Introduction
Though survival rates for pediatric cancer patients have improved dramatically since the early 1960s, cancer remains the leading cause of disease-related death in children and adolescents [1]. Following first-line therapy, approximately 25% of pediatric cancer patients will experience a relapse which generally proves fatal [2, 3]. In addition, nearly two-thirds of pediatric cancer survivors experience chronic, debilitating conditions and even secondary cancers resulting from aggressive treatment paradigms [4]. Despite dose-intensification of chemotherapy and focused efforts on understanding the molecular underpinnings of pediatric tumor types in the hopes of identifying therapeutic targets, survival rates have plateaued in recent years [5]. Therefore, identification and preclinical evaluation of novel targeted therapies in relevant pediatric model systems is necessary to support and better inform subsequent clinical investigation of these agents in pediatric indications.
The vascular endothelial growth factor (VEGF) family consists of five ligands (placental growth factor [PlGF] and VEGF-A, -B, -C, and -D) and three receptors (VEGFR1, VEGFR2, and VEGFR3) [6, 7]. Aberrant activation of VEGFR2 on endothelial cells by tumor cell-secreted VEGF-A drives angiogenesis, the development of new blood vessels from existing vessels. This new blood vessel growth supports tumor progression, local invasion, and metastasis [8, 9]. Inhibition of the VEGF pathway in cancer is thought to not only reduce the total number of vessels to support tumor growth, but also improve the function of vessels within the tumor, thereby more effectively delivering other anti-cancer therapeutics [10]. Because of the multifaceted effects on tumor vessels, the VEGF-A:VEGFR2 signaling axis has been pursued as a therapeutic target in adult solid tumors with approvals across a number of tumor histologies [11–13]. Preclinical and clinical studies have demonstrated that small molecule inhibitors of the VEGF pathway have anti-tumor activity in some pediatric malignancies; however, it is still unclear which pediatric indications may receive the most benefit from anti-VEGFR2 therapy, either alone or in combination with chemotherapy [14–18].
Ramucirumab (Cyramza®, LY3009806) is a fully human monoclonal antibody which specifically binds to and blocks the activation of VEGFR2 by its ligands VEGF-A, -C, and -D [19]. Currently, ramucirumab is FDA-approved for the treatment of advanced gastric or GE junction adenocarcinoma alone or in combination with paclitaxel; metastatic colorectal cancer in combination with FOLFIRI; metastatic non-small cell lung cancer in combination with docetaxel; and as a single agent in hepatocellular carcinoma patients with high alpha fetoprotein levels [20]. For pediatric patients with solid tumors, a dose-finding phase I trial for ramucirumab is currently underway (NCT02564198). Here, we tested ramucirumab or the anti-mouse VEGFR2 antibody DC101 (a ramucirumab surrogate for in vivo studies) in multiple pediatric cancer cell lines and xenograft mouse models with the goal of identifying specific pediatric indications that may respond to ramucirumab-mediated VEGFR2 inhibition.
Results
Pediatric cancer cell lines produce ligands for VEGFR2
To first establish the expression patterns of VEGFR2 and its associated ligands in our pediatric cancer models, we profiled a panel of 11 pediatric cancer cell lines representing neuroblastoma (IMR-32, KELLY, SH-SY5Y), retinoblastoma (Y79), osteosarcoma (HOS, Saos-2, SJSA-1), rhabdomyosarcoma (SJCRH30 [alveolar RMS], RD [embryonal RMS]), malignant rhabdoid tumor (A-204), and Ewing’s sarcoma (RD-ES) for VEGFR2 protein expression (Figure 1A). As expected, VEGFR2 was absent from the majority of cancer cell lines and detected in only 3 out of the 11 cell lines (KELLY, SJCRH30, and RD) at much lower levels than the VEGF-A-stimulated endothelial colony forming cell (ECFC) control.
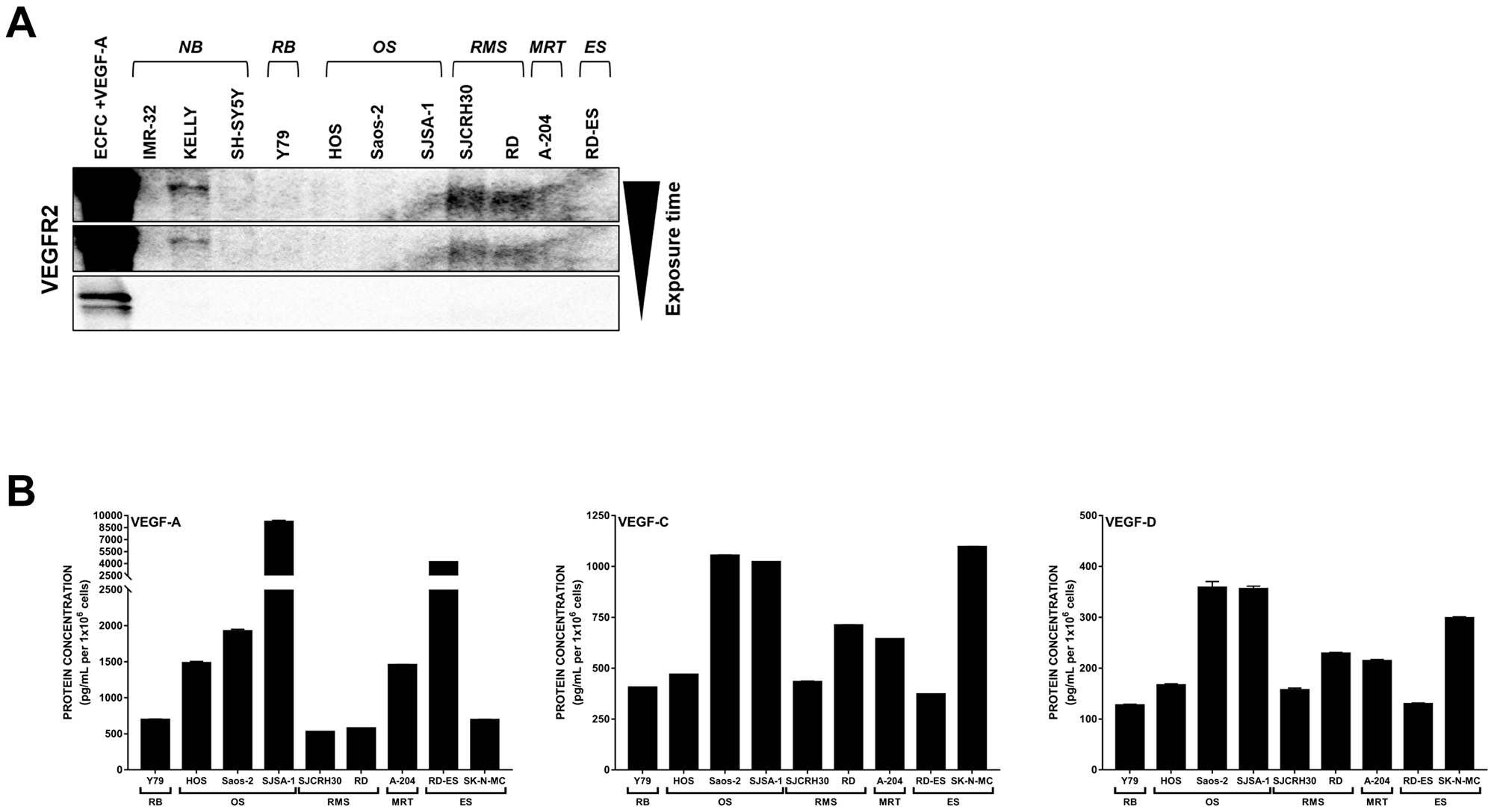
Figure 1: Expression of VEGFR2 and associated VEGFs are detected in pediatric cancer cell lines. (A) Eleven pediatric cancer cell lines were evaluated for endogenous VEGFR2 protein expression. (B) Endogenous levels of VEGF-A (left), -C (middle), and -D (right) protein produced by 9 pediatric cancer cell lines in co-culture conditions were assayed by ELISA. Error bars represent SEM. Note the bro ken y-axis for VEGF-A. Abbreviations: ECFC, endothelial colony forming cell; NB, neuroblastoma; RB, retinoblastoma; OS, osteosarcoma; RMS, rhabdomyosarcoma; MRT, malignant rhabdoid tumor; ES, Ewing’s sarcoma.
Tumor cells can activate VEGFR2 on endothelial cells (and thus promote neovascularization) through production and secretion of VEGF-A, -C, and -D. We previously determined that the neuroblastoma cell lines (IMR-32, KELLY, and SH-SY5Y) produce VEGF-A, -C, and -D in co-culture conditions [21]. We determined that these ligands were also present in media collected from each of the 9 additional non-neuroblastoma cell lines grown in co-culture conditions (Figure 1B). VEGF-A production was the most varied, from over 9000 pg/mL detected in SJSA-1 osteosarcoma media to approximately 525 pg/mL in the SJCRH30 alveolar RMS media. Conversely, VEGF-C levels were generally below 1000 pg/mL and VEGF-D was more uniformly expressed at concentrations below 400 pg/mL across all cell lines tested.
Ramucirumab impedes both VEGF- and tumor-driven cord formation
We next tested the ability of pediatric cancer cell lines to support in vitro endothelial cord formation [22]. As adipocyte derived stem cells (ADSCs) and ECFCs grown together in co-culture conditions were shown to produce a minimal amount of VEGF-A (approximately 40 pg/mL) [22], exogenous VEGF-A was used to drive cord formation in tumor cell-free wells. Indeed, the proangiogenic factors secreted by the panel of pediatric cancer cell lines could promote the formation of cords comparable to those achieved in VEGF-A-driven assays (Figure 2). Inhibition of VEGFR2 using the monoclonal antibody ramucirumab (Cyramza®, LY3009806) significantly blunted cord formation promoted by either tumor cell lines or VEGF-A, as measured by a significant reduction in total tube area (≥65%) with treatment compared to controls (Figure 2). Ramucirumab-mediated reduction in tumor-driven cord formation was not a result of cancer cell death (Supplementary Figure 1), consistent with the lack of target expression in the majority of tumor cell lines tested.
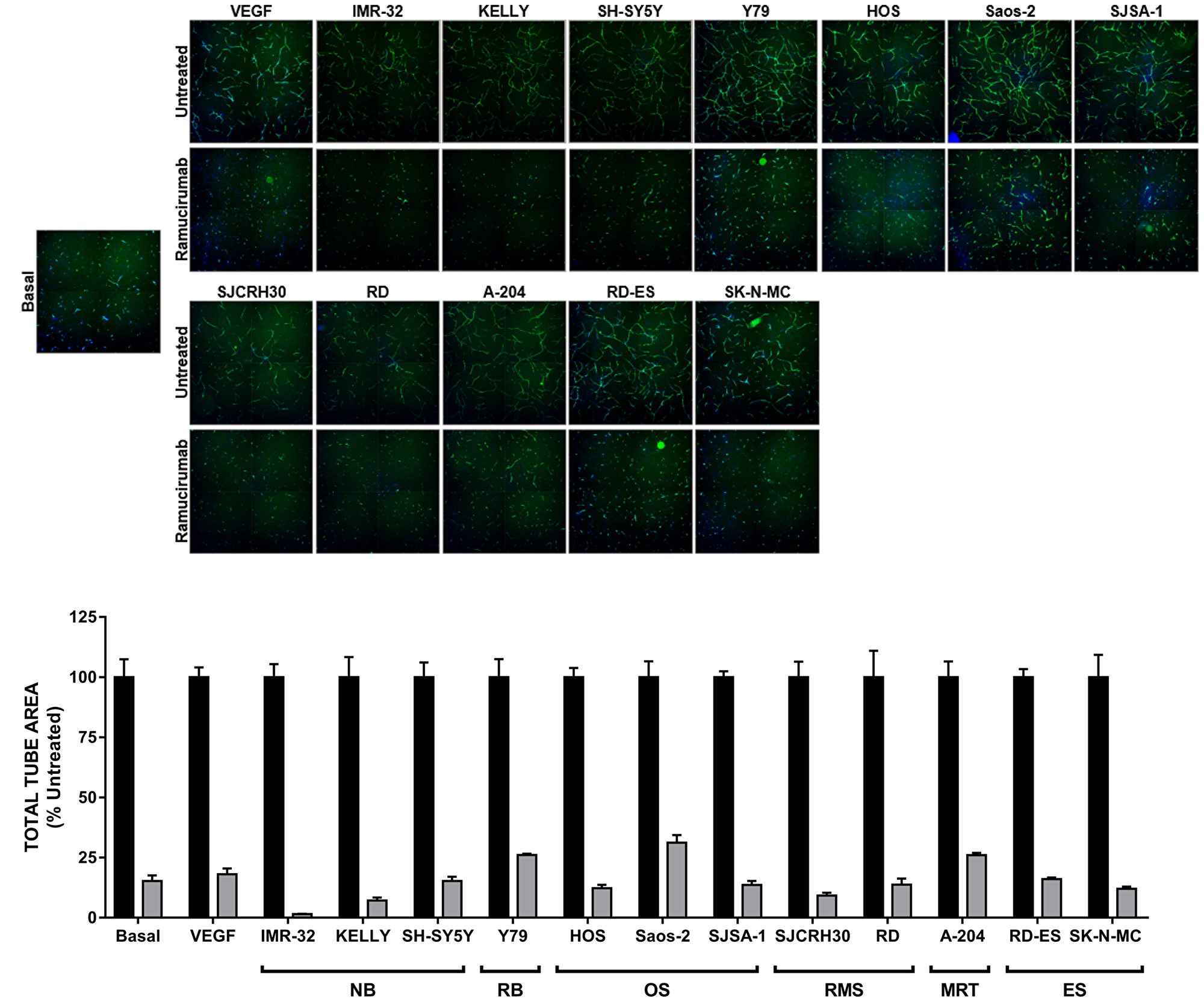
Figure 2: Ramucirumab blocks both VEGF-A- and pediatric tumor cell-driven cord formation in vitro. (Top) Representative images of cords from each condition are shown. (Bottom) Total tube area of VEGF-A- and tumor cell-driven cords is presented, with data for each cell line normalized to its respective untreated control. Black bars: untreated; gray bars: treated with 10 μg/mL ramucirumab. Error bars represent SEM.
DC101 is active as monotherapy or in combination with chemotherapy in a subset of pediatric bone and soft tissue tumor models
As ramucirumab does not cross react with mouse VEGFR2, the rat anti-mouse VEGFR2 antibody DC101 [23] was used to treat 8 cell line-derived (CDX) and 21 patient-derived (PDX) xenograft mouse models representing 10 extracranial pediatric solid tumor types (Table 1 and Supplementary Table 1). Animals were treated with 20 mg/kg DC101 twice weekly for up to 4 weeks, either as a single agent or in combination with chemotherapies typically used for pediatric cancer patients.
Table 1: Summary of in vivo pediatric tumor model studies
| Model | Xenograft type | Tumor type | Analysis day | Chemotherapy | DC101 | Chemo | Combination | Combination effect^ | |
|---|---|---|---|---|---|---|---|---|---|
| Agent | Dose, Schedule | % ΔT/C or regression on analysis day | |||||||
| SJCRH30 | CDX | alveolar RMS | 38 | Doxorubicin | 5 mg/kg, Q7Dx4 | 26 | 57 | 15†‡ | Additive |
| RD | CDX | embryonal RMS | 64 | Doxorubicin | 5 mg/kg, Q7Dx4 | 40 | 20 | 12† | Less than additive |
| CTG-1213 | PDX | embryonal RMS | 26 | Actinomycin D | 0.25 mg/kg, D0, 21 | 46 | 62 | 27‡ | Unknown |
| CTG-0926 | PDX | DSRCT | 28 | Doxorubicin | 5 mg/kg, Q7Dx4 | 60 | 42 | 22†‡ | Unknown |
| CTG-1458 | PDX | DSRCT | 35 | Cyclophosphamide | 100 mg/kg, Q7Dx4 | 50 | 42 | 8†‡ | Additive |
| RD-ES | CDX | Ewing’s sarcoma | 28 | Doxorubicin | 5 mg/kg, Q7Dx4 | 69 | 28 | -30†‡ | Additive |
| CTG-0142 | PDX | Ewing’s sarcoma | 28 | Doxorubicin | 5 mg/kg, Q7Dx4 | 44 | 38 | 11†‡ | Unknown |
| CTG-0785 | PDX | Ewing’s sarcoma | 18 | Doxorubicin | 5 mg/kg, Q7Dx4 | 37 | 46 | 41 | Less than additive |
| CTG-0816 | PDX | Ewing’s sarcoma | 35 | Doxorubicin | 5 mg/kg, Q7Dx4 | 3 | 3 | -49†‡ | Less than additive |
| CTG-0994 | PDX | Ewing’s sarcoma | 21 | Doxorubicin | 5 mg/kg, Q7Dx4 | 19 | 78 | 42‡ | Less than additive |
| CTG-1072 | PDX | hepatoblastoma | 17 | Cisplatin | 5 mg/kg, Q7Dx4 | 55 | 55 | 52 | No effect |
| A-204 | CDX | malignant rhabdoid tumor | 52 | Doxorubicin | 5 mg/kg, Q7Dx4 | 62 | 33 | 31 | No effect |
| IMR-32 | CDX | neuroblastoma | 70 | Doxorubicin | 3 mg/kg, Q7Dx4 | 47 | 30 | 16†‡ | Additive |
| KELLY | CDX | neuroblastoma | 49 | Doxorubicin | 2 mg/kg, Q7Dx3 | 46 | 98 | 48‡ | No effect |
| SH-SY5Y | CDX | neuroblastoma | 52 | Doxorubicin | 5 mg/kg, Q7Dx4 | 25 | 10 | -28 | No effect |
| CTG-0241 | PDX | osteosarcoma | 28 | Doxorubicin | 5 mg/kg, Q7Dx4 | 51 | 109 | 78 | No effect |
| CTG-0242 | PDX | osteosarcoma | 28 | Doxorubicin | 5 mg/kg, Q7Dx4 | 56 | 9 | 21 | No effect |
| CTG-0243 | PDX | osteosarcoma | 16 | Doxorubicin | 5 mg/kg, Q7Dx4 | 229 | 219 | 95†‡ | No effect |
| CTG-1064 | PDX | osteosarcoma | 27 | Doxorubicin | 5 mg/kg, Q7Dx4 | 53 | 157 | 41‡ | No effect |
| Y79 | CDX | retinoblastoma | 44 | Doxorubicin | 5 mg/kg, Q7Dx4 | 16 | 18 | -11 | Unknown |
| CTG-0331 | PDX | synovial sarcoma | 27 | Doxorubicin | 5 mg/kg, Q7Dx4 | 22 | 36 | 7‡ | No effect |
| CTG-1173 | PDX | synovial sarcoma | 52 | Gemcitabine Docetaxel | 60 mg/kg, Q7Dx4 6 mg/kg, Q7Dx4 | 79 | -43 | -64†‡ | Additive |
| CTG-1094 | PDX | undifferentiated sarcoma | 17 | Doxorubicin | 5 mg/kg, Q7Dx4 | 45 | 44 | 53 | No effect |
We observed that single agent DC101 significantly reduced tumor volumes compared to untreated animals in 13 of the 29 (45%) evaluated pediatric cancer models, though the responses were generally limited to tumor growth delay resulting in lower average tumor volume but not stable disease or tumor regression (Table 1). Three RMS models, one alveolar (SJCRH30) and two embryonal (RD and CTG-1213), responded to chemotherapy and DC101 as single agents. When the two drugs were combined, a significantly greater reduction in tumor volume was observed and some individual animals achieved stable disease (Figure 3A, Table 1). Similarly, all three in vivo mouse models of neuroblastoma were sensitive to DC101 monotherapy (Figure 3B, Table 1). While the reduction in KELLY xenograft volumes in the combination treatment arm could be attributed to DC101 alone (limited effects of doxorubicin single agent was observed in this model), improved responses with combination treatment were noted in the other two neuroblastoma models. Individual animals bearing IMR-32 or SH-SY5Y xenografts achieved stable disease or partial response and one animal with an SH-SY5Y tumor demonstrated a complete response when treated with DC101 and doxorubicin.
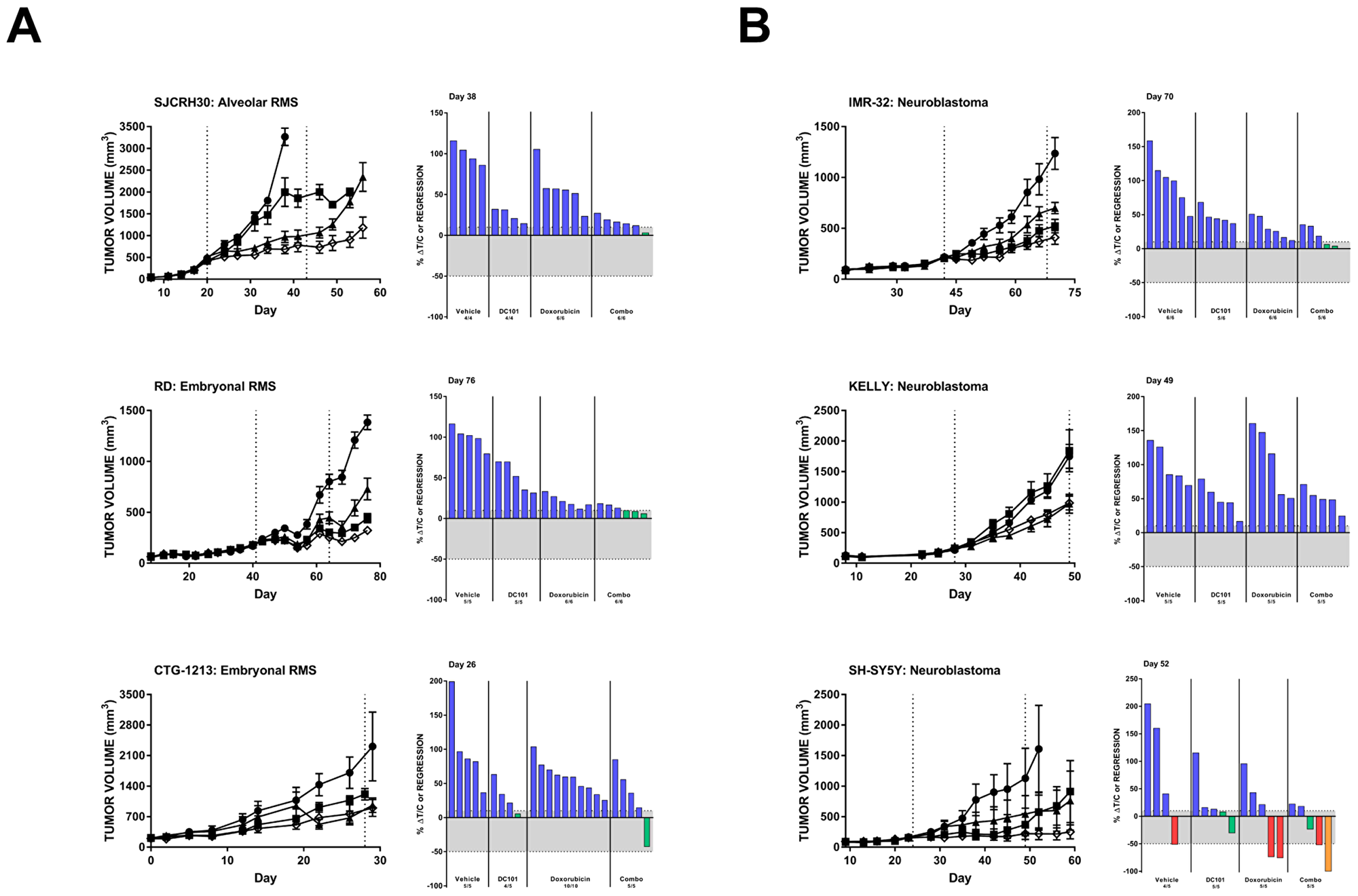
Figure 3: . DC101 monotherapy delays tumor growth in pediatric mouse models of rhabdomyosarcoma (A) and (B) neuroblastoma. All animals were treated with control (●), DC101 (▲), doxorubicin (■), or the combination (◇). Animals were treated when tumors reached approximately 200 mm3. The treatment interval is represented by dotted vertical lines except where treatment began at Day 0 in which case the end of treatment is indicated by a single dotted line at Day 28. Error bars represent SEM. Waterfall plots were generated on the day indicated in the top left corner of the image (generally, the last day the majority of vehicle animals were still evaluable). Blue bars: progressive disease (PD; ≥10% ΔT/C). Green bars: stable disease (SD; <10% ΔT/C and <50% regression). Red bars: partial regression (PR; ≥50% regression and tumor volume ≥ 14 mm3). Orange bars: complete regression (CR; tumor volume < 14 mm3). RMS: rhabdomyosarcoma.
The synovial sarcoma PDX model CTG-1173 was largely nonresponsive to DC101 monotherapy; however, 8/10 animals had tumor regression following gemcitabine/docetaxel treatment. Remarkably, this response was significantly enhanced with the addition of DC101 to gemcitabine/docetaxel (Figure 4A, top; Table 1). Another synovial sarcoma PDX model (CTG-0331) responded to both single agent DC101 and doxorubicin with a tumor growth delay (Figure 4A, bottom). The combination treatment resulted in a pronounced tumor growth delay which was sustained for approximately 3 weeks following the end of treatment. Both desmoplastic small round cell tumor (DSCRT) PDX models responded to single agent treatment with either DC101 or chemotherapy (either cyclophosphamide or doxorubicin) (Figure 4B). Combination treatment of DC101 and cyclophosphamide resulted in stable disease in animals bearing CTG-1458 tumors and DC101 also enhanced the anti-tumor activity of doxorubicin in the CTG-0926 PDX model.
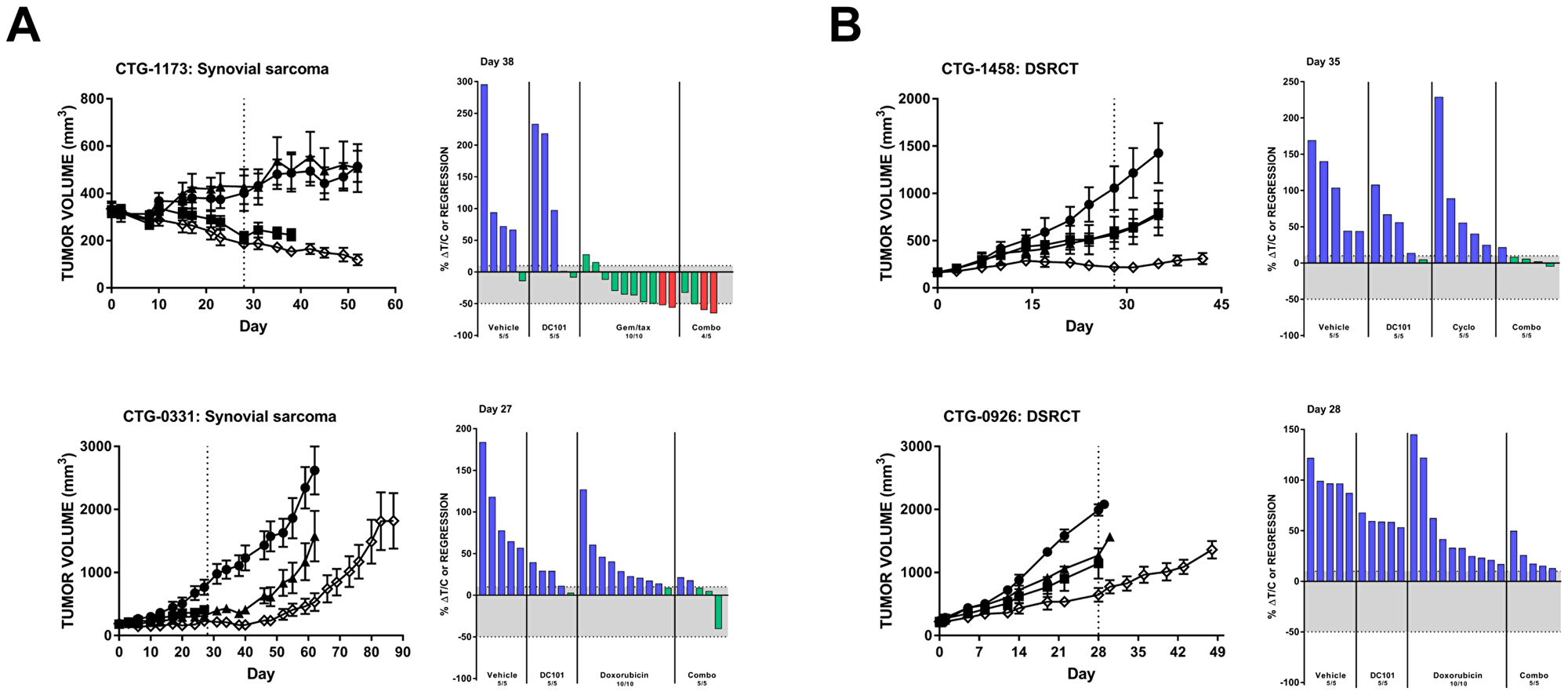
Figure 4: . DC101 enhances the effects of chemotherapy in preclinical models of (A) synovial sarcoma and (B) DSRCT. All animals were treated with control (●), DC101 (▲), chemotherapy (■; noted in waterfall plot), or the combination (◇). Treatment began at Day 0 and ended at Day 28 (dotted vertical line). For each panel, tumor growth curves are shown on the left and waterfall plots on the right. Error bars represent SEM. Waterfall plots were generated on the day indicated in the top left corner of the image (generally, the last day the majority of vehicle animals were still evaluable). Blue bars: progressive disease (PD; ≥10% ΔT/C). Green bars: stable disease (SD; <10% ΔT/C and <50% regression). Red bars: partial regression (PR; ≥50% regression and tumor volume ≥ 14 mm3). Abbreviations: DSRCT, desmoplastic small round cell tumor; Gem/tax, gemcitabine plus docetaxel.
Several Ewing’s sarcoma models responded to in vivo VEGFR2 inhibition (Tables 1 and 2). Significant single agent activity of DC101 was observed in CTG-0994, which was not further enhanced with the addition of doxorubicin (Figure 5A). Stable disease was observed in the CTG-0816 Ewing’s sarcoma PDX model following DC101 monotherapy and combination with doxorubicin resulted in two animals achieving a partial response (>50% regression) (Figure 5B). The combination of DC101 and doxorubicin was superior to either single agent in animals with CTG-0142 Ewing’s sarcoma xenografts (Figure 5C). Six additional PDX models of Ewing’s sarcoma were evaluated in an ‘n of 1’ design (Table 2). One model, CTG-1651, responded to both single agent DC101 and the combination of DC101 with doxorubicin with stable disease (8.4% ΔT/C and 12% regression, respectively). The CTG-1663 model responded to DC101 monotherapy, but this response was not enhanced with the addition of doxorubicin. Three models (CTG-2003, -2113, and -2174) were most sensitive to single agent doxorubicin treatment, while CTG-0143 did not respond to either DC101 or doxorubicin monotherapy and only demonstrated a slight reduction in tumor volume with the combination.
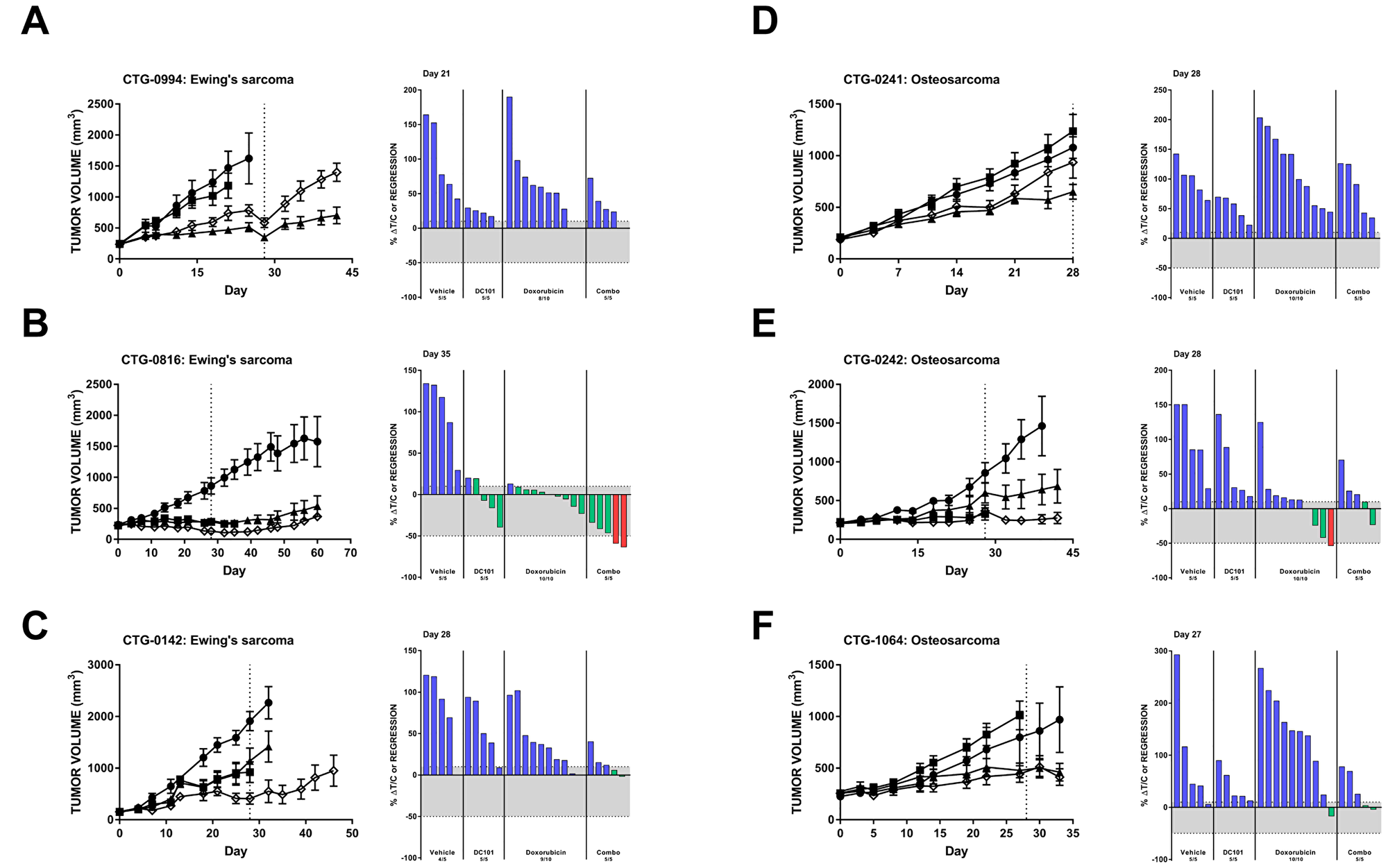
Figure 5: Preclinical models of Ewing’s sarcoma and osteosarcoma respond to DC101 alone and in combination with cytotoxic chemotherapy. All animals were treated with control (●), DC101 (▲), doxorubicin (■), or the combination (◇). Treatment began at Day 0 and ended at Day 28 (dotted vertical line). For each panel, tumor growth curves are shown on the left and waterfall plots on the right. Error bars represent SEM. Waterfall plots were generated on the day indicated in the top left corner of the image (generally, the last day the majority of vehicle animals were still evaluable). Blue bars: progressive disease (PD; ≥10% ΔT/C). Green bars: stable disease (SD; <10% ΔT/C and <50% regression). Red bars: partial regression (PR; ≥50% regression and tumor volume ≥ 14 mm3).
Table 2: Summary of Ewing’s sarcoma ‘n of 1’ study results
| Model | DC101 | Doxorubicin | Combination |
|---|---|---|---|
| % ΔT/C or regression* | |||
| CTG-0143 | 153 | 176 | 66.4 |
| CTG-1651 | 8.4 | 57.4 | −12.1 |
| CTG-1663 | 29.2 | 44 | 20.7 |
| CTG-2003 | 101 | 42.8 | 103 |
| CTG-2113 | 54.6 | 12.3 | 39.6 |
| CTG-2174 | 54.5 | 3.1 | 52.1 |
Additional pediatric tumor models were also interrogated for their response to DC101 with or without chemotherapy (Table 1). Only 1 out of 4 osteosarcoma models (CTG-0241) had a significant reduction in tumor growth with DC101 treatment alone (Figure 5D). Two additional osteosarcoma models demonstrated some reduction in tumor growth with DC101 monotherapy, but these responses were not statistically significant (Figure 5E and 5F). Combination treatment of DC101 and doxorubicin in these models did not significantly reduce tumor growth compared to vehicle and doxorubicin alone. A-204 malignant rhabdoid tumor, Y79 retinoblastoma, CTG-1072 hepatoblastoma, and CTG-1094 undifferentiated sarcoma models responded to DC101 and/or chemotherapy alone, but the combination did not improve response (Table 1).
Two preclinical mouse models respond to DC101 following initial treatment with doxorubicin
We explored the potential for DC101 to act as a maintenance therapy in a subset of PDX models of several histologies (Supplementary Table 1). Following 4 weeks of doxorubicin treatment, animals were split into two groups—one receiving no treatment and one receiving DC101 monotherapy. Two out of 14 models assessed responded to subsequent treatment with DC101 (Supplementary Figure 2). Pronounced inhibition of tumor growth was observed in the CTG-0331 synovial sarcoma model and tumor growth delay was noted in the CTG-0994 Ewing’s sarcoma model when DC101 was administered following completion of the doxorubicin treatment interval.
Collectively, our in vivo data demonstrate that DC101-mediated VEGFR2 inhibition was sufficient to delay tumor growth in alveolar and embryonal RMS, DSRCT, Ewing’s sarcoma, synovial sarcoma, and neuroblastoma. DC101 monotherapy enhanced the duration of tumor growth delay following an initial response to doxorubicin in one model of each synovial sarcoma and Ewing’s sarcoma. We also observed that the concurrent administration of DC101 and chemotherapy improved the anti-tumor response compared to control and chemotherapy alone in mice bearing models of alveolar RMS, embryonal RMS, DSRCT, Ewing’s sarcoma, neuroblastoma, and synovial sarcoma, but not osteosarcoma, hepatoblastoma, rhabdoid, or retinoblastoma.
DISCUSSION
The low incidence of molecular aberrations combined with the relative rarity of pediatric cancer and associated preclinical models often impedes the development and clinical evaluation of novel therapies specifically for pediatric indications [5, 24, 25]. Targeting the tumor microenvironment is a potential avenue to improve patient response to existing regimens consisting of chemotherapy and/or targeted agents with unique mechanisms-of-action. As VEGFR2 is a crucial mediator of tumor angiogenesis and is predominately expressed on the endothelium rather than tumors, it is a promising therapeutic target across different cancer types. Novel agents which target tumor angiogenesis have been assessed clinically across adult and pediatric malignancies, some of which are now FDA-approved for adult indications [12, 26, 27]. However, small molecule inhibitors may block multiple kinases, leading to off-target effects and associated toxicity [28] and to date no VEGFR2 pathway inhibitor has been approved for pediatric indications. In order to identify potential pediatric indications that may benefit from specific VEGFR2 inhibition, we tested ramucirumab or the anti-murine VEGFR2 antibody DC101 in multiple preclinical models of pediatric solid tumors. We demonstrated that ramucirumab abrogated both VEGF-A- and pediatric cancer cell-driven endothelial cord formation in vitro; furthermore, treatment with DC101 improved response to chemotherapy in several xenograft mouse models representing a range of pediatric tumor types with high unmet need.
VEGFR2, the target of ramucirumab and DC101, is primarily expressed on endothelial cells and is largely activated by tumor cell-secreted VEGF, spurring the onset of neovascularization. [29]. Indeed, we observed that the health of pediatric tumor cell lines was not affected by in vitro ramucirumab treatment despite the universal reduction in cord formation across all cell lines tested. This demonstrates the need for combination treatment of VEGFR2 pathway inhibition with a cytotoxic agent to hit both the tumor and the pathogenic tumor microenvironment. We did not expect DC101 alone to promote xenograft regression in our CDX models of pediatric cancer as it only targets the mouse receptor and considered stable disease or tumor growth delay to be indicative of DC101 monotherapy activity. Indeed, multiple pediatric tumor models met these criteria, including alveolar and embryonal RMS, DSRCT, Ewing’s sarcoma, neuroblastoma, and synovial sarcoma and tumor regression was only observed when DC101 was combined with doxorubicin or another cytotoxic agent. Therefore, VEGFR2 inhibition via ramucirumab treatment may provide additional clinical benefit when added to other cytotoxic agents, including those used during pediatric cancer treatment such as vincristine, irinotecan, and temozolomide [5] or targeted agents with unique mechanisms-of-action currently being explored in the pediatric population. Ramucirumab and DC101 were active across multiple pediatric histologies, thus demonstrating the relatively tumor type-agnostic activity of VEGFR2 inhibition. However, there were some pediatric indications such as osteosarcoma that appeared less sensitive to these combinations. The best combination partners for ramucirumab may vary across tumor types and between patients. In addition, tumors can employ one of multiple intrinsic or acquired escape mechanisms to circumvent the anti-tumor activity of VEGF pathway inhibitors [30–32]; however, detailing which of these mechanisms applies to each pediatric tumor model was beyond the scope of this study. Therefore, the efficacy of potential combinations as well as the identification of ramucirumab resistance would be best explored in a clinical setting.
To our knowledge, this preclinical study is the first exploring ramucirumab and DC101 in pediatric cancer models. Our data demonstrate that VEGFR2 inhibition, either as monotherapy or in combination with chemotherapy, promotes anti-tumor responses in several pediatric cancer models and further supports clinical investigation of ramucirumab in combination with other therapies for pediatric cancer patients.
Methods
Cell culture conditions
All cell lines were purchased from the American Type Culture Collection (ATCC) or DSMZ and grown in the media recommended by the respective vendor. Cell lines were maintained at 37° C in 5% CO2.
Test compound
Ramucirumab (LY3009806, Eli Lilly and Company) was diluted in PBS at a stock concentration of 8.9 mg/mL. The anti-murine VEGFR2 antibody DC101 (LY3180389, Eli Lilly and Company) was suspended in phosphate-buffered saline (PBS) for in vivo administration.
Western blot analysis
Cell lysis, SDS-PAGE, and immunoblotting were performed as previously described [21]. Cells pieces were lysed in 1% SDS (Fisher BioReagents, cat#BP2436-200) supplemented with 1x HALT protease and phosphatase inhibitor (ThermoFisher, cat#78441). The following antibodies were purchased from Cell Signaling Technology: VEGFR2 (cat#2479), VEGFR2 Y1175 (cat#2478), ERK1/2 (cat#4695), and ERK1/2 T202/Y204 (cat#4370). For protein assessment following ligand stimulation, cells were incubated with 10 ng/mL VEGF-A (R&D Systems cat#293VE) for 10 minutes prior to harvest and lysate generation.
Quantification of ligand production by cancer cell lines
Pediatric cancer cell lines were grown in adipocyte derived stem cell (ADSC) conditioned media, which are the co-culture conditions for cord formation. Conditioned media was collected at the 48 hour timepoint and analyzed using the MSD Multi-Spot Assay System Angiogenesis Panel 1 (MSD, cat#K15190D) per the manufacturers’ instructions.
In vitro cord formation assays
Cord formation assays were performed as previously described [21, 22, 33]. Ramucirumab was used at a final concentration of 10 μg/mL. VEGF-A (Invitrogen, cat#PHC9394) was used at 10 ng/mL in wells without tumor cells to serve as a positive control for cord formation, as a previous study demonstrated that ADSCs and endothelial colony forming cells grown in co-culture conditions produce approximately 40–50 pg/mL VEGF-A and thus need exogenous ligand for cord formation [22]. In addition, the amount of VEGF-A contributed by ADSCs and ECFCs was assumed constant across all conditions tested.
Cell viability was assessed following completion of the cord formation assay using the CellTiter Glo™ (CTG) Luminescent Cell Viability Assay (Promega, cat# G7571). Transwells containing tumor cells were removed and placed into a white, opaque bottom 96-well plate. Approximate volume was determined by measuring with a pipette, the appropriate volume of CTG reagent was added, and the CTG reagent/lysate solution was transferred to the white plate and read on a SpectraMax plate reader. For each cell line, luminescence was normalized to the average of the DMSO-treated control.
In vivo evaluation of DC101
Experiments involving animals were performed in accordance with American Association for Laboratory Animal Care institutional guidelines. In vivo studies using cell line-derived xenograft (CDX) models were approved by the Eli Lilly and Company Animal Care and Use Committee. In vivo experiments utilizing patient-derived xenograft (PDX) models designated by codes starting with ‘CTG’ were conducted at Champions Oncology (Hackensack, NJ, USA).
To evaluate DC101 and chemotherapy effects on CDX growth, cells were harvested during log phase growth and resuspended in Hank’s Balanced Salt Solution (HBSS). Suspended cells were diluted 1:1 with BD Matrigel Matrix (cat#356234; RD-ES, KELLY, IMR-32, SH-SY5Y) and 5 × 106 cells or 10 × 106 cells (RD-ES and RD) were injected subcutaneously into the right flank of female athymic nude mice. When tumor volumes averaged ~200 mm3, mice were randomized into treatment groups. Animals were given vehicle (20% Captisol™ in water, pH 4), DC101, chemotherapy (doxorubicin, cisplatin, cyclophosphamide, or gemcitabine + docetaxel), or a combination of DC101 and chemotherapy. Combination partners were dependent on the tumor model. DC101 (20 mg/kg, ip) was administered twice weekly for up to 4 weeks. Chemotherapy was given for 4 weeks at the following dose and schedules: doxorubicin, 5 mg/kg (unless otherwise noted) once weekly (Q7D; iv); cisplatin, 4 mg/kg once weekly (Q7D; ip); cyclophosphamide, 100 mg/kg once weekly (Q7D; ip); gemcitabine, 60 mg/kg once weekly (Q7D; ip) + docetaxel, 6 mg/kg once weekly (Q7D; ip). For DC101 maintenance therapy-related studies, animals received doxorubicin for 4 weeks. The doxorubicin group was then split into two equal groups: one received DC101 monotherapy at the same dose/schedule as described above, the other received vehicle control. Studies were terminated when the tumors from the “doxorubicin followed by vehicle” group reached a size which necessitated euthanasia. Additional details, including specific mouse strains and number of animals per arm, are located in Supplementary Table 1.
Tumor volume was transformed to a log scale to equalize variance across time and treatment groups. Log volume data was analyzed with a two-way repeated measures analysis of variance by time and treatment using the MIXED procedures in SAS software (Version 9.3). The correlation model for the repeated measures was Spatial Power. Treated groups were compared to the control group at each time point. The MIXED procedure was also used separately for each treatment group to calculate adjusted means and standard errors at each time point. Combinations were defined as additive if the combination arm was statistically different from both of the single agent arms using the BLISS independence method. Waterfall plots were generated on the day prior to splitting of treatment groups or the last day the majority of vehicle animals were able to be evaluated prior to sacrifice.
Abbreviations
VEGFR1: vascular endothelial growth factor receptor 1; VEGFR2: vascular endothelial growth factor receptor 2; VEGFR3: vascular endothelial growth factor receptor 3; VEGF-A: vascular endothelial growth factor A; VEGF-B: vascular endothelial growth factor B; VEGF-C: vascular endothelial growth factor C; VEGF-D: vascular endothelial growth factor D; PlGF: placental growth factor; RMS: rhabdomyosarcoma; CDX: cell line-derived xenograft; PDX: patient-derived xenograft; DSRCT: desmoplastic small round cell tumor.
CONFLICTS OF INTEREST
All authors are full-time employees of Eli Lilly and Company.
FUNDING
This study was funded by Eli Lilly and Company, Lilly Corporate Center, Indianapolis, Indiana, USA.
References
1. Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL, Anderson RN, Henley SJ, Kohler BA, et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J Natl Cancer Inst. 2017; 109:djx030. https://doi.org/10.1093/jnci/djx030. [PubMed].
2. Ou JY, Spraker-Perlman H, Dietz AC, Smits-Seemann RR, Kaul S, Kirchhoff AC. Conditional survival of pediatric, adolescent, and young adult soft tissue sarcoma and bone tumor patients. Cancer Epidemiol. 2017; 50:150–57. https://doi.org/10.1016/j.canep.2017.08.015. [PubMed].
3. Ceschel S, Casotto V, Valsecchi MG, Tamaro P, Jankovic M, Hanau G, Fossati F, Pillon M, Rondelli R, Sandri A, Silvestri D, Haupt R, Cuttini M. Survival after relapse in children with solid tumors: a follow-up study from the Italian off-therapy registry. Pediatr Blood Cancer. 2006; 47:560–66. https://doi.org/10.1002/pbc.20726. [PubMed].
4. Bhakta N, Liu Q, Ness KK, Baassiri M, Eissa H, Yeo F, Chemaitilly W, Ehrhardt MJ, Bass J, Bishop MW, Shelton K, Lu L, Huang S, et al. The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet. 2017; 390:2569–82. https://doi.org/10.1016/S0140-6736(17)31610-0. [PubMed].
5. Adamson PC. Improving the outcome for children with cancer: development of targeted new agents. CA Cancer J Clin. 2015; 65:212–20. https://doi.org/10.3322/caac.21273. [PubMed].
6. Apte RS, Chen DS, Ferrara N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell. 2019; 176:1248–64. https://doi.org/10.1016/j.cell.2019.01.021. [PubMed].
7. Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016; 15:385–403. https://doi.org/10.1038/nrd.2015.17. [PubMed].
8. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002; 29:15–18. https://doi.org/10.1053/sonc.2002.37263. [PubMed].
9. Oladipupo SS, Kabir AU, Smith C, Choi K, Ornitz DM. Impaired tumor growth and angiogenesis in mice heterozygous for Vegfr2 (Flk1). Sci Rep. 2018; 8:14724. https://doi.org/10.1038/s41598-018-33037-2. [PubMed].
10. Klein D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front Oncol. 2018; 8:367. https://doi.org/10.3389/fonc.2018.00367. [PubMed].
11. Arnold D, Fuchs CS, Tabernero J, Ohtsu A, Zhu AX, Garon EB, Mackey JR, Paz-Ares L, Baron AD, Okusaka T, Yoshino T, Yoon HH, Das M, et al. Meta-analysis of individual patient safety data from six randomized, placebo-controlled trials with the antiangiogenic VEGFR2-binding monoclonal antibody ramucirumab. Ann Oncol. 2017; 28:2932–42. https://doi.org/10.1093/annonc/mdx514. [PubMed].
12. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet. 2016; 388:518–29. https://doi.org/10.1016/S0140-6736(15)01088-0. [PubMed].
13. Fontanella C, Ongaro E, Bolzonello S, Guardascione M, Fasola G, Aprile G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann Transl Med. 2014; 2:123. https://doi.org/10.3978/j.issn.2305-5839.2014.08.14. [PubMed].
14. Wetmore C, Daryani VM, Billups CA, Boyett JM, Leary S, Tanos R, Goldsmith KC, Stewart CF, Blaney SM, Gajjar A. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: a children’s Oncology Group Study ACNS1021. Cancer Med. 2016; 5:1416–24. https://doi.org/10.1002/cam4.713. [PubMed].
15. Maris JM, Courtright J, Houghton PJ, Morton CL, Kolb EA, Lock R, Tajbakhsh M, Reynolds CP, Keir ST, Wu J, Smith MA. Initial testing (stage 1) of sunitinib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008; 51:42–48. https://doi.org/10.1002/pbc.21535. [PubMed].
16. Maris JM, Courtright J, Houghton PJ, Morton CL, Gorlick R, Kolb EA, Lock R, Tajbakhsh M, Reynolds CP, Keir ST, Wu J, Smith MA. Initial testing of the VEGFR inhibitor AZD2171 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008; 50:581–87. https://doi.org/10.1002/pbc.21232. [PubMed].
17. Chisholm JC, Merks JH, Casanova M, Bisogno G, Orbach D, Gentet JC, Thomassin-Defachelles AS, Chastagner P, Lowis S, Ronghe M, McHugh K, van Rijn RR, Hilton M, et al, and European paediatric Soft tissue sarcoma Study Group (EpSSG) and the European Innovative Therapies for Children with Cancer (ITCC) Consortium. Open-label, multicentre, randomised, phase II study of the EpSSG and the ITCC evaluating the addition of bevacizumab to chemotherapy in childhood and adolescent patients with metastatic soft tissue sarcoma (the BERNIE study). Eur J Cancer. 2017; 83:177–84. https://doi.org/10.1016/j.ejca.2017.06.015. [PubMed].
18. Berthold F, Hömberg M, Proleskovskaya I, Mazanek P, Belogurova M, Ernst A, Sterba J. Metronomic therapy has low toxicity and is as effective as current standard treatment for recurrent high-risk neuroblastoma. Pediatr Hematol Oncol. 2017; 34:308–19. https://doi.org/10.1080/08880018.2017.1373314. [PubMed].
19. Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int J Cancer. 2002; 97:393–99. https://doi.org/10.1002/ijc.1634. [PubMed].
20. Chau I, Peck-Radosavljevic M, Borg C, Malfertheiner P, Seitz JF, Park JO, Ryoo BY, Yen CJ, Kudo M, Poon R, Pastorelli D, Blanc JF, Chung HC, et al. Ramucirumab as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib: patient-focused outcome results from the randomised phase III REACH study. Eur J Cancer. 2017; 81:17–25. https://doi.org/10.1016/j.ejca.2017.05.001. [PubMed].
21. Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A, Stancato LF. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017; 23:4354–63. https://doi.org/10.1158/1078-0432.CCR-16-2876. [PubMed].
22. Falcon BL, Swearingen M, Gough WH, Lee L, Foreman R, Uhlik M, Hanson JC, Lee JA, McClure DB, Chintharlapalli S. An in vitro cord formation assay identifies unique vascular phenotypes associated with angiogenic growth factors. PLoS One. 2014; 9:e106901. https://doi.org/10.1371/journal.pone.0106901. [PubMed].
23. Prewett M, Huber J, Li Y, Santiago A, O’Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999; 59:5209–18. [PubMed].
24. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, Johann PD, Balasubramanian GP, Segura-Wang M, Brabetz S, Bender S, Hutter B, Sturm D, et al, and ICGC PedBrain-Seq Project, and ICGC MMML-Seq Project. The landscape of genomic alterations across childhood cancers. Nature. 2018; 555:321–27. https://doi.org/10.1038/nature25480. [PubMed].
25. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, Zhou X, Li Y, Rusch MC, Easton J, Huether R, Gonzalez-Pena V, Wilkinson MR, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018; 555:371–76. https://doi.org/10.1038/nature25795. [PubMed].
26. Zirlik K, Duyster J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol Res Treat. 2018; 41:166–71. https://doi.org/10.1159/000488087. [PubMed].
27. Ronca R, Benkheil M, Mitola S, Struyf S, Liekens S. Tumor angiogenesis revisited: regulators and clinical implications. Med Res Rev. 2017; 37:1231–74. https://doi.org/10.1002/med.21452. [PubMed].
28. Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006; 6:714–27. https://doi.org/10.1038/nrc1913. [PubMed].
29. Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012; 2:a006593. https://doi.org/10.1101/cshperspect.a006593. [PubMed].
30. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int J Mol Sci. 2018; 19:E1232. https://doi.org/10.3390/ijms19041232. [PubMed].
31. Zarrin B, Zarifi F, Vaseghi G, Javanmard SH. Acquired tumor resistance to antiangiogenic therapy: mechanisms at a glance. J Res Med Sci. 2017; 22:117. https://doi.org/10.4103/jrms.JRMS_182_17. [PubMed].
32. Falcon BL, Chintharlapalli S, Uhlik MT, Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol Ther. 2016; 164:204–25. https://doi.org/10.1016/j.pharmthera.2016.06.001. [PubMed].
33. Falcon BL, O’Clair B, McClure D, Evans GF, Stewart J, Swearingen ML, Chen Y, Allard K, Lee LN, Neote K, McEwen DP, Uhlik MT, Chintharlapalli S. Development and characterization of a high-throughput in vitro cord formation model insensitive to VEGF inhibition. J Hematol Oncol. 2013; 6:31. https://doi.org/10.1186/1756-8722-6-31. [PubMed].