
INTRODUCTION
Breast cancer of the most common malignancy among women in Europe and worldwide. Genetic factors play a substantial role in breast cancer etiology [1]. Specific inherited mutations in BRCA1 and BRCA2 increase the risk of female breast cancer. Together, BRCA1 and BRCA2 mutations account for about 5 to 10 percent of all breast cancers [2]. Individuals with BRCA1 mutations are predominantly predisposed to estrogen receptor (ER)-negative breast cancer, whereas other known susceptibility loci for breast cancer are stronger associated with ER-positive tumors [1]. Patients with ER-negative tumors have a worse short-term prognosis [3] and a weaker association with reproductive risk factors [4]. Furthermore, Li-Fraumeni patients with germline mutations of TP53 have an increased risk of developing breast cancer.
It is important to clarify the molecular mechanism of breast cancer development which can help to detect breast cancer at an early stage, and to study single nucleotide polymorphisms that affect the pathways which could be relevant for tumor formation and/or progression. Numerous studies with large patient cohorts such as the Breast Cancer Association Consortium (BCAC) have identified single nucleotide polymorphisms that are associated with the onset of breast cancer [5–8]. Genes that encode proteins with functions in the DNA damage response, such as TP53 and its key inhibitors MDM2 and MDMX [9], are most likely candidates to harbor allelic variants that influence breast cancer susceptibility.
The p53-pathway is essential for the cells’ adequate response to stress. In detail, p53 regulates transcriptional programs important in suppressing tumor formation and progression as well as the cellular response to certain therapies by regulating cell-cycle-arrest, cell death, metabolic processes, DNA repair, and others [10]. MDM2 and MDMX have indispensable roles in regulating the activity and the levels of p53 in both unstressed cells and following genotoxic stress [11]. By binding to p53, both MDM2 and MDMX inhibit p53’s function as a transcription factor. Furthermore, MDM2 also mediates the proteasomal degradation of p53 [12] by serving as an E3 ligase [13]. On the other hand, p53 mediates transcription of the MDM2 and MDMX gene through p53-sensitive promoters thereby forming a fine-tuned negative-feedback loop [14–16].
Several aspects of the p53-MDM2-MDMX-axis are particularly relevant to human cancer. The TP53 gene is mutated in nearly half of all sporadic human cancers; however, there are great differences among tumor types [17]. Importantly, tumors expressing wild-type p53 are often characterized by overexpression of MDM2 or MDMX, due to gene amplification or other mechanisms, particularly in breast cancer [18, 19]. Varying transcription levels, and the subsequent biological outcome, can be explained by the impact of different alleles of single nucleotide polymorphisms (SNPs) in the MDM2 and MDMX gene. In this context, Bond et al. have identified a SNP in the p53-sensitive P2-promoter of the MDM2 gene – designated as SNP309 – which affects the risk of developing cancer [20] in a gender-specific and hormone-dependent manner [21]. Subsequently, however, conflicting results have been published regarding the association between SNP309G and increased cancer-risk, specifically in Caucasian populations. For example, Knappskog et al. could not reproduce these findings in the context of breast and ovarian cancer development [22]. Interestingly, the group described a second SNP 24 bp upstream of SNP309 in the MDM2 P2 promoter (SNP285) and showed that the increased transcription caused by the G-allele of SNP309 is reduced by the C-allele of SNP285, whereby SNP285C and SNP309G are inherited as haplotype. Therefore, the risk of breast and ovarian cancer is decreased in SNP285C/SNP309G-haplotype carriers [22].
Similarly to MDM2, MDMX is often amplified and overexpressed in tumors (reviewed in [23]). As described for MDM2, specific SNPs in the MDMX gene may affect the activity of p53 and the tumor-risk [24]. Recently, we identified a SNP (SNP34091, rs4245739) in the 3′-UTR of the MDMX gene. The C-allele of rs4245739 creates a binding site for hsa-miR-191 and hsa-miR-887 which causes downregulation of the MDMX expression in the respective tissue [25]. In ovarian cancer, the A/A-genotype is associated with a significantly shortened disease-free and overall survival dependent on the ER-status of the tumor [25]. Several genome-wide association studies (GWAS) revealed that the 1q32-locus, including SNP34091 (rs4245739), is a susceptibility locus in ER-negative breast cancer [26] and triple-negative breast cancer [27, 28].
In the study presented here, we assessed the impact of SNPs in the TP53, MDM2, and the MDMX gene on the age-at-diagnosis and event-free survival in a prospective cohort of German breast cancer patients (n = 815) and correlated these data with (i) the TP53-mutational status, (ii) the hormone receptor and HER2 status as surrogate markers for the intrinsic subtypes as well as (iii) clinic-pathological data. Our data clearly indicate allele-specific effects of SNPs in critical regulators of p53 on the age-at-diagnosis as well as the event-free survival, depending on the ER-status, respectively on the intrinsic subtypes as well as the TP53-mutational status. Our findings provide evidence for the distinct etiological pathways in invasive ER-negative and ER-positive breast cancers.
RESULTS
TP53 mutational status
In a subset of 257 patients from the cohort the TP53 mutational status was determined by NGS. Here, in 16.7% of the patients (43/257) mutations in the TP53 were detected. All mutations were located in exons 4 through 8 and affected the amino acid sequence of p53. 79.1% (34/43) were missense mutations and 20.9% (9/43) were nonsense mutations which a single nucleotide exchange leading to a premature stop codon (Table 1, Figure 1). According to the TP53 mutations database [29] all mutations that were detected in our cohort have already been described in the context of breast cancer.
Table 1: Summary of TP53 mutations in breast cancer
Case |
Histology |
Exon |
Codon |
Conserved region |
Mutation Type |
Change |
wt |
mt |
wt-AA |
mt-AA |
|---|---|---|---|---|---|---|---|---|---|---|
#188 |
basal |
4 |
107 |
NC |
Nonsense |
C>G |
TAC |
TAG |
Tyr |
STOP |
#229 |
basal |
4 |
110 |
NC |
Missense |
G>C |
CGT |
CCT |
Arg |
Pro |
#161 |
basal |
4 |
111 |
NC |
Missense |
T>C |
CTG |
CCG |
Leu |
Pro |
#277 |
Lum B |
5 |
126 |
NC |
Missense |
T>A |
TAC |
AAC |
Tyr |
Asn |
#287 |
HER2 |
5 |
127 |
Co |
Missense |
C>T |
TCC |
TTC |
Ser |
Phe |
#158 |
HER2 |
5 |
132 |
Co |
Missense |
A>G |
AAG |
AGG |
Lys |
Arg |
#225 |
HER2 |
5 |
143 |
Co |
Missense |
G>A |
GTG |
ATG |
Val |
Met |
#396 |
Lum B |
5 |
157 |
Co |
Missense |
G>A |
GTC |
ATC |
Val |
Ile |
#306 |
basal |
5 |
157 |
Co |
Missense |
G>T |
GTC |
TTC |
Val |
Phe |
#361 |
Lum A |
5 |
163 |
Co |
Nonsense |
C>A |
TAC |
TAA |
Tyr |
STOP |
#516 |
HER2 |
5 |
163 |
Co |
Missense |
A>G |
TAC |
TGC |
Tyr |
Cys |
#146 |
basal |
5 |
165 |
NC |
Nonsense |
C>T |
CAG |
TAG |
Gln |
STOP |
#339 |
Lum B |
5 |
173 |
Co |
Missense |
G>T |
GTG |
TTG |
Val |
Leu |
#209 |
Lum B |
5 |
173 |
Co |
Missense |
T>C |
TGT |
GCG |
Val |
Ala |
#226 |
Lum A |
5 |
175 |
Co |
Missense |
G>A |
CGC |
CAC |
Arg |
His |
#224 |
Lum A |
5 |
175 |
Co |
Missense |
G>A |
CGC |
CAC |
Arg |
His |
#404 |
Lum A |
5 |
176 |
Co |
Missense |
G>T |
TGC |
TTC |
Cys |
Phe |
#344 |
Lum B |
5 |
177 |
Co |
Missense |
C>T |
CCC |
TCC |
Pro |
Ser |
#208 |
Lum B |
5 |
178 |
Co |
Missense |
C>A |
CAC |
AAC |
His |
Asn |
#368 |
Lum B |
5 |
179 |
Co |
Missense |
A>G |
CAT |
CGT |
His |
Arg |
#425 |
Lum A |
5 |
183 |
NC |
Nonsense |
C>G |
TCA |
TGA |
Ser |
STOP |
#528 |
Lum B |
5 |
185 |
NC |
Missense |
A>G |
AGC |
GGC |
Ser |
Gly |
#451 |
basal |
6 |
192 |
NC |
Nonsense |
C>T |
CAG |
TAG |
Gln |
STOP |
#297 |
HER2 |
6 |
194 |
Co |
Missense |
T>G |
CTT |
CGT |
Leu |
Arg |
#267 |
basal |
6 |
197 |
NC |
Missense |
T>A |
GTG |
GAG |
Val |
Glu |
#345 |
Lum A |
6 |
213 |
Co |
Nonsense |
C>T |
CGA |
TGA |
Arg |
STOP |
#309 |
basal |
6 |
213 |
Co |
Nonsense |
C>T |
CGA |
TGA |
Arg |
STOP |
#174 |
Lum A |
6 |
213 |
Co |
Nonsense |
C>T |
CGA |
TGA |
Arg |
STOP |
#541 |
basal |
6 |
213 |
Co |
Nonsense |
C>T |
CGA |
TGA |
Arg |
STOP |
#408 |
Lum A |
6 |
214 |
Co |
Missense |
A>G |
CAT |
CGT |
His |
Arg |
#268 |
Lum B |
6 |
220 |
NC |
Missense |
A>G |
TAT |
TGT |
Tyr |
Cys |
#480 |
Lum B |
7 |
232 |
NC |
Missense |
A>T |
ATC |
TTC |
Ile |
Phe |
#220 |
Lum A |
7 |
241 |
Co |
Missense |
C>G |
TCC |
TGC |
Ser |
Cys |
#228 |
Lum B |
7 |
245 |
Co |
Missense |
G>A |
GGC |
AGC |
Gly |
Ser |
#148 |
Lum B |
7 |
249 |
Co |
Missense |
A>G |
AGG |
GGG |
Arg |
Gly |
#450 |
Lum A |
7 |
258 |
NC |
Missense |
G>A |
GAA |
AAA |
Glu |
Lys |
#159 |
Lum B |
7 |
259 |
NC |
Missense |
G>T |
GAC |
TAC |
Asp |
Tyr |
#360 |
Lum B |
7 |
259 |
NC |
Missense |
A>T |
GAC |
GTC |
Asp |
Val |
#302 |
basal |
8 |
273 |
NC |
Missense |
G>A |
CGT |
CAT |
Arg |
His |
#344 |
Lum B |
8 |
276 |
Co |
Missense |
C>G |
GCC |
GGC |
Ala |
Gly |
#332 |
basal |
8 |
282 |
Co |
Missense |
C>T |
CGG |
TGG |
Arg |
Trp |
#316 |
Lum B |
8 |
294 |
NC |
Nonsense |
del |
FS |
|||
#383 |
Lum B |
8 |
306 |
NC |
Nonsense |
del |
FS |
Abbreviations: basal – triple-negative (ductal); HER2 – HER2-positive (non-luminal); Lum A – luminal A-like; Lum B – luminal B-like (HER2-independent); NC – non-conserved region of the TP53 gene; Co – highly conserved region of the TP53 gene; FS – frameshift; del – deletion.
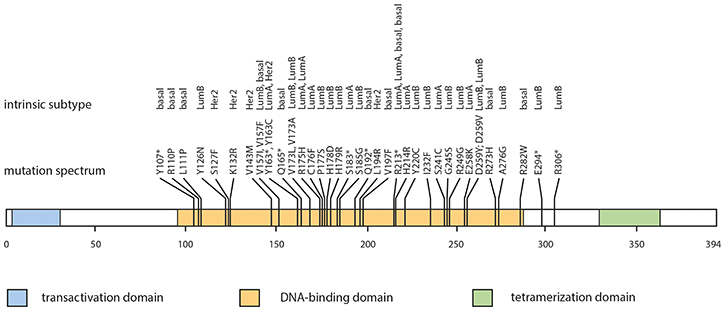
Figure 1: TP53 mutation spectrum according to intrinsic subtype. Mutation plot of somatic TP53 mutations (missense and nonsense mutations) in breast cancer. Abbr.: R110P – wild-type amino acid, codon, altered amino acid; Y107* - wild-type amino acid, altered codon, STOP; Bas – triple-negative (ductal); Her – HER2-positive (non-luminal); LuA – luminal A-like; LuB – luminal B-like.
Depending on the intrinsic cancer subtype frequency of TP53 mutations varied from 11.9% (9 out of 84) in Luminal A-like tumors, 20% (17 out of 85) in Luminal B-like (HER2-independent), 37.9% (11 out of 29) in triple-negative tumors and 55.5% (5 out of 9) in HER2-positive (non-luminal) tumors. Statistic evaluation revealed that TP53 mutational status was associated with HER2-overexpession (p = 0.018, binary-logistic regression) and the lack of ER- and PR-expression (p < 0.001; p < 0.001, respectively; binary-logistic regression; Supplementary Table 1).
Impact of TP53 germline variations in TP53 mutated and unmutated breast cancer
In our study, the percentages harboring the three different genotypes of the Arg72Pro-SNP were recorded to be: G/G 49.2% (406/815); G/C 43.8% (351/815), and C/C 7.0% (51/815). The genotype frequencies were found to be in Hardy-Weinberg-equilibrium (p > 0.95) and were comparable to the distribution in European individuals of the 1000 Genomes Project [30]. Correlation with the clinical data revealed that presence of the C-allele was significantly associated with higher histological grade of the primary tumor (OR = 1.615; p = 0.032; Supplementary Table 2). However, there was no association of the Arg72Pro-SNP with the age-at-diagnosis (p = 0.198; variance test).
By separating the patients according to their TP53 mutational status, we found that in patients whose tumors were wild-type for the TP53 gene the homozygous Arg-allele (91/210) was significantly associated with a later age-at-diagnosis compared with patients who carried at least one Pro-allele (66.2 vs. 59.7 yrs.; p < 0.001; log-Rank-test; Figure 2). This effect was not observed in patients with a mutated TP53 gene (62.3 vs. 60.3 yrs.; p = 0.827; log-Rank-test).
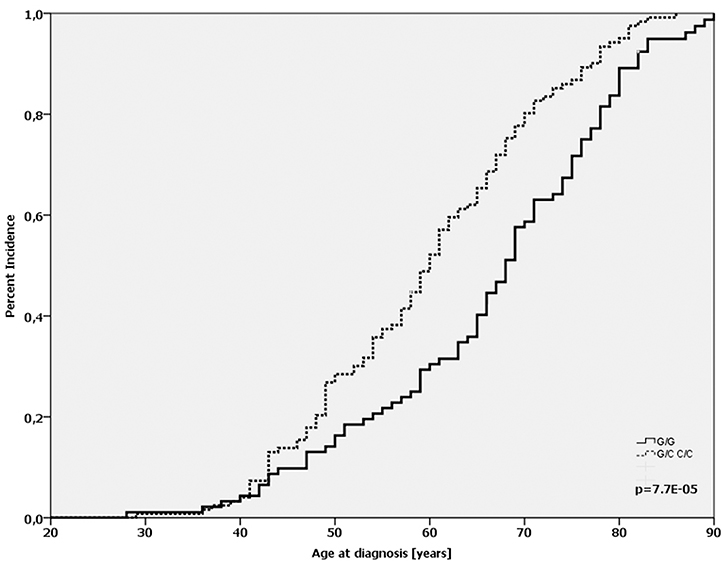
Figure 2: Age-at-diagnosis of the first breast cancer for patients with the different genotypes of the TP53 Arg72Pro-SNP (G/G vs. G/C+C/C) and TP53 wild-type gene.
We next explored potential associations of the respective genotypes of the TP53 Arg72Pro with differential times to recurrence and survival after diagnosis. As mentioned above, the TP53 Arg72Pro-SNP is not associated with the age-at-diagnosis in our breast cancer patient cohort. The same applies for the impact of the respective alleles on the event-free survival (Supplementary Table 3). There was no difference regarding the time to local or distant recurrence between patients with different genotypes of the TP53 Arg72Pro-SNP (p = 0.356, log-Rank-test). In addition, the TP53 mutational status also did not affect the event-free survival time. Although patients with TP53 wild-type tumors had an average event-free survival of 78 months compared with 70.6 months for patients with TP53 mutated tumors, the difference was not significant (p = 0.311; log-Rank-test).
MDM2 - SNP285 and SNP309 (rs2279744 and rs117039649)
The allelic status of MDM2 SNP309 was successfully determined in 815 cases. The T/T genotype was detected in 45.3% of the patients (369 of 815), 42.9% (350 of 815) were heterozygous (T/G), and 11.8% (96 of 815) of the patients were homozygous for the G-allele (G/G). The genotype frequencies MDM2 SNP309 were in Hardy-Weinberg-equilibrium (p > 0.9) and comparable to those found in European individuals of the 1000 Genomes Project [30]. The distribution of the genotypes did not differ among tumor grades and histological subtypes. We also did not detect an association with the ER- and PR-status of the patient’s tumor (p = 0.177, p = 0.74, respectively; binary-logistic regression; Supplementary Table 4).
The G-allele of SNP309 has been shown to be associated with an earlier age-at-diagnosis in numerous tumor types, including breast cancer (Review in [9]). Therefore, we compared the age when the tumor was diagnosed in the patients of our cohort according to their genotype. There was no difference regarding the age-at-diagnosis for patients with the respective genotypes of SNP309. Breast cancer patients with the T/T genotype were diagnosed at 61.9 years, T/G patients at 62.6 years, and patients who carried the homozygous G-allele at 61.6 years, respectively (p = 0.826, log-Rank-test; Supplementary Table 5).
Since it has been shown that SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner [21], we analyzed patients with ER-negative (n = 126) and ER-positive (n = 689) tumors separately. This revealed that the average age-at-diagnosis of patients with the T/T-T/G genotype whose tumors were ER-negative was 55.4 years compared with 62.1 years for G/G-patients (p = 0.091; log-Rank-test). Surprisingly, there was no difference regarding the age-at-diagnosis in patients with ER-positive tumors (62.3 vs. 62.5 yrs; p = 0.986; log-Rank-test). The distribution of the SNP309 genotypes was comparable between ER-negative and ER-positive tumors.
Next, we looked for an association between SNP309, the TP53 mutational status and the age-at-diagnosis. We found that in patients with a mutated TP53 gene, the T/T-status of SNP309 was significantly associated with an earlier age-at-diagnosis compared with T/G-G/G-patients (56.9 vs. 65.2 yrs; p = 0.04; log-Rank-test; Supplementary Table 5). In patients with TP53 wild-type tumors, no difference was observed (T/T: 62 yrs., T/G-G/G: 62.8 yrs.; p = 0.668; log-Rank-test). A striking difference of 14.7 years regarding the age-at-diagnosis was observed in a subgroup of patients (n = 23) whose tumors were ER-positive and harbored a mutated TP53 gene. Patients with a homozygous T-allele were diagnosed on average at the age of 53.5 years compared with 68.2 years for T/G-G/G-patients (p = 0.002; log-Rank-test).
In order to assess the potential impact of SNP309 on the luminal vs. non-luminal subtypes of breast cancer, we compared the frequency of SNP309 genotypes in the respective subgroups, which were surrogate definitions of intrinsic subtypes determined by immunohistochemistry (Table 2). We observed a slightly higher frequency of the T/T-genotype in triple-negative (ductal) tumors (54.3%) compared with luminal A-like, luminal B-like (HER2-independent), and HER2-positive (non-luminal) tumors (44.9%, 43.5%, 45.9%, respectively), though this association was not significant. We next explored, whether the genotypes of SNP309 are associated with the age-at-diagnosis within the histopathologically determined subtypes. In luminal A-like and luminal B-like (HER2-independent) breast cancer, the different alleles of SNP309 had no impact on the age-at-diagnosis (p = 0.966, p = 0.911, respectively; log-Rank-test). In the HER2-positive (non-luminal) subtype, however, the T/T-genotype of SNP309 (n = 17) was associated with a significantly earlier age-at-diagnosis compared with the T/G-G/G-genotypes (57.8 vs. 63.3 yrs; p = 0.016; log-Rank-test). In contrast, in the triple-negative (ductal) subgroup the G/G-patients (n = 8) had an average age-at-diagnosis of 51 years compared with 63 years for SNP309T carriers (p = 0.004; log-Rank-test).
Table 2: Surrogate definitions of intrinsic subtypes
Subtype |
Characteristics |
|---|---|
Luminal-A-like |
HR positive |
Luminal-B-like (HER2-independent) |
HR positive |
HER2 positive |
HR negative |
triple negative tumors |
HR negative |
Furthermore, we assessed the potential impact of MDM2 SNP285 alone or in combination with MDM2 SNP309. Our patient cohort consisted exclusively of Caucasian women and the distribution of respective alleles matched to published frequencies observed in people cohorts from Norway and the Netherlands [22]. 92.6% (764 of 825) of the cases were homozygous for the G-allele, 6.9% (57 of 825) were heterozygous G/C, and 0.5% (4 of 825) carried the homozygous C-allele The genotype frequencies MDM2 SNP285 were in Hardy-Weinberg-equilibrium (p > 0.95). The C/C genotype was associated with an increased tumor grade (OR = 1.67; p = 0.044; Supplementary Table 6). Comparing the age of onset revealed that patients with SNP285 C/C (n = 4) were diagnosed on average at the age of 44.2 (95% CI: 37.9 – 48.1), whereas the average age-at-diagnosis of G/G patients was 62.4 and 62.7 for G/C patients, respectively (p = 0.0007; log-Rank-test; Figure 3, Supplementary Table 7). It must be emphasized that only four patients carried the C/C genotype (see above); nonetheless, the difference is highly significant. Next, we evaluated the combination of SNP285 and SNP309 on the age of onset among breast cancer patients. The MDM2 SNP 309 G/G genotype was observed in 11.8% of patients (96 of 815) and only 4 of these showed the homozygous SNP285C/309G haplotype. Also in this subgroup SNP285 C/C dramatically reduced the age of onset only among patients with a SNP309 G/G status (44.2 years vs. 61.4 years in all patients with SNP 309 GG). There was neither a correlation of the C/C gene status with the immunohistochemically determined intrinsic subtype (p = 0.336; chi-square-test) nor with the ER-status of the tumor (p = 0.15, chi-square-test). Despite this low age-at-diagnosis, only one patient was diagnosed with recurrence after 18 months. No event was detected in the remaining C/C-patients.
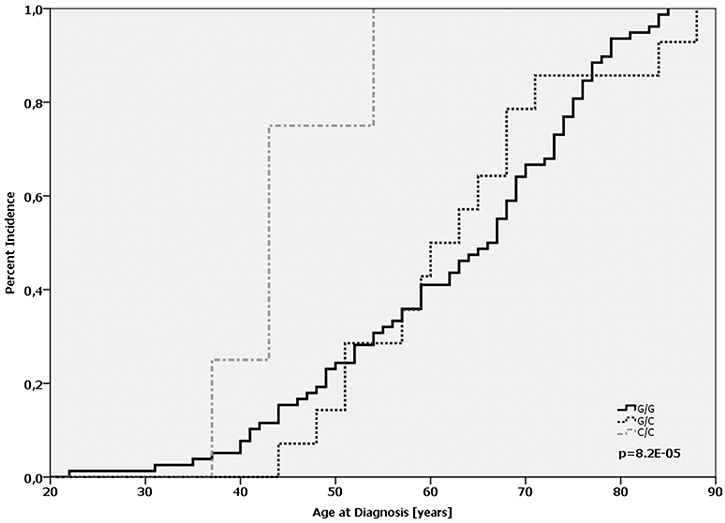
Figure 3: Age-at-diagnosis of the first breast cancer for patients with the different genotypes of MDM2 SNP285 (G/G vs. G/C vs. C/C).
The comparison of the event-free survival for patients with the different genotypes of the MDM2 SNP309 revealed that none of the alleles was associated with the time span until the occurrence of local or distant recurrence (p = 0.357; log-Rank-test). In addition, we could not find any association of the alleles of SNP309 with the time to recurrence when we separated the patients according to their status of ER- (p = 0.487; log-Rank-test), and HER2-expression (p = 0.329; log-Rank-test) as well as TP53 mutation (p = 0.303; log-Rank-test). Interestingly, patients with triple-negative (ductal) tumors and a homozygous T-allele of SNP309 had an event-free survival of 58.9 months compared with 66.2 months for patients with at least one G-allele, though the difference was not significant (p = 0.227; log-Rank-test). In contrast, the respective genotypes of SNP309 were not associated with either shortened or prolonged event-free survival within luminal A-like, luminal B-like (HER2-independent), and HER2-positive subtypes.
MDMX SNP31826
The allelic status of MDMX SNP31826 was analyzed in 815 cases. The percentages harboring the three different genotypes were recorded to be 40.6% (331 of 815), 51.8% (422 of 815), and 7.6% (62 of 815), respectively. The genotype frequencies MDMX SNP31826 were in Hardy-Weinberg-equilibrium (p > 0.95) and comparable to those found in European individuals of the 1000 Genomes Project [30]. There were no significant associations of MDMX SNP31826 and clinico-pathological parameters such as tumor grading, histological subtype and hormone receptor status (Supplementary Table 8). However, patients with the homozygous A-allele were characterized by a 1.3-fold elevated risk for infiltrations of lymphatic vessels (p = 0.045; binary-logistic regression; Supplementary Table 8).
In our cohort, there was no statistically significant difference regarding the average age-at-diagnosis between the MDMX genotypes (p = 0.284; log-Rank-test; Supplementary Table 9). For the homozygous wild-type (G/G), the heterozygote (G/A) and the homozygous variant (A/A) the average age-at-diagnosis was 62.6, 61.5 and 63.3 years, respectively. In the subgroup of ER-positive tumors (n = 689), no difference regarding the age-at-diagnosis was observed (p = 0.28; log-Rank-test; Supplementary Table 9). In ER-negative tumors (n = 126), however, there was a left shift in the cumulative incidence curve corresponding to a 10.6 years earlier age-at-diagnosis for patients with the A/A-genotype (n = 6) compared with the G/G-genotype (n = 53); however, the difference was not significant (p = 0.121; log-Rank-test). Notably, the difference was 11.2 years when ER-positive and ER-negative patients with the A/A-genotype were compared (64.4 vs. 53.2 yrs; p = 0.025, log-Rank-test; Figure 4). Interestingly, women with the homozygous A-allele were at least 40 years old when they were diagnosed with breast cancer. Taken together, these results underline the data published by Kulkarni et al., that MDMX SNP31826 is significantly linked to an earlier onset of disease in ER-negative breast cancer [31].
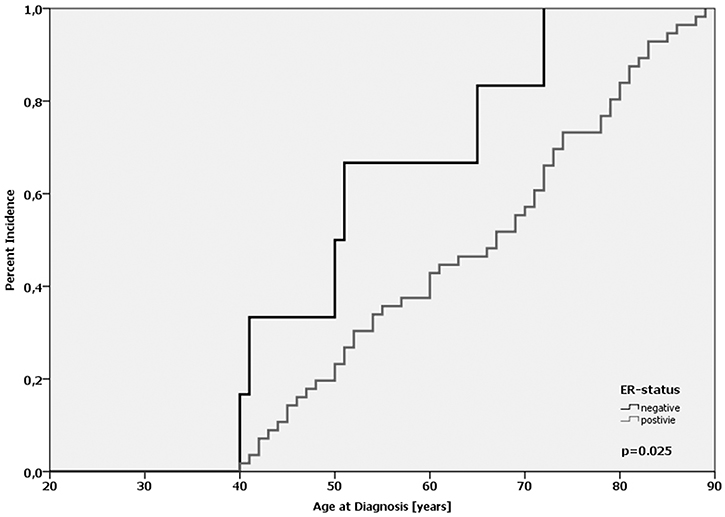
Figure 4: Age-at-diagnosis of the first breast cancer for patients with the A/A-genotype of MDMX SNP31826 and different ER-expression status.
The TP53 mutational status did not associate with a different age-of-diagnosis in patients with the respective MDMX SNP31826 alleles. In patients whose tumors harbor a wild-type TP53 gene, the average age-at-diagnosis was 61.5, 63.1 and 61.3 years for the homozygous wild-type (G/G), the heterozygote (G/A) and the homozygous variant (A/A) (p = 0.734; log-Rank-test), and 58 and 62.3 years for the homozygous wild-type (G/G) and the heterozygote (G/A) allele in p53-mutated tumors (p = 0.729; log-Rank-test; Supplementary Table 9). Furthermore, we identified a subgroup of patients – albeit small – that is characterized by a notable early age-of-diagnosis. Since the risk for breast cancer is related to age at menopause, we analyzed the age-of-diagnosis in patients before the age of 55. Our data revealed that the average age-at-diagnosis of patients with ER-negative, p53-mutated tumors and the G/G-genotype of MDMX SNP31826 was 37 years compared with 50 years for G/A-patients (p = 0.001; log-Rank-test). This was not observed in patients with ER-positive tumors, TP53-mutated tumors (p = 0.998; log-Rank-test) nor in patients with TP53-wild-type tumors irrespective of the ER-status (data not shown).
The average event-free survival time for patients with luminal B-like (HER2-independent) tumors and a homozygous A-allele of MDMX SNP31826 was 62.3 months compared with 70.6 months for patients with a homozygous G-allele (p = 0.012; log-Rank-test; Figure 5B). In contrast, there was no difference for patients with luminal A-like tumors and the respective genotypes (p = 0.723; log-Rank-test; Figure 5A). Interestingly, in triple-negative (ductal) tumors, no events occurred in the subgroup of patients with homozygous A-allele. The average event-free survival for all triple-negative (ductal) patients was 63.3 months. There was no impact of the respective MDMX SNP31826 alleles on the event-free survival within other subgroups, e.g. patients with different ER- or PR-expression status.
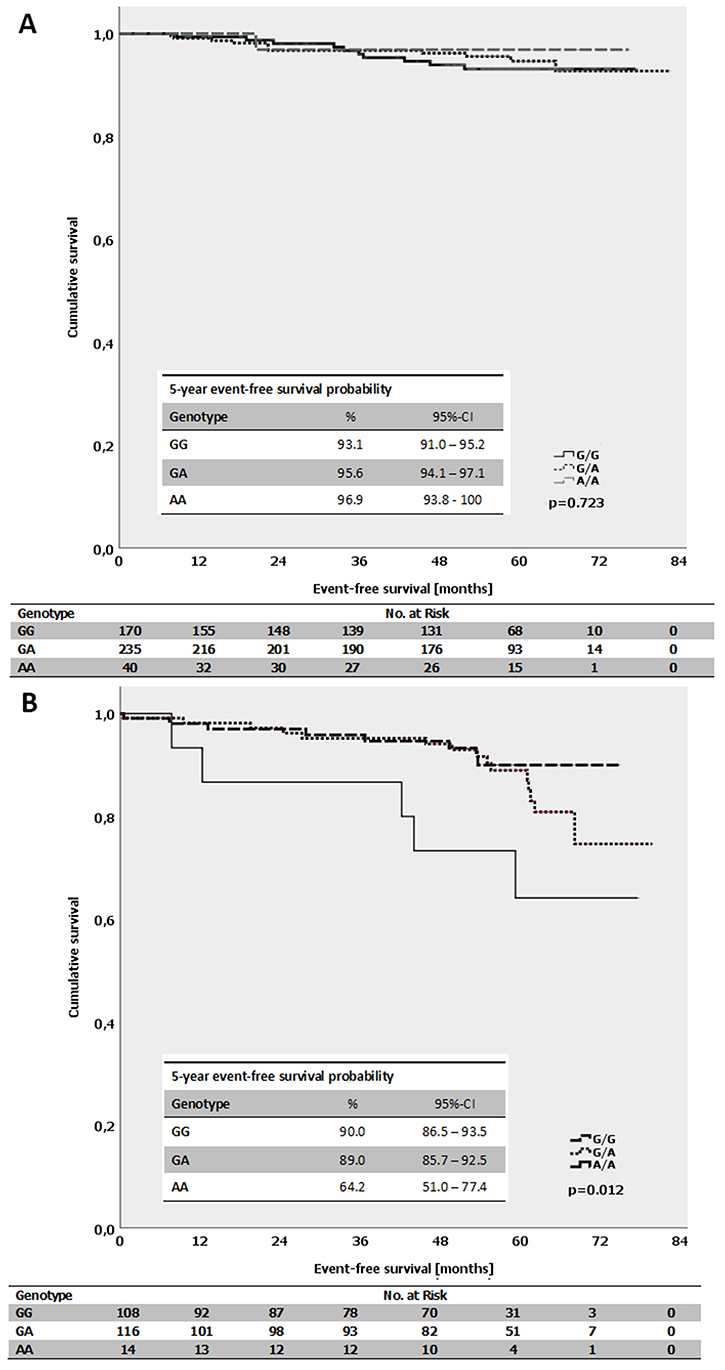
Figure 5: (A) Kaplan-Meier survival estimates for the event-free survival for patients with the different genotypes of the MDMX SNP31826 (G/G vs. G/A vs. A/A) in patients with luminal A-like subtype. (B) Kaplan-Meier survival estimates for the event-free survival for patients with the different genotypes of the MDMX SNP31826 (G/G vs. G/A vs. A/A) in patients with luminal B-like subtype (HER2-independent).
With respect to the influence of the respective alleles of MDMX SNP31826 on the age-at-diagnosis and event-free survival, there were no significant differences in our patient cohort (Supplementary Table 9).
MDMX SNP34091
The allelic status of MDMX SNP34091 was determined in 815 breast cancer patients. The wild-type A/A-genotype accounted for 52.3% of the patients (426 of 815), 36.6% were heterozygous A/C (332 of 815), and 7% were homozygous for the C-allele (57 of 815). This is comparable to the allelic distribution found in healthy individuals (57.4%, 36.6%, 6%) [25]. The genotype frequencies MDMX SNP34091 were in Hardy-Weinberg-equilibrium (p > 0.5). The 1q32 locus where the MDMX gene resides has been shown to be a risk-factor of ER-negative breast cancer [26, 28]. We therefore asked whether the impact of the respective alleles of MDMX SNP34091 might be dependent on the ER-expression status of the tumor. We show that the A/A-genotype is associated with a 1.8-fold increased risk to develop an ER-positive tumor compared with genotypes with at least one C-allele (p = 0.042; binary-logistic regression, Supplementary Table 10).
MDMX SNP34091 A/A+A/C patients were diagnosed on average at the age of 62.2 years, whereas C/C patients at the age of 64.6 years. This difference, however, was not significant (p = 0.222; log-Rank-test; Supplementary Table 11). When the patients were separated according to their ER-status of the tumor, no significant impact of the respective alleles of MDMX SNP34091 on the age-at-diagnosis was observed (ER-negative: p = 0.633; ER-positive: 0.219; log-Rank-test, Supplementary Table 11). Interestingly, among pre-menopausal patients whose tumors were ER-negative the C/C-genotype was significantly associated with an earlier age-at-diagnosis compared with ER-positive patients (40 vs. 46.4 yrs; p = 0.001; log-Rank-test). In contrast, no difference regarding the age-at-diagnosis was observed in pre-menopausal A/A-patients (p = 0.325; log-Rank-test) and in post-menopausal patients (A/A: p = 0.762; C/C: p = 0.816; log-Rank-test).
By evaluating the impact of the alleles of MDMX SNP34091 on the event-free survival in breast cancer patients, we found that A/A-patients had an average time until recurrence of 77.1 months compared with 67.3 months for C/C-patients (p = 0.219; log-Rank-test; Supplementary Table 11). A notable difference was observed in the luminal B-like (HER2-independent) but not in the luminal A-like and triple-negative (ductal) subtype. In luminal B-like breast cancer patients (HER2-independent), the C/C-genotype was associated with a significantly shortened event-free survival of only 54.8 months, compared with 74 months for A/A-patients (p < 0.001; log-Rank-test; Figure 6B). In contrast, the respective alleles of MDMX SNP34091 did not affect the event-free survival in patients with luminal A-like tumors (p = 0.806; log-Rank-test; Figure 6A). The A/A-genotype was also significantly associated with a prolonged event-free survival compared with C/C-patients in the subgroup of ER-positive (luminal) tumors (A/A: 79 months, C/C: 68 months; p = 0.04; log-Rank-test). However, while the event-free survival in the ER-negative subgroup (non-luminal) was shorter in general, the average time to recurrence was comparable between the A/A- and C/C-patients (A/A: 64 months, C/C: 60 months; p = 0.844; log-Rank-test).
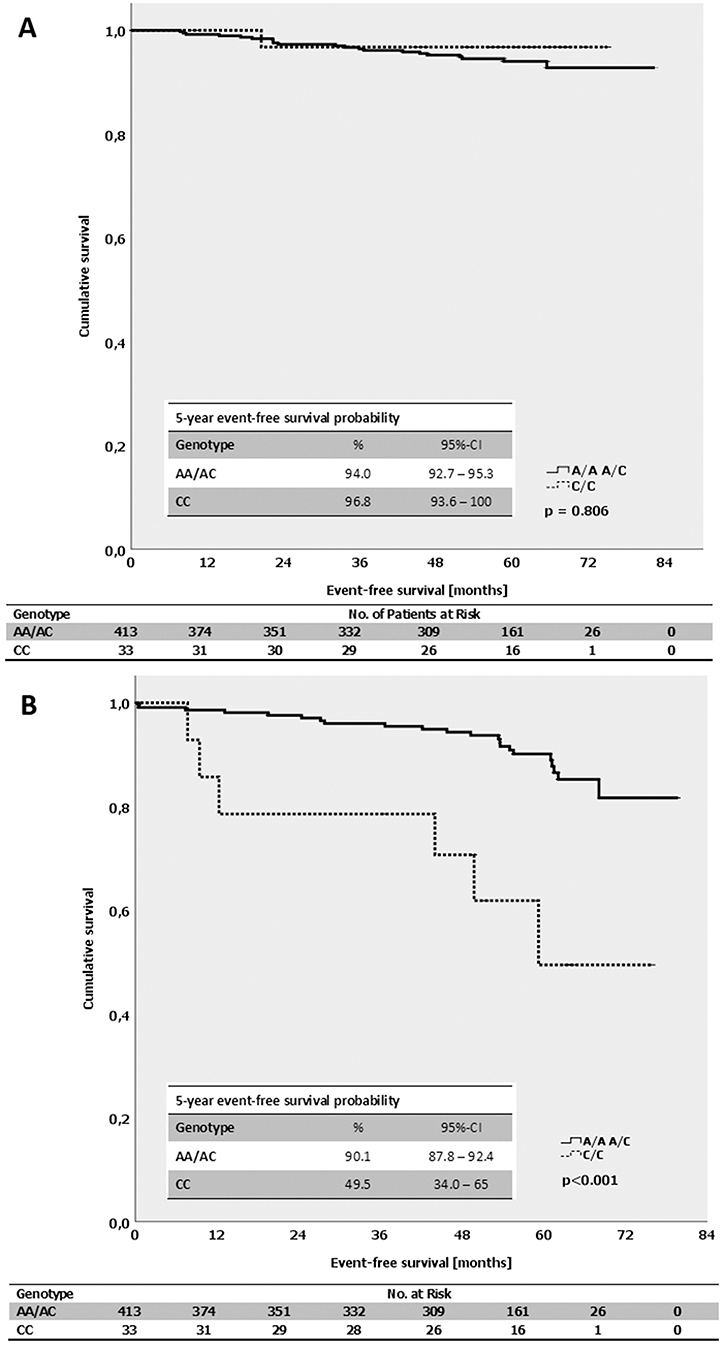
Figure 6: (A) Kaplan-Meier survival estimates for the event-free survival for patients with the different genotypes of the MDMX SNP34091 (A/A+A/C vs. C/C) in patients with luminal A-like subtype. (B) Kaplan-Meier survival estimates for the event-free survival for patients with the different genotypes of the MDMX SNP34091 (A/A+A/C vs. C/C) in patients with luminal B-like subtype (HER2-independent).
To further define the influence of MDMX SNP34091 on age-at-diagnosis and event-free survival, we performed subgroup analysis stratified by the St. Gallen risk criteria which are briefly described in Table 3 according to Goldhirsch et al. [32]. Classification of patients with low, intermediate and high risk is based on clinical (age) and histopathological parameters like nodal status, hormone receptor status, and TNM [32]. Our results show that average age-at-diagnosis of patients with the C/C-genotype in the low-risk group was 51.7 years compared with 71.6 years for patients whose tumors were classified as “high risk” (p = 0.022; log-Rank-test; Supplementary Table 11). There was no difference regarding the age-at-diagnosis for A/A- and A/C-patients and the St. Gallen risk groups (p = 0.118; p = 0.310, respectively; log-Rank-test). Unexpectedly, the C/C-genotype in the high-risk group was associated with the shortest event-free survival time of 55.8 months compared with 68.2 months for patients in the intermediate risk-group. In addition, A/A-patients within the intermediate risk group had an event-free survival of 78.7 months, however, the difference was not significant (p = 0.130; log-Rank-test). No event-free survival time could be calculated for patients within the low risk-group since all cases were censored.
Table 3: Risk assessment according to the St. Gallen risk criteria
Risk level |
Characteristics |
|---|---|
Low risk |
all of the following criteria: |
Intermediate risk |
pN0 and at least one of the following criteria: |
High risk |
pN > 1 |
DISCUSSION
In this descriptive, case-only study design of breast cancer patients, we examined the association of SNPs at the 1q32- and 12q13-loci and in the TP53 gene with the onset and progression of breast cancer in a large German patient cohort with extensive clinical and pathological annotations. The study design employing solely breast cancer tissue probes was chosen, since no pathogenetic point mutations with matching exchanges in the localizations under study have been described until now. While the TP53 Arg72Pro-SNP and the MDM2 SNP309 have been widely studied in numerous tumor types [33], this is – to our knowledge – the first comprehensive study determining the effect of SNPs at the 1q32-locus on the onset and event-free survival in breast cancer.
Since the breast is one of the most dynamic tissues of the body, the tight control of cell proliferation during breast morphogenesis is important. Both members of the MDM family – MDM2 and MDMX – are necessary for the development and function of healthy breasts [reviewed in [34]]. The main function is dynamic negative regulation of the tumor suppressor p53. In the absence of stress, in healthy tissue, p53 levels are kept low by MDM2 and MDMX [reviewed in [35]]. In response to stress signals, however, p53 is relieved from the inhibitory effects of the MDM proteins, resulting in the accumulation of p53 and allowing it to activate appropriate pathways to ensure genomic integrity. Consistently, in normal cells of the breast duct, p53 is barely detectable while MDM2 and MDMX are at high levels [36]. It has been shown that imbalances in the expression levels of p53, MDM2 and MDMX result in the disruption of the finely tuned regulatory feedback loop. This, subsequently, might increase the risk for breast cancer [37]. Different allelic variants of the MDM2 and MDMX gene have been shown to associate with altered mRNA and protein expression levels in tumors [20, 25].
The G-allele of SNP309 has been linked to early age-at-diagnosis in numerous cancer types, for example in women with soft tissue sarcomas and colorectal carcinomas [20, 21] as well as patients with ER-positive ovarian carcinomas [38]. In breast cancer, however, no association with an increased risk or earlier age-at-diagnosis was detected in a meta-analysis comprising 5,836 cases [33] and another German study [39, 40]. This is in line with the results presented here. There was no correlation of SNP309 with the age-at-diagnosis and event-free survival in our patient cohort, even after the patients were separated according to their ER-expression status. It can be concluded that the effect of the respective alleles of SNP309 is very small, and is only present in subgroups. A strength of this study is the availability of information of all study participants from pathology and clinical reports, and therefore our statistical evaluations included subtype analysis. Interestingly, the wild-type T-allele of SNP309 was associated with earlier age-at-diagnosis in ER-negative intrinsic subtypes (HER2-positive [non-luminal], triple-negative [ductal]) but not in ER-positive subtypes (luminal A-like-and luminal B-like [HER2-independent]), as well as, in patients with TP53-mutated but not in TP53-wild-type tumors. However, there was no effect of MDM2 SNP309 on the event-free survival of the patients in none of subgroups.
The conflicting results regarding the impact of the MDM2 SNP309 G-allele can partly be explained by a second distinct SNP in the MDM2 P2 promoter. Knappskog et al. discovered a SNP, MDM2 SNP285, which is located 24 bp upstream from MDM2 SNP309. The C-allele of SNP285 is located on the G-allele of SNP309, forming a SNP285C/309G haplotype [22]. It has been shown that the G-allele of SNP309 increased the affinity of the transcription factor Sp1 to the MDM2 P2 promoter, leading to elevated MDM2 expression and increased cancer risk [20]. SNP285C, in contrast, significantly reduces the Sp1-binding the P2-promoter and subsequently transcription of the MDM2 gene [22]. Consistent with this finding, individuals harboring the SNP285 C-allele had a decreased risk of breast and ovarian cancer, while the most profound effect was found in breast cancer patients with homozygous SNP309 G-allele. In contrast, in our study the C/C-genotype was associated with an extremely earlier age-at-diagnosis for patients harboring the G/G-genotype of SNP309. There was no effect of the heterozygous SNP285 C-allele (n = 57) on the age-at-diagnosis irrespective of the SNP309 allele-status. This is surprising since as one would expect at least slight effects of the C-allele in individuals with SNP285GC-SNP309TG+GG-genotype as it has been observed by Knappskog et al. [22]. On the other hand, however, the MDM2 SNP285CC/309GG-genotype is associated with a favorable outcome, since only one patient was diagnosed with recurrence after 18 months. Interestingly, all MDM2 SNP285 C/C patients were in the St. Gallen intermediate-risk group. Therefore, it seems reasonable to assume the SNP285 status is an additional factor for risk stratification for patients in the intermediate risk group.
Like SNP309 in the MDM2 gene, SNPs in the MDMX gene are apparently correlated with early age-at-diagnosis and risk of breast cancer dependent on the ER-expression status [26, 27, 31, 41]. In our patient cohort, the homozygous A-allele of MDMX SNP31826 associates with an 11 years earlier age-at-diagnosis only in ER-negative breast cancers. This confirms results published by Kulkarni et al. [31]. In addition, the effect that the A-allele of SNP31826 is correlated with early tumor onset is restricted to the triple-negative (ductal) subtype and was not observed in patients luminal A-like or luminal B-like (HER2-independent) breast cancer. Previous data suggested that MDM2 SNP309 is functionally active in the presence of estrogen signaling [20, 21] while the effects of MDMX SNP34091 are dominant in the absence of hormone signaling [25]. Recent studies, however, revealed that ERα physically interacts with both the MDM2 and the MDMX gene, and that ERα expression correlates with MDM2 and MDMX gene expression independent of p53 [42]. Furthermore, the authors demonstrate that expression of MDM2 and MDMX mRNA is elevated in ER-positive breast cancer samples, such as luminal A-like and luminal B-like (HER2-independent), compared with ER-negative subtypes (HER2-positive [non-luminal], triple-negative [ductal]). This suggests that effect of the MDMX SNPs becomes more dominant in the absence of active hormone signaling, and that subtle alterations of MDMX expression due to the different alleles may have greater impact in tumors with lack of ERα expression. Given the fact that MDM2 and MDMX have well-characterized functions in breast cancer formation and progression (reviewed in [34]), the ERα-mediated, p53-independent upregulation of MDM2 and MDMX will likely enhance these tumor-promoting processes [42]. This establishes a regulatory feedback-loop between MDM2, MDMX, and ERα. It is conceivable that specific allelic variants of MDM2 and MDMX add another layer to this fine-tuned crosstalk in breast cancer. Furthermore, these data, collectively, could explain the different effect of MDM2 and MDMX SNPs in the distinct breast cancer subtypes.
Three interesting observations have been made regarding the impact of the MDMX SNP34091 on the age-at-diagnosis and event-free survival of breast cancer patients. Firstly, while there was no difference regarding the age-at-diagnosis of the respective genotypes for patients with ER-positive tumors, we found that C/C patients with ER-negative tumors were diagnosed either at the age of 40 or above the age of 70. Secondly, a notable difference of the event-free survival was detected only for patients with luminal B-like (HER2-independent) tumors but no other intrinsic subtypes. The C/C-genotype was associated with a significantly shortened event-free survival time of 54 months in these patients. Luminal B-like tumors are characterized by a greater percentage of TP53 mutations, MDM2 amplification, and higher proliferation compared to luminal A-like tumors [43]. This could, at least partly, explain the subtype-specific influence of the MDMX SNP34091 C/C genotype. Furthermore, C/C-patients had the shortest event-free survival time (51 months) in the St. Gallen low risk group. This is on average two years shorter than A/A-patients. The underlying mechanisms of the association of the MDMX SNP34091 C-allele with early tumor onset and shorter event-free survival have yet to be determined. Nonetheless, our data suggest to further elucidate its role as a risk factor in tumor prevention and as a predictor of recurrent disease.
Breast cancer is a heterogeneous disease. Attempts have been taken to define clinical and/or molecular subtypes of the disease and to elucidate driver events that are selected for during tumorigenesis. Curtis et al. analyzed copy number variations, SNPs and gene expression in a large cohort of breast cancer patients [44]. It has been shown that both inherited and acquired somatic alterations were associated with expression in 40% of the genes. The list of identified driver genes included both MDM2 and MDMX. Furthermore, the chromosomal regions of the MDM2 and MDMX gene are amplified in the majority of the integrated clusters identified by Curtis et al. [44]. It is conceivable that the amplification of the genes is the main reason for the overexpression of MDM2 and MDMX, and, therefore, the impact of different alleles is predominantly observable in tumors devoid of inherited or somatic copy number aberrations. High levels of MDM2 have been detected in 38% of human breast cancer cases [45]. This cannot be explained by gene amplification alone. Numerous other mechanisms contribute to increased MDM2 expression and/or activity in breast cancer, such as ERα-expression, downregulation of p14ARF, TGF1-b1-expression, as well as, expression of MDMX isoforms (reviewed in [34]).
In conclusion, we have found that specific allelic variants of the p53 tumor suppressor pathway are associated with earlier age-at-diagnosis and shortened event-free survival in subgroups of breast cancer patients. These results gained by these easily accessible and measurable biomarkers may be used in the future to identify individuals with an increased risk of developing breast cancer and to predict the responsiveness of conventional and targeted therapies. Especially, the early onset of pre-menopausal breast cancer is thought to be strongly associated with genetic predisposition. Targeted sequencing of variants in other known breast cancer susceptibility genes or genome-wide sequencing of germline variations [26, 27, 46] may provide additional clues to identify individuals with increased risk of early-onset breast cancer. Our findings need to be confirmed in independent cohorts in a subtype-specific manner and functional studies are mandatory to determine the molecular mechanisms underlying the observed effects.
MATERIALS AND METHODS
Patients and tissues samples
Human primary breast cancer tumors were collected at the Martin-Luther-University Halle-Wittenberg as part of the multicenter prospective PiA study (Prognostic assessment in routine Application, NCT 01592825) of unselected patients with operable and histopathological confirmed invasive breast cancer. The fresh-frozen tumor samples were derived from five different certified breast cancer centers in Germany (Martin-Luther University Halle-Wittenberg, Hospital Fuerth, Hospital St. Elisabeth & St. Barbara Halle [Saale], Breast Center Hildesheim, Breast Center Goslar [Harz]). The study was approved by the ethics committee of the Martin-Luther University Halle-Wittenberg (15.09.2009 and 10.3.2010 for patient recruitment, 15.09.2016 for this sub protocol). The PiA-study is in accordance with the Declaration of Helsinki. All patients gave their written informed consent. For tumor and patients characteristics refer to Table 4. Breast cancer subtypes were defined using histopathological information like receptor status according to the St. Gallen classification (Tables 2, 3) and by von Minckwitz and colleagues [47, 48].
Table 4: Summary of clinicopathologic data of patients with breast cancer
Characteristics |
Patients (815) |
||
|---|---|---|---|
No. (%) |
|||
age at diagnosis |
median |
62.2 yrs. |
|
< 55 yrs. |
270 |
(33.1) |
|
>/= 55 yrs. |
545 |
(66.9) |
|
T stage (primary tumor) |
pT1 |
420 |
(51.5) |
pT2 |
349 |
(42.8) |
|
pT3 |
39 |
(4.8) |
|
pT4 |
7 |
(0.9) |
|
N stage (lymph node) |
N0 |
501 |
(61.5) |
N1 |
231 |
(28.3) |
|
N2 |
51 |
(6.3) |
|
N3 |
3 |
(3.9) |
|
M stage (distant metastases) |
M0 |
815 |
(100.0) |
M1 |
0 |
(0.0) |
|
lymph vessel invasion |
L0 |
592 |
(72.6) |
L1 |
223 |
(27.4) |
|
grade |
G1 |
94 |
(11.5) |
G2 |
508 |
(62.3) |
|
G3 |
211 |
(26.2) |
|
histology |
ductal invasive |
647 |
(79.4) |
lobular invasive |
123 |
(15.1) |
|
other |
45 |
(5.5) |
|
HR-status |
negative |
118 |
(14.5) |
positive |
697 |
(85.5) |
|
ER-status |
negative |
126 |
(15.5) |
positive |
689 |
(84.5) |
|
PR-status |
negative |
247 |
(30.3) |
positive |
568 |
(69.7) |
|
HER2-status |
negative |
702 |
(86.1) |
positive |
113 |
(13.9) |
|
Abbreviations: HR – hormone receptor; ER – estrogen receptor; PR – progesterone receptor; HER2 – human epidermal growth factor receptor 2.
DNA isolation and genotyping
Genomic DNA of native tumor material was isolated using tris/triton X-100 buffer for lysis followed by centrifugation. DNA of the cell debris was isolated with the QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany). Five single nucleotide polymorphisms were subsequently analyzed in genes that encode proteins which are important members of the p53-pathway: p53 - rs1042522, MDM2 – rs2279744 [20], rs117039649 [22]; MDMX – rs1563828 [24], and rs4245739 [25]. To determine the allelic status of rs1042522 (p53), rs2279744 and rs117039649 (MDM2), the genomic DNA was amplified by PCR with specific primers (p53 fw: 5′-CGTTCTGGTAAGGACAAGGGT-3′, p53 rev: 5′-AAGAAATGCAGGGGGATACGG-3′; MDM2 fw: 5′-CGGGAGTTCAGGGTAAAGGT-3′; MDM2 rev: 5′-AAAGCTGAGTCAACCTGC-3′). PCR products were gel purified and subsequently analyzed by direct sequencing using the BigDye Terminator Cycle Sequencing 3.1 Kit (Applied Biosystems, Darmstadt, Germany) according to the manufacturer’s intructions. The SNPs in the MDMX gene (rs1563828, rs4245739) were determined with a custom-made Taqman Assay by Applied Biosystems on a Rotorgene 3000 cycler (Qiagen, Hilden, Germany).
TP53 next generation sequencing
In a subset of 257 samples all coding exonic and flanking intronic regions of the human TP53 gene were amplified from genomic DNA with Platinum™ Taq DNA polymerase (Life Technologies) by multiplex PCR using two primer pools with 12 non-overlapping primer pairs each, yielding approximately 180 bp amplicons. Each sample was tagged with a unique 8-nucleotide barcode combination using twelve differently barcoded forward and eight differently barcoded reverse primer pools. Barcoded PCR products from up to 96 samples were pooled, purified and an indexed sequencing library was prepared using the NEBNext® ChIP-Seq Library Prep Master Mix Set for Illumina in combination with NEBNext® Multiplex Oligos for Illumina (New England Biolabs). The quality of sequencing libraries was verified on a Bioanalyzer DNA High Sensitivity chip (Agilent) and quantified by digital PCR. 2 x 250 bp paired-end sequencing was carried out on an Illumina MiSeq (Illumina) according to the manufacturer’s recommendations at a mean coverage of 150x.
Read pairs were demultiplexed according to the forward and reverse primers and subsequently aligned using the Burrows-Wheeler Aligner against the Homo sapiens Ensembl reference (rev. 79). Overlapping mate pairs where combined and trimmed to the amplified region. Coverage for each amplicon was calculated via SAMtools (v1.1) [50]. To identify putative mutations, variant calling was performed using SAMtools in combination with VarScan2 (v2.3.9) [51]. Initially, SAMtools was used to create pileups with a base quality filter of 15. Duplicates, orphan reads, unmapped and secondary reads were excluded. Subsequently, Varscan2 was applied to screen for SNPs and InDels separately, using a low-stringency setting with minimal variant frequency of 0.1, a minimum coverage of 20 and a minimum of 10 supporting reads per variant to account for cellular and clonal heterogeneity. Minimum average quality was set to 20 and a strand filter was applied to minimize miscalls due to poor sequencing quality or amplification bias. The resulting list of putative variants was compared against the IARC TP53 (R17) database to check for known TP53 cancer mutations.
Statistical analyses
All statistical analyses were done by using SPSS software (version 21.0, IBM, SPSS, Inc., Chicago, IL, USA). P < 0.05 was determined as criterion for statistical significance for all executed statistical tests. If possible, the 95% confidence interval was calculated to declare the statistical precision of an observed effect. Furthermore, the mean age-at-diagnosis in the study cohort was examined related to the genotype. The t-test was used to compare two genotypes, the F-test to compare all three genotypes. Hazard and survival curves were generated using the Kaplan-Meier-analysis. In order to determine the event-free survival time, local, regional as well as distant recurrence, and death from breast cancer were included as “events” according to Hudis et al. [49]. The standardized definitions for efficacy endpoints (STEEP) were used as endpoints definitions.
Author contributions
FB, MB, CW, MV, EK, TS and CT conceived the study and designed the study; MB, AN and KP carried out the experimental work; JB, EW and KFB performed histologic analysis, immunohistochemical analysis and provision of fresh-frozen and formalin-fixed tumor tissue; FB and MB carried out analysis and interpretation of data; FB, MB, AN, MN and EK performed bioinformatical and statistical analysis; CW, MV, EK, TS and CT provided technical and material support; VH, TL, CU, SP, JJ, CT and EK obtained informed consent from patients, performed surgery, collected clinical information and obtained follow-up information on events and survival; FB and MB prepared figures and tables. FB and MB carried out the drafting of manuscript. All authors reviewed and approved the manuscript.
ACKNOWLEDGMENTS
We appreciate the contributions of other members of our laboratories: Beate Heydel, Kathrin Stückrath, Dieter Berger. The authors wish to thank Jill Bargonetti for critical reading of the manuscript.
Ethics statement
The study was approved by the Ethics Committee of the medical faculty of the Martin-Luther University Halle-Wittenberg and all patients gave written consent for the use of material for research purposes.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Supplementary Materials.
CONFLICTS OF INTEREST
The authors declare no conflicts of interests.
FUNDING
This study received intramural research funding through the Roux-Program of the Martin-Luther-University Halle-Wittenberg, PiA trial number 25/36 (2012) and 21/14 (2010). Universitätsklinikum Giessen & Marburg (UKGM) to AN; Rhön Klinikum AG to TS.
REFERENCES
1. Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. 2010; 4:174–91.
2. Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: new genetic developments, new therapeutic avenues. Hum Genet. 2008; 124:31–42.
3. Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, Cheang MC, Gelmon K, Nielsen TO, Blomqvist C, Heikkila P, Heikkinen T, Nevanlinna H, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010; 7:e1000279.
4. Yang XR, Chang-Claude J, Goode EL, Couch FJ, Nevanlinna H, Milne RL, Gaudet M, Schmidt MK, Broeks A, Cox A, Fasching PA, Hein R, Spurdle AB, et al. Associations of breast cancer risk factors with tumor subtypes: a pooled analysis from the Breast Cancer Association Consortium studies. J Natl Cancer Inst. 2011; 103:250–63.
5. Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley KA, Scollen S, Baynes C, Ponder BA, Chanock S, Lissowska J, Brinton L, Peplonska B, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007; 39:352–8.
6. Dunning AM, Michailidou K, Kuchenbaecker KB, Thompson D, French JD, Beesley J, Healey CS, Kar S, Pooley KA, Lopez-Knowles E, Dicks E, Barrowdale D, Sinnott-Armstrong NA, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016; 48:374–86.
7. Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock SJ, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, et al. A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res. 2007; 67:2951–6.
8. Seibold P, Schmezer P, Behrens S, Michailidou K, Bolla MK, Wang Q, Flesch-Janys D, Nevanlinna H, Fagerholm R, Aittomaki K, Blomqvist C, Margolin S, Mannermaa A, et al. A polymorphism in the base excision repair gene PARP2 is associated with differential prognosis by chemotherapy among postmenopausal breast cancer patients. BMC Cancer. 2015; 15:978.
9. Stracquadanio G, Wang X, Wallace MD, Grawenda AM, Zhang P, Hewitt J, Zeron-Medina J, Castro-Giner F, Tomlinson IP, Goding CR, Cygan KJ, Fairbrother WG, Thomas LF, et al. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat Rev Cancer. 2016; 16:251–65.
10. Mello SS, Attardi LD. Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol. 2018; 51:65–72.
11. Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001; 29:92–5.
12. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997; 387:299–303.
13. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997; 420:25–7.
14. Barak Y, Gottlieb E, Juven-Gershon T, Oren M. Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994; 8:1739–49.
15. Zauberman A, Flusberg D, Haupt Y, Barak Y, Oren M. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 1995; 23:2584–92.
16. Phillips A, Teunisse A, Lam S, Lodder K, Darley M, Emaduddin M, Wolf A, Richter J, de Lange J, Verlaan-de Vries M, Lenos K, Bohnke A, Bartel F, et al. HDMX-L is expressed from a functional p53-responsive promoter in the first intron of the HDMX gene and participates in an autoregulatory feedback loop to control p53 activity. J Biol Chem. 2010; 285:29111–27.
17. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010; 2:a001008.
18. Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004; 24:5835–43.
19. Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG. Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res. 2001; 61:1839–42.
20. Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004; 119:591–602.
21. Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, Bartel F, Taubert H, Wuerl P, Hait W, Toppmeyer D, Offit K, Levine AJ. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res. 2006; 66:5104–10.
22. Knappskog S, Bjornslett M, Myklebust LM, Huijts PE, Vreeswijk MP, Edvardsen H, Guo Y, Zhang X, Yang M, Ylisaukko-Oja SK, Alhopuro P, Arola J, Tollenaar RA, et al. The MDM2 promoter SNP285C/309G haplotype diminishes Sp1 transcription factor binding and reduces risk for breast and ovarian cancer in Caucasians. Cancer Cell. 2011; 19:273–82.
23. Karni-Schmidt O, Lokshin M, Prives C. The Roles of MDM2 and MDMX in Cancer. Annu Rev Pathol. 2016; 11:617–44.
24. Atwal GS, Kirchhoff T, Bond EE, Montagna M, Menin C, Bertorelle R, Scaini MC, Bartel F, Bohnke A, Pempe C, Gradhand E, Hauptmann S, Offit K, et al. Altered tumor formation and evolutionary selection of genetic variants in the human MDM4 oncogene. Proc Natl Acad Sci U S A. 2009; 106:10236–41.
25. Wynendaele J, Bohnke A, Leucci E, Nielsen SJ, Lambertz I, Hammer S, Sbrzesny N, Kubitza D, Wolf A, Gradhand E, Balschun K, Braicu I, Sehouli J, et al. An illegitimate microRNA target site within the 3’ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010; 70:9641–9.
26. Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, Orr N, Rhie SK, Riboli E, Feigelson HS, Le Marchand L, Buring JE, Eccles D, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013; 45:392–8, 8e1–2.
27. Purrington KS, Slager S, Eccles D, Yannoukakos D, Fasching PA, Miron P, Carpenter J, Chang-Claude J, Martin NG, Montgomery GW, Kristensen V, Anton-Culver H, Goodfellow P, et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis. 2014; 35:1012–9.
28. Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013; 73:2025–30.
29. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53: 10.1002/humu.23035.
30. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010; 467:1061–73.
31. Kulkarni DA, Vazquez A, Haffty BG, Bandera EV, Hu W, Sun YY, Toppmeyer DL, Levine AJ, Hirshfield KM. A polymorphic variant in human MDM4 associates with accelerated age of onset of estrogen receptor negative breast cancer. Carcinogenesis. 2009; 30:1910–5.
32. Goldhirsch A, Ingle JN, Gelber RD, Coates AS, Thurlimann B, Senn HJ, Panel m. Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Ann Oncol. 2009; 20:1319–29.
33. Schmidt MK, Reincke S, Broeks A, Braaf LM, Hogervorst FB, Tollenaar RA, Johnson N, Fletcher O, Peto J, Tommiska J, Blomqvist C, Nevanlinna HA, Healey CS, et al. Do MDM2 SNP309 and TP53 R72P interact in breast cancer susceptibility? A large pooled series from the breast cancer association consortium. Cancer Res. 2007; 67:9584–90.
34. Haupt S, Vijayakumaran R, Miranda PJ, Burgess A, Lim E, Haupt Y. The role of MDM2 and MDM4 in breast cancer development and prevention. J Mol Cell Biol. 2017; 9:53–61.
35. Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007; 26:1306–16.
36. Haupt S, Buckley D, Pang JM, Panimaya J, Paul PJ, Gamell C, Takano EA, Lee YY, Hiddingh S, Rogers TM, Teunisse AF, Herold MJ, Marine JC, et al. Targeting Mdmx to treat breast cancers with wild-type p53. Cell Death Dis. 2015; 6:e1821.
37. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013; 13:83–96.
38. Bartel F, Jung J, Bohnke A, Gradhand E, Zeng K, Thomssen C, Hauptmann S. Both germ line and somatic genetics of the p53 pathway affect ovarian cancer incidence and survival. Clin Cancer Res. 2008; 14:89–96.
39. Wilkening S, Bermejo JL, Hemminki K. MDM2 SNP309 and cancer risk: a combined analysis. Carcinogenesis. 2007; 28:2262–7.
40. Economopoulos KP, Sergentanis TN. Differential effects of MDM2 SNP309 polymorphism on breast cancer risk along with race: a meta-analysis. Breast Cancer Res Treat. 2010; 120:211–6.
41. Gansmo LB, Romundstad P, Birkeland E, Hveem K, Vatten L, Knappskog S, Lonning PE. MDM4 SNP34091 (rs4245739) and its effect on breast-, colon-, lung-, and prostate cancer risk. Cancer Med. 2015; 4:1901–7.
42. Swetzig WM, Wang J, Das GM. Estrogen receptor alpha (ERα/ESR1) mediates the p53-independent overexpression of MDM4/MDMX and MDM2 in human breast cancer. Oncotarget. 2016; 7:16049–69. https://doi.org/10.18632/oncotarget.7533.
43. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
44. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–52.
45. Yu Q, Li Y, Mu K, Li Z, Meng Q, Wu X, Wang Y, Li L. Amplification of Mdmx and overexpression of MDM2 contribute to mammary carcinogenesis by substituting for p53 mutations. Diagn Pathol. 2014; 9:71.
46. Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, Soucy P, Fredericksen Z, Barrowdale D, Dennis J, Gaudet MM, Dicks E, Kosel M, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet. 2013; 9:e1003212.
47. Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ, Panel m. Strategies for subtypes--dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011; 22:1736–47.
48. von Minckwitz G, Untch M, Blohmer JU, Costa SD, Eidtmann H, Fasching PA, Gerber B, Eiermann W, Hilfrich J, Huober J, Jackisch C, Kaufmann M, Konecny GE, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012; 30:1796–804.
49. Hudis CA, Barlow WE, Costantino JP, Gray RJ, Pritchard KI, Chapman JA, Sparano JA, Hunsberger S, Enos RA, Gelber RD, Zujewski JA. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J Clin Oncol. 2007; 25:2127–32.
50. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–9.
51. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–76.