation of P-4EBP1) and repression of its transcription (through downregulation of P-STAT3). Since the HIF-1α targets, VEGF-A and VEGF-C, are involved in tumor angiogenesis and lymphangiogenesis, we further assessed the effects of everolimus on potential crosstalk between tumor and vascular endothelial and lymphatic cells. Our results revealed that everolimus significantly inhibited tumor angiogenesis and lymphangiogenesis in HN31 and FaDu xenografts, potentially via a paracrine effect. Tumor cells secrete VEGF-A and VEGF-C in the tumor microenvironment which promotes tumor angiogenesis and lymphangiogenesis. VEGF-A mediated angiogenesis plays a pivotal role in tumor recurrence and growth [25–27], whereas VEGF-C mediated lymphangiogenesis is crucial for invasion and metastasis [28–30]. Therefore, we postulate that the inhibition of tumor angiogenesis and lymphangiogenesis by everolimus prevents the recurrence of TP53 mutant HNSCC tumors. Finally, we have shown that everolimus substantially reduces cell proliferation, in vitro network formation, and migration of the endothelial cell line HMEC-1 with concomitant downregulation of the STAT3/HIF-1α/VEGF pathway.
mTOR is well established as a positive regulator of HIF-1α expression and its transcriptional activity [36–39]. The downregulation of HIF-1α target genes by mTORi prevented the growth of renal cell carcinoma [36]. However, the underlying mechanisms for everolimus-induced inhibitory effect on tumor growth have not been elucidated in TP53 mutant HNSCC. Considering the molecular context of TP53 mutant HNSCC, mutant 53 protein causes sustained activation of the mTOR pathway to upregulate HIF-1α, which has been implicated in the expansion of residual cancer stem cells in colorectal cancer [40]. This residual cancer stem cell that survives definitive therapy leads to tumor recurrence [41–44]. Therefore, we postulate that the inhibition of HIF-1α-mediated angiogenesis by everolimus plays a pivotal role in preventing tumor recurrence of TP53 mutant HNSCC. However, HIF-1α target genes are also involved in other critical aspects of cancer biology, including cell survival, chemotherapy and radiation resistance, immortalization, immune evasion, metastasis, and metabolism. The intervention of these oncological processes might also halt the progression of TP53 mutant HNSCC, which has not been explored in this current study. Previous work from our lab has shown that everolimus induces autophagy-dependent cell death (ADCD) in TP53 mutant HNSCC through tumor cell-intrinsic mechanisms [35]. This study demonstrated that everolimus inhibits tumor angiogenesis and lymphangiogenesis through the downregulation of HIF-1α within the tumor microenvironment. Therefore, we conclude that prevention of TP53 mutant HNSCC tumor growth by everolimus consists of multifaceted mechanisms that involve modulation of both tumor cells and the tumor microenvironment. Further studies are required to elucidate the underlying detailed mechanisms.
Materials and Methods
Cell culture
Three HPV-negative HNSCC cell lines with TP53 mutations were used in this study: 1) HN31 (C176F and A161S), kindly provided by Dr. Jeffrey Myers at The University of Texas M.D. Anderson Cancer Center, 2) FaDu (R248L) procured from American Type Culture Collection, and 3) UPCI-SCC114 (SCC114) (R248Q), kindly provided by Dr. Susanne Gollin at the University of Pittsburgh. Information on TP53 mutational status and disease background of cell lines are described previously [35]. These cell lines have Evolutionary Action (EA) scores of more than 75, therefore considered as high risk TP53 mutations [45]. All cell lines were maintained in Dulbecco-modified Eagle medium (Corning, Corning, NY, USA), 10% fetal bovine serum (R&D Systems, Minneapolis, MN, USA), antibiotic/antimycotic (Hyclone, Logan, UT, USA), glutamine (Sigma-Aldrich, St. Louis, MO, USA), sodium pyruvate (Sigma-Aldrich, St. Louis, MO) and non-essential amino acids (Sigma-Aldrich, St. Louis, MO, USA). HMEC-1 and HMEC-1A, human endothelial and lymphatic endothelial cell lines, respectively, were kindly provided by Dr. J. Steven Alexander at LSU Health Sciences Center-Shreveport and maintained in MCDB 131 medium (Sigma-Aldrich, St. Louis, MO, USA), supplemented with 20 mM HEPES, 1 ug/ml hydrocortisone, 10 ng/ml EGF and 10% fetal bovine serum. Cells were grown in monolayers and maintained in humidified 5% CO2 atmosphere at 37°C.
Western blot
Cells were grown in their respective medium and treated with 100 nm of everolimus (Selleckchem, Houston, TX) for 24 hours. The cells were lysed using 1X cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) containing a Protease Inhibitor Cocktail (Roche Molecular Biochemicals, Germany) and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO, USA) [46]. Briefly, protein lysates (30–50 μg) were denatured using Laemmli Sample Buffer then loaded, run in precast gels, and transferred to PVDF membranes using the Trans-Blot Turbo Transfer System (Bio-Rad, Hercules, CA, USA). After blocking with either BSA (3%) or milk (5%), membranes were incubated with primary antibody overnight at 4°C. The primary antibodies used were HIF-1α, P-mTOR S2448, mTOR, P-S6 S235/236, P-STAT3 Y705, P-STAT3 S727, GAPDH (Cell Signaling Technology, Danvers, MA, USA), VEGF-A, and VEGF-C (Abcam, Cambridge, UK). The membranes were next incubated with either an anti-rabbit HRP-conjugated secondary antibody (R&D Systems, Minneapolis, MN, USA), or an anti-mouse HRP-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA, USA) for one hour. The chemiluminescent signal was developed using SuperSignal Chemiluminescent Substrates (Thermo Fisher Scientific, Waltham, MA, USA), and captured with a ChemiDoc XRS+ System (Bio-Rad, Hercules, CA, USA). Acquired images were analyzed and quantified using Image J software.
Cell viability assay
HNSCC cell lines were seeded in 96-well tissue culture plates at a density of 1000–2000 cells/well for 24 hours. Cells were then treated with everolimus (10 and 100 nm) for 72 hours. For HMEC cell lines, 8000 cells/well were seeded in a 96-well plate for 24 hours, followed by 72 hours of treatment with everolimus. After respective treatments, cell viability was measured using the CellTiter 96® Aqueous cell proliferation assay according to the manufacturer’s instructions (Promega Corporation, Madison, WI, USA). Cell viability was quantified and expressed as percent control.
HNSCC xenograft model
The mouse xenograft experiment was conducted in compliance with the Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee guidelines under the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals. 1 × 106 FaDu cells or 2 × 106 HN31 cells were injected subcutaneously into both flanks of 6-8 weeks old female athymic nude mice (Charles River Laboratories, Shrewsbury, MA, USA). Tumors were measured using a digital caliper and volume calculated using the formula [(length × width2)/2]. Mice were randomized to two groups of 20 mice each. To mimic minimal residual disease in patients, treatment started on day five before the appearance of tumors. The experimental group received 5 mg/kg of everolimus dissolved in 1% CMC-Na (Sigma-Aldrich, St. Louis, MO, USA) by oral gavage daily, whereas control mice received 1% CMC-Na. Tumor volume and body weight were measured three times per week. After three weeks, mice were sacrificed, the tumors excised, and tumor lysates were prepared using RIPA buffer (Cell Signaling Technology, Danvers, MA, USA). Tumor lysates were then analyzed by Western blot.
Immunohistochemistry and microvascular density quantification
The xenograft tissue was fixed in zinc-formalin and paraffin embedded. 4 μm FFPE tissue sections were deparaffinized, dehydrated, processed with antigen retrieval buffer, and incubated with primary antibodies. For CD31 staining, citrate buffer (pH 6.0), and LYVE-1, EDTA buffer (pH 9.0) was used for antigen retrieval following the manufacturer’s protocol using a pressure cooker. The slides were then incubated with either anti-CD31 (Cell Signaling Technology, Danvers, MA, USA), or anti-LYVE1 (R&D Systems, Minneapolis, MN, USA) antibody for 24 hours at 4°C. After incubation with biotinylated secondary antibody (Vector Lab, Burlingame, CA, USA for 30 min, slides were incubated with streptavidin-biotin-peroxidase (Vector Lab, Burlingame, CA, USA). DAB substrate was used to visualize positive staining. Images were acquired with the Olympus VS200 Research Slide Scanner under 40× magnification. Microvascular density was measured using the Aperio Microvessel Analysis Algorithm (Aperio, Vista, CA, USA).
ELISA
The amount of VEGF-A secreted in the cell culture medium was measured using VEGFA Human ELISA Kit (Abcam, Cambridge, UK) according to the manufacturer’s protocol. Briefly, FaDu, SCC114 and HN31 cell lines were seeded overnight in a 6-well plate and treated with 100nM of everolimus for 24 hours. Cell medium was collected and centrifuged at 500 RCF. 50 ul of supernatant was used to determine VEGF-A levels secreted in the medium.
Endothelial cell proliferation assay
Exponentially growing HMEC-1 cells were plated at a density of 8 × 103 cells per well in 96-well plates and grown in a complete MCDB131 medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10% FBS for 24 hours. After 24 hours, cells were treated with either 100 nm of everolimus or 100 ng/mL of VEGF (R&D Systems, Minneapolis, MN, USA) or a combination of everolimus and VEGF for 72 hours. Cell viability was measured using the CellTiter 96® Aqueous cell proliferation assay according to the manufacturer’s instructions (Promega, Madison, WI, USA). The cell viability was quantified and expressed as percent control.
In vitro angiogenesis (tube morphology) assay
2.5 × 103 cells in 200 μl of MCDB 131 medium supplemented with 0.5% FBS and either everolimus, VEGF, or both were added to the wells of 96 well plates pre-coated with growth factor–depleted matrigel (7 μg/mL; Becton Dickinson, Bedford, MA, USA). After 16 hours, the medium was removed gently without disturbing newly formed tubules. Images were captured at 40× magnification on a Leica microscope. The image was analyzed using angioanalyzer plugins to measure the number of branch points and segments. Five random 40× images were used for each well. Each experiment was done in triplicate and repeated twice.
Endothelial cell migration assay
Migration assays were done in transwell tissue culture plates (6.5 mm and 4-μm pore size, Becton Dickinson and Co., Franklin Lakes, NJ, USA). The bottom of the transwell chamber was coated with 10 mg/mL of collagen I (Sigma-Aldrich, St. Louis, MO, USA) for 30 minutes. 50 × 103 cells were seeded into each well and allowed to migrate for 6 hours. The cells were then fixed with methanol and stained with crystal violet. Migrated cells were counted in five random 10× fields. Each experiment was done in triplicate and repeated twice. The results are expressed as mean number and number of cells +/− SE.
Abbreviations
HNSCC: Head and Neck Squamous Cell Carcinoma; MRD: Minimal Residual Disease Model; mTORi: mTOR Inhibitor; HMEC: Human Microvascular Endothelial Cells; ELISA: Enzyme Linked Immunosorbent Assay; FFPE: Formalin-Fixed, Paraffin-Embedded.
ACKNOWLEDGMENTS
We thank Dr. Jeffrey N. Myers from MD Anderson Cancer Center (Houston, TX) for providing the HN31 cell line, Dr. Susanne Gollin from University of Pittsburgh (Pittsburgh, PA) for the UPCI-SCC114 cell line, Dr. J. Steven Alexander from LSU Health Sciences Center-Shreveport for HMEC cell lines, and the Feist-Weiller Cancer Center Upper Aero-Digestive Tract Cancer Focus Group for their support.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
Ethical statement
All animal work was conducted under approved Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee protocols following the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
FUNDING
Supportive funds were provided by the Feist-Weiller Cancer Center Upper Aerodigestive Tract Focus Group.
References
1. Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008; 371:1695–709. https://doi.org/10.1016/S0140-6736(08)60728-X. [PubMed].
2. Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008; 100:261–69. https://doi.org/10.1093/jnci/djn011. [PubMed].
3. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333:1154–57. https://doi.org/10.1126/science.1206923. [PubMed].
4. Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517:576–82. https://doi.org/10.1038/nature14129. [PubMed].
5. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, Cortez E, Xie TX, Zhang D, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013; 3:770–81. https://doi.org/10.1158/2159-8290.CD-12-0537. [PubMed].
6. Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ. Gain of function mutations in p53. Nat Genet. 1993; 4:42–46. https://doi.org/10.1038/ng0593-42. [PubMed].
7. van Oijen MG, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000; 6:2138–45. [PubMed].
8. Cordani M, Oppici E, Dando I, Butturini E, Dalla Pozza E, Nadal-Serrano M, Oliver J, Roca P, Mariotto S, Cellini B, Blandino G, Palmieri M, Di Agostino S, Donadelli M. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol Oncol. 2016; 10:1008–29. https://doi.org/10.1016/j.molonc.2016.04.001. [PubMed].
9. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005; 102:8204–9. https://doi.org/10.1073/pnas.0502857102. [PubMed].
10. Nathan CO, Amirghahari N, Abreo F, Rong X, Caldito G, Jones ML, Zhou H, Smith M, Kimberly D, Glass J. Overexpressed eIF4E is functionally active in surgical margins of head and neck cancer patients via activation of the Akt/mammalian target of rapamycin pathway. Clin Cancer Res. 2004; 10:5820–27. https://doi.org/10.1158/1078-0432.CCR-03-0483. [PubMed].
11. Nathan CO, Franklin S, Abreo FW, Nassar R, De Benedetti A, Glass J. Analysis of surgical margins with the molecular marker eIF4E: a prognostic factor in patients with head and neck cancer. J Clin Oncol. 1999; 17:2909–14. https://doi.org/10.1200/JCO.1999.17.9.2909. [PubMed].
12. Day TA, Shirai K, O’Brien PE, Matheus MG, Godwin K, Sood AJ, Kompelli A, Vick JA, Martin D, Vitale-Cross L, Callejas-Varela JL, Wang Z, Wu X, et al. Inhibition of mTOR Signaling and Clinical Activity of Rapamycin in Head and Neck Cancer in a Window of Opportunity Trial. Clin Cancer Res. 2019; 25:1156–64. https://doi.org/10.1158/1078-0432.CCR-18-2024. [PubMed].
13. Nathan CO, Hayes DN, Karrison T, Harismendy O, Flores JM, Moore-Medlin T, Vokes EE, Gutkind JS, Neupane P, Mills G, Sargi Z, Seiwert T, Grilley-Olson J, et al. A Randomized Multi-institutional Phase II Trial of Everolimus as Adjuvant Therapy in Patients with Locally Advanced Squamous Cell Cancer of the Head and Neck. Clin Cancer Res. 2022; 28:5040–48. https://doi.org/10.1158/1078-0432.CCR-21-4290. [PubMed].
14. Ganly I, Soutar DS, Brown R, Kaye SB. p53 alterations in recurrent squamous cell cancer of the head and neck refractory to radiotherapy. Br J Cancer. 2000; 82:392–98. https://doi.org/10.1054/bjoc.1999.0932. [PubMed].
15. Wang ZB, Peng XZ, Chen SS, Ning FL, Du CJ, Wang K, Ma W, Cheng YF. High p53 and MAP1 light chain 3A co-expression predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mol Med Rep. 2013; 8:41–46. https://doi.org/10.3892/mmr.2013.1451. [PubMed].
16. Frech S, Hörmann K, Riedel F, Götte K. Lymphatic vessel density in correlation to lymph node metastasis in head and neck squamous cell carcinoma. Anticancer Res. 2009; 29:1675–79. [PubMed].
17. Lee BS, Jang JY, Seo C, Kim CH. Crosstalk between head and neck cancer cells and lymphatic endothelial cells promotes tumor metastasis via CXCL5-CXCR2 signaling. FASEB J. 2021; 35:e21181. https://doi.org/10.1096/fj.202001455R. [PubMed].
18. Ekshyyan O, Moore-Medlin TN, Raley MC, Sonavane K, Rong X, Brodt MA, Abreo F, Alexander JS, Nathan CA. Anti-lymphangiogenic properties of mTOR inhibitors in head and neck squamous cell carcinoma experimental models. BMC Cancer. 2013; 13:320. https://doi.org/10.1186/1471-2407-13-320. [PubMed].
19. Patel V, Marsh CA, Dorsam RT, Mikelis CM, Masedunskas A, Amornphimoltham P, Nathan CA, Singh B, Weigert R, Molinolo AA, Gutkind JS. Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res. 2011; 71:7103–12. https://doi.org/10.1158/0008-5472.CAN-10-3192. [PubMed].
20. Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, Marini A, Antonov AA, Le Quesne J, Baena Acevedo JD, Dudek K, Sozzi G, Pastorino U, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci U S A. 2018; 115:E10869–78. https://doi.org/10.1073/pnas.1808314115. [PubMed].
21. Amelio I, Melino G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem Sci. 2015; 40:425–34. https://doi.org/10.1016/j.tibs.2015.04.007. [PubMed].
22. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012; 33:207–14. https://doi.org/10.1016/j.tips.2012.01.005. [PubMed].
23. Atala A. Re: mTORC1 Drives HIF-1α and VEGF-A Signalling via Multiple Mechanisms Involving 4E-BP1, S6K1 and STAT3. J Urol. 2016; 195:524. https://doi.org/10.1016/j.juro.2015.10.121. [PubMed].
24. Dodd KM, Yang J, Shen MH, Sampson JR, Tee AR. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. 2015; 34:2239–50. https://doi.org/10.1038/onc.2014.164. [PubMed].
25. Cao Y, E G, Wang E, Pal K, Dutta SK, Bar-Sagi D, Mukhopadhyay D. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res. 2012; 72:3912–18. https://doi.org/10.1158/0008-5472.CAN-11-4058. [PubMed].
26. Gacche RN. Compensatory angiogenesis and tumor refractoriness. Oncogenesis. 2015; 4:e153. https://doi.org/10.1038/oncsis.2015.14. [PubMed].
27. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000; 14:34–44. https://doi.org/10.1101/gad.14.1.34. [PubMed].
28. Miyahara M, Tanuma J, Sugihara K, Semba I. Tumor lymphangiogenesis correlates with lymph node metastasis and clinicopathologic parameters in oral squamous cell carcinoma. Cancer. 2007; 110:1287–94. https://doi.org/10.1002/cncr.22900. [PubMed].
29. Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001; 7:192–98. https://doi.org/10.1038/84643. [PubMed].
30. Thelen A, Jonas S, Benckert C, Weichert W, Schott E, Bötcher C, Dietz E, Wiedenmann B, Neuhaus P, Scholz A. Tumor-associated lymphangiogenesis correlates with prognosis after resection of human hepatocellular carcinoma. Ann Surg Oncol. 2009; 16:1222–30. https://doi.org/10.1245/s10434-009-0380-1. [PubMed].
31. Ferris RL, Flamand Y, Weinstein GS, Li S, Quon H, Mehra R, Garcia JJ, Chung CH, Gillison ML, Duvvuri U, O’Malley BW Jr, Ozer E, Thomas GR, et al. Phase II Randomized Trial of Transoral Surgery and Low-Dose Intensity Modulated Radiation Therapy in Resectable p16+ Locally Advanced Oropharynx Cancer: An ECOG-ACRIN Cancer Research Group Trial (E3311). J Clin Oncol. 2022; 40:138–49. https://doi.org/10.1200/JCO.21.01752. [PubMed].
32. Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007; 357:2552–61. https://doi.org/10.1056/NEJMoa073770. [PubMed].
33. Zhou G, Liu Z, Myers JN. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J Cell Biochem. 2016; 117:2682–92. https://doi.org/10.1002/jcb.25592. [PubMed].
34. Nathan CO, Amirghahari N, Rong X, Giordano T, Sibley D, Nordberg M, Glass J, Agarwal A, Caldito G. Mammalian target of rapamycin inhibitors as possible adjuvant therapy for microscopic residual disease in head and neck squamous cell cancer. Cancer Res. 2007; 67:2160–68. https://doi.org/10.1158/0008-5472.CAN-06-2449. [PubMed].
35. Alam MM, Marin Fermin J, Spiller PT, Burnett C, Rong X, Moore-Medlin T, Maxwell CO, Khandelwal AR, Nathan CO. Rapalogs induce non-apoptotic, autophagy-dependent cell death in HPV-negative TP53 mutant head and neck squamous cell carcinoma. Mol Carcinog. 2022; 61:33–44. https://doi.org/10.1002/mc.23357. [PubMed].
36. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002; 22:7004–14. https://doi.org/10.1128/MCB.22.20.7004-7014.2002. [PubMed].
37. Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007; 282:20534–43. https://doi.org/10.1074/jbc.M611782200. [PubMed].
38. Brugarolas J, Kaelin WG Jr. Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell. 2004; 6:7–10. https://doi.org/10.1016/j.ccr.2004.06.020. [PubMed].
39. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004; 10:594–601. https://doi.org/10.1038/nm1052. [PubMed].
40. Matsumoto K, Arao T, Tanaka K, Kaneda H, Kudo K, Fujita Y, Tamura D, Aomatsu K, Tamura T, Yamada Y, Saijo N, Nishio K. mTOR signal and hypoxia-inducible factor-1 alpha regulate CD133 expression in cancer cells. Cancer Res. 2009; 69:7160–64. https://doi.org/10.1158/0008-5472.CAN-09-1289. [PubMed].
41. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009; 106:13820–25. https://doi.org/10.1073/pnas.0905718106. [PubMed].
42. Dave B, Mittal V, Tan NM, Chang JC. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012; 14:202. https://doi.org/10.1186/bcr2938. [PubMed].
43. Gomez-Casal R, Bhattacharya C, Ganesh N, Bailey L, Basse P, Gibson M, Epperly M, Levina V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol Cancer. 2013; 12:94. https://doi.org/10.1186/1476-4598-12-94. [PubMed].
44. Quint K, Tonigold M, Di Fazio P, Montalbano R, Lingelbach S, Rückert F, Alinger B, Ocker M, Neureiter D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int J Oncol. 2012; 41:2093–102. https://doi.org/10.3892/ijo.2012.1648. [PubMed].
45. Osman AA, Neskey DM, Katsonis P, Patel AA, Ward AM, Hsu TK, Hicks SC, McDonald TO, Ow TJ, Alves MO, Pickering CR, Skinner HD, Zhao M, et al. Evolutionary Action Score of TP53 Coding Variants Is Predictive of Platinum Response in Head and Neck Cancer Patients. Cancer Res. 2015; 75:1205–15. https://doi.org/10.1158/0008-5472.CAN-14-2729. [PubMed].
46. Ekshyyan O, Rong Y, Rong X, Pattani KM, Abreo F, Caldito G, Chang JK, Ampil F, Glass J, Nathan CO. Comparison of radiosensitizing effects of the mammalian target of rapamycin inhibitor CCI-779 to cisplatin in experimental models of head and neck squamous cell carcinoma. Mol Cancer Ther. 2009; 8:2255–65. https://doi.org/10.1158/1535-7163.MCT-08-1184. [PubMed].