
Introduction
With an estimated 234,000 new cases and 154,000 deaths in 2018, lung cancer is the leading cause of cancer deaths in the United States, accounting for one-quarter of all cancer deaths [1]. Approximately, 80% of lung cancer deaths are associated with smoking, which confers a 25-fold increase in relative risk. Smoking-associated lung cancer has one of the highest mutational burdens of all cancers [2]. Despite advancements in molecularly targeted therapies for patients harboring actionable genetic abnormalities such as mutations in EGFR, ALK, RET, or BRAF, the majority of lung cancers lack identifiable driver oncogenes or harbor mutations in KRAS, TP53, and other genetic abnormalities that are not actionable targets [2-4].
The KrasLSL-G12D; p53fl/fl mouse model was developed as a powerful model for studying lung adenocarcinomas [5, 6]. Tumor protein p53 (TP53 or p53) (46%) and Kirsten rat sarcoma viral oncogene homolog (KRAS) (33%) are the most commonly mutated genes in human lung adenocarcinomas with a co-occurrence rate of ~15% [2, 7, 8]. Because neither genetic alteration is clinically actionable, chemotherapy remains the mainstay of treatment in patients with oncogenic KRAS driver mutations. Consequently, chemoresistance to genotoxic agents such as docetaxel and cisplatin remains an important clinical problem facing lung cancer patients.
The ABL kinases were initially identified as oncogenes in the context of patients with chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL) who presented with BCR-ABL1 fusion proteins due to a chromosomal translocation of ABL1 to the Breakpoint Cluster Region (BCR) gene sequences [9]. Recent studies have identified a potential role for ABL kinases in solid tumors [10, 11]. Data from The Cancer Genome Atlas (TCGA) showed alterations of ABL1 and ABL2 in human lung adenocarcinomas, including copy number enhancement of ABL2 and somatic mutations in ABL1 in 1-2% of patients [2]. Further, ABL2 genetic variations were identified in lung cancer in never smokers who were exposed to radon [12]. Importantly, increased ABL kinase activity has been detected in lung cancer cells without genomic alterations [13], and inactivation of ABL kinases suppresses lung cancer metastasis following intracardiac injection of NSCLC lines into immune-deficient mice [14].
Here, we demonstrate for the first time in the context of a KrasLSL-G12D; p53fl/fl (KP) mouse model of lung cancer that ABL kinase inhibition sensitizes primary lung adenocarcinomas to treatment with the chemotherapeutic agent, docetaxel. Further, we found that Abl inhibition promoted differentiation of the KP lung tumors, which was associated with increased cell death in the presence of docetaxel. Sensitization of lung tumors with Abl allosteric inhibitors to sub-therapeutic doses of chemotherapy would significantly decrease the deleterious side effects of chemotherapy and enhance response rates in patients with lung adenocarcinomas.
RESULTS
Inhibition of ABL kinases impairs KrasG12D/+; p53-/- driven lung tumors
To evaluate whether ABL kinases play a role in the progression of primary lung adenocarcinomas, we evaluated whether pharmacological inhibition of the ABL kinases impaired tumor growth in an autochthonous KrasLSL-G12D; p53fl/fl (KP) mouse model [6]. Intranasal delivery of an adenovirus containing a Cre-recombinase expressing construct results in the activation of oncogenic Kras and loss of p53 in infected cells. Consequently, spontaneous lung adenocarcinomas form throughout the lung approximately 8-12 weeks after viral delivery. Treatment with the Abl allosteric inhibitor, GNF5, and/or the chemotherapeutic agent, docetaxel, was initiated once tumor formation was confirmed by µCT scans performed at 8 weeks following infection (total tumor volume per mouse was < 1 cm3 with an average tumor diameter of < 3mm). GNF5 binds specifically to the myristoyl-binding site in the kinase domain of the ABL kinases, and is an Abl-specific inhibitor unlike the commonly used ATP-binding site inhibitors such as imatinib and nilotinib, which are known to interact with numerous other protein kinases [15]. We found that combination treatment with GNF5 and docetaxel markedly decreased tumor progression compared to vehicle control-treated mice or mice singly-treated with GNF5 or docetaxel alone (Figure 1A-1C).
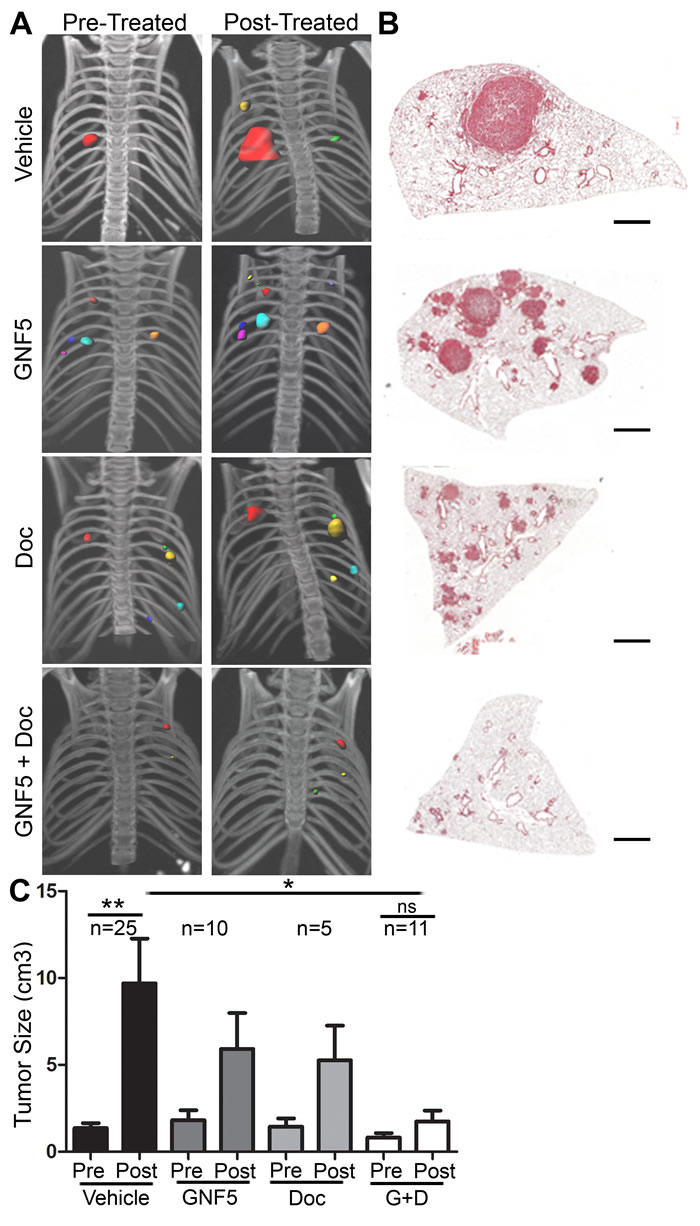
Figure 1: Inhibition of ABL kinases impairs KrasG12D/+; p53-/- driven lung tumors. Treatments began 8 weeks after Adeno-Cre infection of LSL-KrasG12D/+; p53fl/fl mice. A. 3D-reconstructions of µ-CT scans of mice before and after 14 days of treatment with vehicle, GNF5 (100 mg/kg b.i.d.), docetaxel (10 mg/kg, twice weekly), or GNF5 (100 mg/kg) + docetaxel (10 mg/kg, twice weekly). B. H&E sections of lungs from mice in each treatment group 10 weeks after adenoviral induction. Scale bar = 2,000 µm. C. Quantification of total tumor volume in bilateral lungs evaluated by µ-CT scans taken before (8 weeks) and after (10 weeks) treatment with vehicle, GNF5 alone, docetaxel alone, or combination therapy shows that combination treatment significantly impairs tumor growth in mice compared to vehicle treated mice. Graphs depict means and S.E.M. of “n” mice, where “n” represents each individual animal used.
Combination treatment with Abl inhibitor and docetaxel decreases cell proliferation and increases cell death in KrasG12D/+; p53-/- driven lung tumors
To evaluate the mechanism by which combination treatment with the Abl allosteric inhibitor, GNF5, and docetaxel inhibits lung adenocarcinoma growth in vivo, we harvested mouse lungs treated with vehicle control, GNF5 alone, docetaxel alone, or combination therapy two weeks after initiating treatment (12 weeks after delivery of adenovirus). Staining of lung tissue sections with the proliferation marker, Ki67, showed a profound decrease in Ki67 expression by immunohistochemistry (IHC) and immunofluorescence (IF) in double-treated mice compared to vehicle control mice or mice treated with GNF5 or docetaxel alone (Figure 2A-2C). We also found a significant decrease (~50% reduction) in expression of phospho-histone H3, a marker expressed specifically in mitotic cells, in mice treated with combination therapy compared to vehicle control treated mice (Supplementary Figure 1A and B).
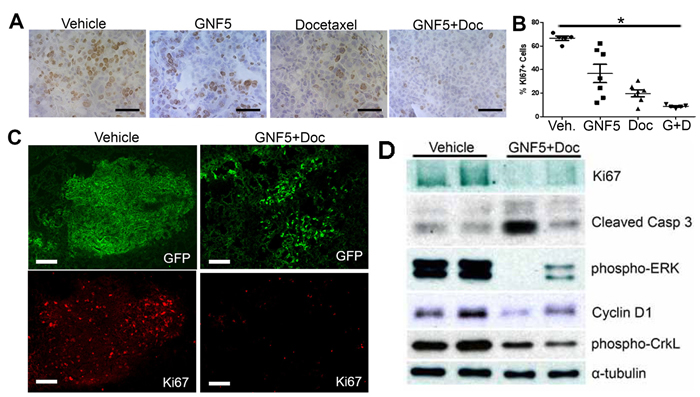
Figure 2: Inhibition of ABL kinases in the presence of docetaxel decreases cell proliferation and increases cell death in KrasG12D/+; p53-/- driven lung tumors. Treatments began 10 weeks after Adeno-Cre infection of Rosa26-fGFP; LSL-KrasG12D/+; p53fl/fl mice. Mouse lungs were harvested at 12 weeks after Adeno-Cre infection. A. IHC for Ki67+ cells in sections of mouse lungs from mice treated with vehicle control, docetaxel, GNF5, or combination (docetaxel+GNF5) treatment showing a decrease in Ki67 staining, particularly in the combination therapy group. Scale bar = 50 µm. B. Quantification of IHC staining for Ki67 (n = 5-7 tumors per group). C. Immunofluorescence staining for Ki67 showing a decrease in the percentage of Ki67+ (red) tumor cells (labeled with farnesylated GFP, green) in mice treated with combination therapy of GNF5 and docetaxel compared to vehicle treated mice. Scale bar = 150 µm. D. Immunoblotting of lysates showed a decrease in Ki67 expression and increase in cleaved caspase 3 expression in mice given combination therapy compared to control mice and a corresponding decrease in pERK and cyclin D1, which are downstream targets of oncogenic Kras. Phospho-CrkL is a marker for ABL kinase activity while GAPD is a loading control. Graphs depict means and S.E.M.
Cell proliferation and apoptosis were evaluated in lung tumors derived from KrasLSL-G12D; p53fl/fl mice expressing the Rosa26-fGFP reporter, which allowed for isolation of GFP+ lung tumor cells by FACS analysis 12 weeks after delivery of adenovirus expressing Cre recombinase (Figure 2). Immunoblotting of protein lysates from the isolated GFP+ cells showed a marked decrease in Ki67 expression, as well as decreased levels of the Kras downstream targets cyclin D1 and phospho-ERK in mice treated with GNF5 and docetaxel (Figure 2D). To evaluate apoptosis, protein lysates from the isolated GFP+ cells were immunoblotted for cleaved caspase 3. We found an increase in expression of cleaved caspase 3 in a subset of GNF5 and docetaxel double-treated mice compared to vehicle control mice (Figure 2D). The increase in cleaved caspase 3 was primarily detected in double-treated tumors with greater inhibition of the cell proliferation markers phospho-Erk and cyclin D1 (Figure 2D). These data suggested that combination treatment with GNF5 and docetaxel inhibits cell proliferation and increases cell death in KrasG12D/+; p53-/- driven lung adenocarcinomas.
Inhibition of ABL kinases sensitizes primary KrasG12D/+ mouse-derived organoids and cell lines to treatment with docetaxel
Next we evaluated the effects of ABL kinase inhibition on the growth of organoid cultures of primary tumors derived from KrasG12D/+ mice. To this end, SPC (Sftpc)-CreERT2; KrasLSL-G12D; Rosa26-tdTomato mice were treated with tamoxifen to induce tumor formation specifically in Type II alveolar epithelial cells expressing the SPC (Sftpc) driver [16]. Tomato+ cells were isolated from mice after four weeks post-treatment and grown in Matrigel in the presence of primary mouse fibroblasts (GFP+) to assess tumor organoid formation (Figure 3A). Organoids were treated with vehicle control, GNF5 alone, docetaxel alone, or combination therapy starting three days after plating, and organoids were then grown for two weeks. We observed a decrease in the size of organoids treated with the combination therapy compared to vehicle- or single agent-treated organoids (Figure 3B-3C). By comparison, we observed no decrease in size or morphology of primary lung alveolospheres isolated from SPC (Sftpc)-CreERT2; Rosa26-tdTomato mice, expressing wild-type Kras, treated with GNF5 (Supplementary Figure 2A).
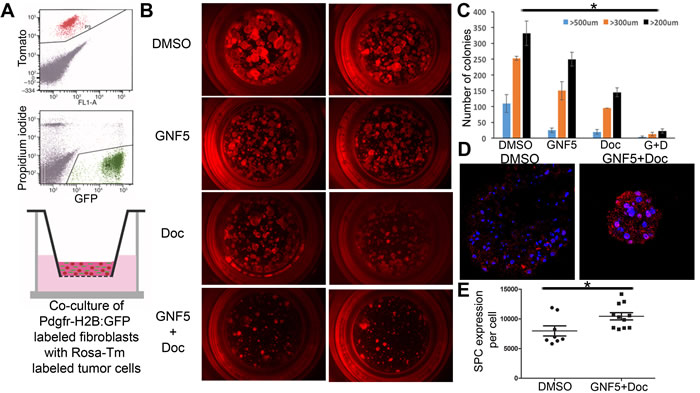
Figure 3: Inhibition of ABL kinases sensitizes primary KrasG12D/+ derived organoid cultures to treatment with docetaxel in 3D tumor sphere assays. A. Model: SPC (Sftpc)-CreERT2; KRASLSL-G12D; Rosa26-tdTomato mice were given tamoxifen to induce tumor formation. Tomato+ cells were then isolated from mice after tumor formation and grown in Matrigel in transwell inserts in the presence of primary mouse fibroblasts (derived from PDGFRα-H2B: GFP mice) to induce tumor organoid formation. B and C. Organoids were treated with vehicle (DMSO), GNF5, docetaxel (DOC), or combination treatment (GNF5 + DOC) for 2 weeks and assessed for organoid size 2 weeks after treatment (B); quantification revealed a significant reduction in organoid size in response to combination treatment compared to vehicle, GNF5, or docetaxel alone (C). Graphs depict means and S.E.M. D.-E. Immunofluorescence staining of organoids treated with GNF5 and docetaxel show increased expression of the Type II cell marker, SPC, compared to vehicle control treated mice. Quantification is provided in (E). Scale bar = 10 µm. Graphs depict means and S.E.M.
Several primary cell lines derived from KrasG12D/+; p53-/- mouse lung tumors were also evaluated in growth/viability assays using Cell-Titer-Glo in the presence or absence of the Abl allosteric inhibitor with and without docetaxel. We found significantly reduced cell growth in the combination treatment group compared to treatment with GNF5 or docetaxel alone (Supplementary Figure 3A). These data are consistent with in vivo data showing that ABL kinase inhibition sensitizes primary lung tumors to treatment with docetaxel (Figure 1).
Inhibition of ABL kinases sensitizes KrasG12D/+; p53-/- lung adenocarcinomas to treatment with docetaxel by promoting cell differentiation
To further define the mechanisms by which ABL kinase inhibition sensitizes KrasG12D/+; p53-/- driven mouse lung tumors to docetaxel treatment, the expression of a variety of lung epithelial cell differentiation markers was evaluated following treatment with the ABL kinase inhibitor, GNF5, with and without docetaxel. A defining characteristic of adenocarcinomas is their progressive de-differentiation from low grade, glandular structures with well-differentiated tumor cells to high grade tumors that lack defined tissue morphology with poorly-differentiated tumor cells [17]. Thus, we hypothesized that ABL kinase inhibition may inhibit or reverse lung tumor de-differentiation.
We first evaluated whether combining Abl inhibition with chemotherapies altered the expression of differentiation marker genes in vivo. KrasLSL-G12D; p53fl/fl mice were crossed with mice expressing the Rosa26-fGFP reporter for isolation of GFP+ tumor cells by FACS. These mice were used for drug treatment following 10 weeks post-delivery of adenovirus-Cre (rather than 8 weeks after adenovirus-Cre as shown in Figure 1) in order to increase the size of the tumors in the combination therapy group and permit the isolation of sufficient quantities of protein for analysis of differentiation markers. This model is also more clinically relevant, as patients with lung adenocarcinomas typically present with moderate to poorly differentiated tumors [18]. Notably, µCT scans of mouse lungs before (10 weeks after viral induction) and after 2 weeks of drug treatment (12 weeks after viral induction) showed a three-fold reduction in tumor size for mice treated with both GNF5 and docetaxel compared to vehicle control treated mice (Figure 4A). Analysis of protein lysates from GFP+ tumors revealed an increase in terminal differentiation markers such as the ciliated marker, acetylated α-tubulin, and a corresponding decrease in the basal cell marker keratin 5 (Krt5) in mice treated with GNF5, docetaxel and combination therapy group compared to the vehicle control group (Figure 4B). Further, expression of the secretory cell marker CC10/Scgb1a1 was markedly enhanced by GNF5 and co-treatment of GNF5 + docetaxel (Figure 4B). Consistent with these results, RT-qPCR analysis of isolated GFP+ tumor cells isolated from mice co-treated with GNF5 and docetaxel showed a significant increase in expression of terminal differentiation markers (Type II cell marker, SPC, and secretory cell marker, CC10) with a concomitant decrease in expression of basal stem-cell markers including p63 (Figure 4C). We also found a corresponding increase in SPC protein expression by IF analysis of mouse lung adenocarcinomas in response to the combination therapy of GNF5 with docetaxel (Supplementary Figure 4).
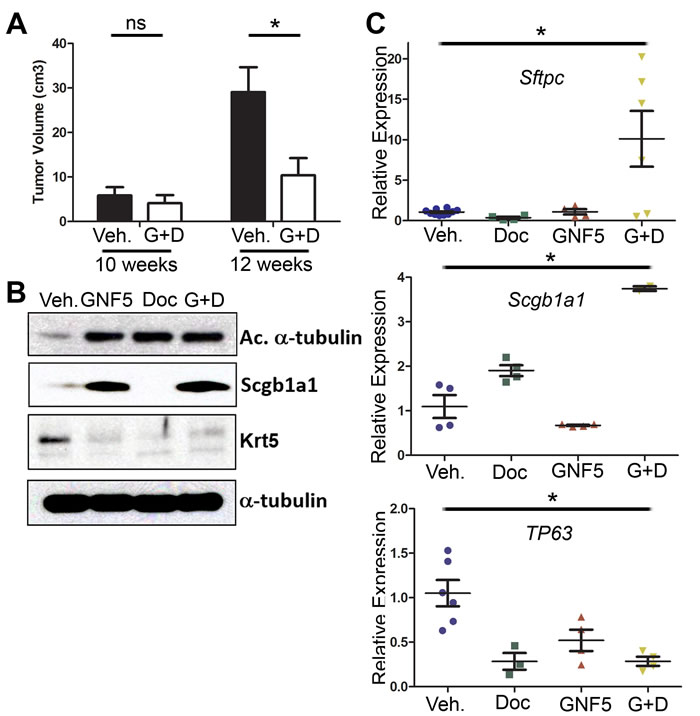
Figure 4: Combination treatment of GNF5 and docetaxel induces lung tumor cell differentiation in vivo. GFP+ cells were isolated from KRASLSL-G12D; p53fl/fl; Rosa26-fGFP mice two weeks after treatment with vehicle, docetaxel, GNF5, or combination treatment and 12 weeks after induction of tumors with adenovirus. A. Quantification of total tumor volume in bilateral lungs evaluated by µ-CT scans taken before (10 weeks) and after (12 weeks) treatment with vehicle or combination therapy (GNF5 + docetaxel) shows that combination treatment significantly impairs tumor growth in mice compared to vehicle treated mice (n = 11 mice per group). B. Western blot analysis showed an increase in expression of the ciliated cell marker, acetylated α-tubulin, and the secretory cell marker, Scgb1a1, with a corresponding decrease in expression of the basal cell marker, keratin 5, in mice treated with GNF5 and docetaxel. C. RT-qPCR analysis for indicated cell treatment groups shows an increase in expression of terminal cell markers (Sftpc: Type II cell marker and Scgb1a1: secretory cell marker) with a corresponding decrease in expression of the basal cell marker, TP63, in mice treated with the combination therapy compared to control or mice treated with GNF5 or docetaxel alone (n = 4-6 mice per group, each RT-PCR assay performed in triplicate). Graphs depict means and S.E.M.
Consistent with these in vivo phenotypes, we found an increase in expression of terminally differentiated cell markers in two different in vitro assays in response to co-treatment with GNF5 and docetaxel. Analysis of tumor organoid cultures derived from SPC (Sftpc)-CreERT2; KrasLSL-G12D; Rosa26-tdTomato mice, showed that organoids from KRAS+ mice treated with the combination treatment of GNF5 and docetaxel had significantly higher expression of SPC compared to vehicle control-treated organoids from KRAS+ mice (Figure 3D-3E). Notably, vehicle control-treated organoids derived from KRAS+ mice demonstrated reduced expression of the Type II cell marker, SPC, compared to normal lung alveolospheres isolated from control KRAS-negative mice (Supplementary Figure 2B). Moreover, primary lung adenocarcinoma cell lines derived from KrasLSL-G12D; p53fl/fl lung tumors also showed an increase in expression of differentiation markers (acetylated α-tubulin) and a decrease in expression of basal cell markers (Krt5) after co-treatment with GNF5 and docetaxel compared to vehicle control or single-agent treatment in vitro (Supplementary Figure 3B). These in vitro data are consistent with the in vivo mouse data in showing that treatment with GNF5 and docetaxel promotes cell differentiation of lung adenocarcinomas.
ABL kinases promote Yap1 signaling in KrasG12D/+; p53-/- driven lung tumors
The Hippo pathway transcriptional regulatory factor Yap1 has been shown to regulate the proliferation and differentiation of lung epithelial cells during development and regeneration [19, 20]. Yap1 regulates organ size and morphology during development [21, 22]. Nuclear Yap1 is associated with pro-proliferative and anti-differentiation responses in lung airway cells and other tissues during development and tumorigenesis, and regulates expression of genes implicated in cell proliferation, including c-Myc [23-25]. In contrast, cytoplasmic Yap1 is associated with pro-differentiation and anti-proliferative responses [25]. Multiple cancer types are characterized by accumulation of nuclear Yap1, which is prevalent in highly proliferative, aggressive tumors and is associated with poor outcomes [26-28].
We evaluated Yap1 expression in KrasG12D/+; p53-/- driven mouse lung tumors by immuno-staining of lung tissue sections from mice treated with vehicle control or the combination therapy of GNF5 and docetaxel. High-level expression of nuclear Yap1 in vehicle control treated mice was markedly reduced in mice treated with the combination therapy, which promoted a punctate pattern of Yap1 cytosolic localization (Figure 5A-5B). Notably, total Yap1 protein expression was unchanged in vehicle control treated mice compared to mice in the combination therapy group (Figure 5C).
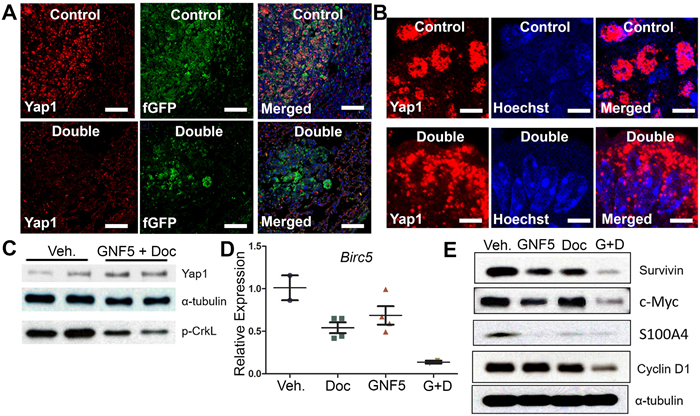
Figure 5: Combination treatment of GNF5 and docetaxel induces cytoplasmic localization of Yap1 and decreases expression of downstream transcription targets of Yap1 compared to vehicle control treated lung adenocarcinomas. GFP+ cells were isolated from KRASG12D/+; p53-/-; Rosa26-fGFP mice two weeks after treatment with vehicle, docetaxel, GNF5, or combination treatment and 12 weeks after induction of tumors with adenovirus. A. Immunofluorescence staining for Yap1 shows a decrease in Yap1 nuclear localization in mice treated with GNF5 and docetaxel compared to vehicle control treated mice. Scale = 75 µm. B. Higher magnification images are provided to show sub-cellular localization of Yap1 in control and combination treatment mice. Scale = 7.5 µm. C. Western blot analysis showed no significant difference in total Yap1 protein expression in double treated mice compared to vehicle treated control mice. Phospho-CrkL expression is a marker of ABL kinase activity. D. RT-qPCR analysis showed a decrease in mRNA transcript expression of the downstream Yap1 target, Birc5, which encodes the protein survivin, in mice treated with both GNF5 and docetaxel compared to vehicle control treated mice or mice treated with GNF5 or docetaxel alone (n = 3-4 mice per group, each RT-qPCR assay performed in triplicate). E. Western blot analysis showed a decrease in protein expression of downstream transcriptional targets of Yap1, including survivin, c-Myc, S100A4, and cyclin D1. Graphs depict means and S.E.M.
To determine whether the decrease in nuclear Yap1 staining resulted in decreased expression of Yap1 transcriptional targets, we isolated protein lysates from GFP+ lung adenocarcinoma cells derived from KrasG12D/+; p53-/-; Rosa26-fGFP mice treated with vehicle control, GNF5 alone, docetaxel alone, or combination therapy and lysates were analyzed by Western blotting for known Yap1-regulated targets. We found a profound decrease in the expression of Yap1 targets including Survivin, c-Myc, S100A4, and Cyclin D1 in mice treated with GNF5 and docetaxel (Figure 5E). We also detected a corresponding decrease in Survivin/Birc5 mRNA transcript levels in lung tumors isolated from KrasG12D/+; p53-/-; Rosa26-fGFP mice treated with combination therapy (Figure 5D). These data suggest that combination treatment with an Abl allosteric inhibitor and docetaxel inhibits nuclear Yap1 signaling in these tumors.
DISCUSSION
Chemotherapy is a first-line treatment in the majority of patients with lung adenocarcinomas that often fails due to intrinsic or acquired resistance. Thus, combination therapies that sensitize tumors to chemotherapy might have a profound impact in the treatment of these patients. Here we show that ABL kinase inhibition sensitizes KrasG12D+/-; p53-/- tumors to treatment with docetaxel such that sub-therapeutic doses of docetaxel confer reduced cell growth and increased cell death both in vivo and in vitro in the presence of Abl allosteric inhibitors.
Tumors frequently co-opt pathways implicated in development and regeneration to promote cell growth and inhibit cell death. As adenocarcinomas progress from less aggressive, well-differentiated tumors with glandular morphology reflecting their cell of origin (low grade tumors) to aggressive, poorly-differentiated tumors that lack any identifiable morphology (high grade tumors), they undergo a de-differentiation process that includes reduced expression of markers characteristic of their cell of origin and increased expression of basal cell markers or other ectopic protein expression markers [29]. Some lung tumors lose their epithelial morphology through an epithelial-mesenchymal transition (EMT). We have previously shown that ABL kinases are upregulated in metastatic lung tumors and promote differentiation of lung epithelial cells in the context of bacterial pneumonia [30]. We hypothesized that inhibition of the ABL kinases might promote differentiation of lung adenocarcinomas from more de-differentiated, basal-like tumors to differentiated tumors expressing terminal cell differentiation markers such as SPC. The data presented here support this hypothesis using an autochthonous KrasG12D/+; p53-/- lung cancer mouse model, lung cancer organoid 3D cultures and primary 2D cell cultures. We found increased expression of pro-differentiation markers and decreased expression of basal cell markers in lung adenocarcinoma-bearing mice or lung tumor cells treated with the ABL kinase inhibitor, GNF5, compared to vehicle control treated mice.
ABL kinase inhibition may drive differentiation of lung cancer cells through modulation of the Hippo pathway. Nuclear Yap1 promotes proliferative pathways in cells and translocation of Yap1 to the cytoplasm is associated with a switch from proliferative to pro-differentiation signaling [21, 31]. Accumulating data suport roles for Yap1 in tumor progression and chemoresistance [32, 33]. KRAS-mutated human non-small cell lung cancers are associated with increased nuclear YAP1, and downregulation of YAP1 has been shown to synergize with the chemotherapeutic agent, cisplatin [34]. Nuclear YAP1 was shown to promote cancer cell survival by activating Survivin expression [35]. Several distinct mechanisms have been reported to regulate YAP1-mediated regulation of therapy resistance in lung cancer cells including acquistion of an EMT phenotype and overexpression and/or activating mutations of receptor tyrosine kinases [36]. Activation of Yap1 and various other oncogenic pathways converge to activate MYC and drive chemoresistance [37]. Inhibition of ABL kinases would blunt signaling to Yap1 and other factors linked to chemoresistance, and might be exploited as a potential strategy to treat lung tumors. Because of the high toxicity profiles of compounds that target Yap1 [38], the use of Abl allosteric inhibitors may be a promising new therapeutic strategy for lung adenocarcinoma patients.
MATERIALS AND METHODS
Mouse experiments
KrasLSL-G12D; p53fl/fl mice were kindly provided by Dr. David Kirsch at Duke University. To induce lung tumors, mice were given 30 µL virus dilution in DMEM followed by 30 µL of DMEM intranasally in 6-week-old mice under isofluorane anesthesia. The virus dilution was prepared by diluting 44 µL viral stock (VVC U of Iowa-5; University of Iowa Vector Core Facility) in 500 µL DMEM with 2.5 µL 2M calcium chloride. Mouse tumors were evaluated by conducting complete body µCT scans 8 or 10 weeks after viral induction for tumor inhibition and tumor regression studies, respectively. In mice in which tumors were identified, treatment with GNF5 (100 mg/mL b.i.d. via oral gavage), docetaxel (10 mg/kg twice/week as indicated via intraperitoneal injection), or combination treatment was initiated for two weeks. After two weeks, follow-up complete body µCT scans were performed to evaluate change in tumor size. Analysis of tumor size was then performed using Imaris Version 8 software.
Drug preparation
GNF5 (N-(2-Hydroxyethyl)-3-(6-(4-(trifluoromethoxy)phenylamino) pyrimidin-4-yl) benzamide) was synthesized at the Duke University’s Small Molecule Synthesis Facility, and validated by LC-MS and 1H-NMR/FT-IR spectra and with cell-based assays that confirm potencies and cell signaling inhibitory activities. For in vivo experiments, GNF5 was prepared in a suspension with 0.5% methylcellulose and 0.5% Tween-80 at a concentration of 10 mg/mL, and mice were treated with 100 mg/kg b.i.d. via oral gavage.
Docetaxel (LC Laboratories: D-1000) stock solution was prepared by dissolving the powder at a concentration of 30 mg/mL in ethanol. Working dilutions were prepared fresh each day by diluting the stock solution 1:10 in 5% sucrose water containing 10% Tween 80. The working dilutions were filtered using 0.4 µm filters and given to mice intra-peritoneally at a dose of 10 mg/kg twice weekly.
Preparation of lung tissue sections
Mice were euthanized after two weeks of treatment either 10 or 12 weeks after viral induction. The aorta was dissected to reduce intravascular blood volume, and the lungs were exposed through an incision across the diaphragm and the bottom half of the sternum. The trachea was exposed and cannulated with a sterile 22-gauge Abbocath-T catheter to inflation fix the mouse lungs. Fixation with 4% paraformaldehyde solution in PBS was performed for 15 minutes at room temperature by suspending fixation solution in an apparatus 40 cm above the mice. The entire mediastinum including the trachea, lungs, and heart were then carefully excised and immediately placed in 4% paraformaldehyde for 4 hours on a rotator at 4°C. Specimens were then washed three times for 15 minutes each with PBS. The left lungs were transferred to 70% ethanol overnight, paraffin embedded at the Duke University Immunohistopathology Core Facility, and cut to 5 µm thick sections. The right lungs were sucrose protected (30% sucrose in water), frozen in Optimal Cutting Temperature (OCT) compound at -80°C, and cut to 5 µm thick sections.
Hematoxylin and eosin staining and quantification
Tissue sections were deparaffinized, rehydrated, and incubated with hematoxylin staining reagent for 10 minutes followed by treatment of acid alcohol, Scott’s water, and an eosin secondary counter-stain for 1 minute each. Slides were cleared by xylene, mounted with mounting medium, and analyzed on a Zeiss Axio Imager upright microscope using a 10x objective. Entire left lung coronal sections at approximately the same depth were obtained for each mouse of each experimental condition. Lung sections were imaged and stitched together using the Zeiss Zen software to produce whole left lung images. The fraction of tumor area was determined using the particle analysis function on FIJI. A one-way ANOVA followed by a post-hoc Tukey test was performed to evaluate the differences in tumor area between each group. No image adjustments were applied to H&E images prior to quantification.
Immunofluorescence
Left lung tissue sections were deparaffinized, rehydrated, and heat inactivated (BioCare Medical Decloaking Chamber). Right lung tissue sections were thawed at room temperature for 15 minutes. Both deparaffinized and frozen sections were then washed in PBS and blocked in 3% goat serum in PBS with 0.05% Tween-20 for one hour. Sections were incubated with primary antibodies in blocking solution overnight at 4°C in a humidified chamber at concentrations indicated below. Sections were then washed with PBS followed by incubation with the appropriate secondary antibody in blocking solution for one hour at room temperature. Sections were then washed with PBS, incubated with the nuclear stain, Hoechst33342, and washed again with PBS before mounting using aqueous mounting media (Dako-S3025). Antibodies for immunofluorescence and IHC experiments included: Anti-SPC (Millipore-AB3786) at a 1:1000 dilution, anti-GFP (Aves Labs-GFP-1020) at a 1:1000 dilution, anti-pHH3 (Sigma-Aldrich-H9908) at a 1:1000 dilution, and anti-Yap1 (Abnova-H00010413-M01) at 1:250 dilution. Secondary antibodies with fluorescent labels were purchased from ThermoFisher (anti-mouse, anti-rabbit, and anti-rat Alexa Fluors 488, 561, and 647) and used at 1:1000 dilutions.
Quantification of immunofluorescence experiments
All cell counts were evaluated for entire left lung sections using a 10x objective on the Zeiss AxioImager microscope using Zen software at the Duke Light Microscope Core Facility. Cell counts were validated, and representative high-resolution images were taken using 40x and 100x objectives on the Leica SP8 confocal microscope. Stitched images were quantified using the Analyze Particle feature in Fiji software to determine total cell counts across the entire lung for each mouse. No image adjustments were applied to 10x objective stitched images prior to quantification. Representative high-resolution images displayed in the figures were modified along a linear scale for ease of viewing due to differences in sensitivities of antibodies.
FACS
Mice were euthanized after two weeks of treatment either 10 or 12 weeks after viral induction. The aorta was dissected to reduce intravascular blood volume, and the lungs were exposed through an incision across the diaphragm and the bottom half of the sternum. The trachea was exposed and cannulated with a sterile 22-gauge Abbocath-T catheter to inflation fix the mouse lungs with enzyme solution containing 4.5 mg Type I collagenase (ThermoFisher: 17100017), 100 µL elastase (4U/mL) (Worthington Biochemical Corporation: LS002279), 300 µL dispase (5U/mL) (BD Biosciences: 354235), and 200 µL Dnase I (0.33U/mL) (Roche: 10104159001). Lungs were dissociated by enzyme digestion by first separating them from surrounding tissue, cutting into small pieces using scissors, and incubating at 37°C in the enzyme solution with vigorous shaking every five minutes. Once homogenized, 8mL DMEM-F12 solution containing 10% FBS was added to neutralize the enzyme solution. The cell suspension was then filtered through a 70 µm strainer, centrifuged, and resuspended in red blood cell lysis buffer (ThermoFisher: 00-4333-57). 8mL DMEM-F12 solution containing 10% FBS was added to neutralize the lysis buffer. The cell suspension was then centrifuged, resuspended in 1mL DMEM-F12 containing 2% BSA, and filtered using a 40 µm cell strainer for FACS. FACS was performed using a BD-DiVa system at the Duke Flow Cytometry Core Facility. RNA or protein was collected from isolated GFP-labeled cells using an RNA isolation kit (GE-25050071) or RIPA buffer digestion (containing protease and phosphatase inhibitors), respectively.
RT-PCR
RNA was isolated from cells using an RNA isolation kit (GE-25050071), and complementary DNA was synthesized using oligo(dT) primers and Moloney murine leukemia virus reverse transcriptase (Invitrogen). Real-time PCR was performed using iQ SYBR Green Supermix (BioRad-1708882). The primers used were as follows: mouse Scgb1a1, ATCGCCATCACAATCACTACTG (forward) and CAGTCTCTTCAGCTGGGTGC (reverse); mouse Gapdh, AGGTCGGTGTGAACGGATTTG (forward) and TGTAGACCATGTAGTTGAGGTCA (reverse); mouse Sftpc, AACGCCTTCTCATCGTGGT (forward) and TAGATATAGTAGAGTGGTAGCT (reverse); mouse Birc5, AAGGAATTGGAAGGCTGGG (forward) and TTCTTGACAGTGAGGAAGGC (reverse); mouse TP63, CCCACAGACTGCAGCATTG (forward) and GAGATGAGGAGGTGAGGAGAAG (reverse). Analysis was performed using a BioRad CFX384 real-time machine and CFX Manager software. PCR assays were performed in duplicate. The expression of each gene was normalized to that of Gapdh (mouse).
Immunoblotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50mM Tris-HCl, pH 7.5, 150mM NaCl, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate with protease and phosphatase inhibitors. Cell debris was removed by microcentrifugation, and protein concentration was quantified using the DCTM Protein Assay (Bio-Rad Laboratories). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis (BioRad-1610183) and transferred onto 0.2 µm pore nitrocellulose membranes. Membranes were probed with the appropriate antibodies overnight at 4°C at dilutions indicated below, thoroughly washed, and then incubated with secondary HRP-tagged antibodies (Jackson Laboratory) for one hour at room temperature. Blots were incubated using chemiluminescent reagents (ThermoFisher-34580) and developed using x-ray film (GE-45001).
| Antibody |
Source |
Catalog number |
| Ki67 |
BD pharmingen |
550609 |
| c-Myc |
Cell Signaling |
5605 |
| Cleaved Caspase 3 |
Cell Signaling |
9661 |
| p-CrkL (y207) |
Cell Signaling |
3181 |
| Cyclin D1 |
Cell Signaling |
2978 |
| S100A4 |
abcam |
Ab27957 |
| Survivin |
Cell Signaling |
2808 |
| p-Erk (T202/Y204) |
Cell Signaling |
4376 |
| Acetyl-α-tubulin |
Cell Signaling |
5335 |
| Keratin 5 |
BioLegend |
905501 |
| Scgb1a1 (CC10) |
Millopore |
07-623 |
| α-tubulin |
Millipore-Sigma |
05-829 |
Tumor spheroid assay
6–8 weeks old KrasLSL-G12D/+; Sftpc-CreERT2; LSL-TdTomato mice were administrated with Tamoxifen (T5648; Sigma-Aldrich) at 20 mg/ml stock solution in corn oil and given via intraperitoneal injection of 0.25 mg/g. 4 weeks after Tamoxifen injection, KrasLSL-G12D/+; Sftpc-CreERT2; LSL-TdTomato and PDGFRα-eGFP mouse lungs [39] were dissociated using enzymatic digestion as described above. TdTomato positive cells and GFP positive cells were sorted using a BD-DiVa system at the Duke Flow Cytometry Core Facility. Viable Tdtomato single positive cells (5,000 cells) were mixed with PDGFRα-GFP positive fibroblasts (100,000 cells) in 1:1 Matrigel/MTEC plus media for a total volume of 100 μL in 24-well (0.4-μm) Transwell filter inserts. 500 μL MTEC plus medium was placed in the lower chamber and changed every other day. Cultures were treated with GNF5 (5 µM) and/or docetaxel (1nM) and maintained in a humidified 37°C incubator in 5% CO2. After two weeks, tumor sphere sizes were evaluated for each group.
Viability and proliferation assay
KrasG12D/+; p53-/- mouse cell lines were graciously provided by Kwok-Kin Wong (New York University) and Jonathan Kurie (MD Anderson). Cells (2x103) were seeded in 96 well plates in triplicates, drugs were added at the indicated concentrations the following day and measured each day using Celltiter-glo (Promega).
Statistical analysis
The number of animals in each group is indicated in the figures and/or figure legends. Differences among groups for immunofluorescence and H&E experiments were assessed using one-way ANOVA followed by post-hoc Tukey tests. All quantification for these experiments was performed using stitched images for the entire left lung ( > 100,000 cells counted per image). Representative high-resolution images from each animal group are indicated in the figures. Results are presented as means +/- standard error of measurement. Individual group testing was performed only when statistical significance was achieved from the ANOVA. Real-time RT-qPCR experiments were all performed in triplicate in three separate mouse samples. “*”, “**”, and “***” indicate p-values, < 0.05, < 0.01, and < 0.001, respectively. Bar graphs represent means with standard errors of measurement.
Abbreviations
ANOVA: analysis of variance; CC10: Clara cell 10 protein; FACS: fluorescent activated cell sorting; GFP: green fluorescent protein; H&E: Hemotoxylin & Eosin; HRP: horse radish peroxidase; µCT: X-ray microtomography; PDGFR: platelet derived growth factor receptor; RIPA: radioimmunoprecipitation assay; RT-qPCR: Real time quantitative polymerase chain reaction; SPC: surfactant protein C.
Author contributions
A.K. designed the study, performed experiments, interpreted results, and wrote the paper. J.G. designed the study and performed the experiments. C.M.M. performed experiments and interpreted results. X.X. performed the tumor sphere assay experiments. A.M.P. conceived the project, analyzed the data, and wrote the paper.
Acknowledgments
We would like to thank Dr. David Kirsch (Duke University) for graciously providing the KrasG12D/+; p53fl/fl mice, and Lorraine Da Silva Campos (Duke University) for technical help with the µCT scans of the mouse lungs. We thank Dr. Kwok-Kin Wong (New York University) and Dr. Jonathan Kurie (MD Anderson) for graciously providing the KP mouse derived tumor cell lines. We thank Dr. Emily Riggs (Duke University) for breeding and maintaining the mouse colonies. We thank Dr. Brigid Hogan (Duke University) and Dr. Mark Onaitis (UCSD) for generating and providing the KrasLSL-G12D/+; Sftpc-CreERT2; LSL-TdTomato and PDGFRα-eGFP mice.
Conflicts of Interest
The authors declare no competing interests
Funding
A.K. receives support from National Heart, Lung, and Blood Institute grant, F30HL126448, and the training grant, T32GM007171. C.M.M. receives support from the National Cancer Institute grant, F31CA224952. This work was supported by National Institute of Health grant R01CA195549 and Free to Breathe Metastasis Research Grant to A.M.P.
REFERENCES
1. Society AC. Cancer Facts & Figures 2018. Atlanta: American C ancer Society; 2018.
2. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ, Danilova L, Ding L, Getz G, Hammerman PS, et al, and Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543–50. https://doi.org/10.1038/nature13385.
3. Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar AF, Minna JD. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989; 246: 491-4.
4. McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, Hodges E, Rosebrock AP, Bronson RT, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A. 2016; 113: E6409-17. https://doi.org/10.1073/pnas.1613601113.
5. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001; 15: 3243-8. https://doi.org/10.1101/gad.943001.
6. DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009; 4: 1064-72. https://doi.org/10.1038/nprot.2009.95.
7. Shajani-Yi Z, de Abreu FB, Peterson JD, Tsongalis GJ. Frequency of Somatic TP53 Mutations in Combination with Known Pathogenic Mutations in Colon Adenocarcinoma, Non-Small Cell Lung Carcinoma, and Gliomas as Identified by Next-Generation Sequencing. Neoplasia. 2018; 20: 256-62. https://doi.org/10.1016/j.neo.2017.12.005.
8. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, Lito P, Berger M, Solit DB, Hellmann M, Kris MG, Rudin CM, Ni A, et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res. 2018; 24: 334-40. https://doi.org/10.1158/1078-0432.CCR-17-1841.
9. Wong S, Witte ON. The BCR-ABL story: bench to bedside and back. Annu Rev Immunol. 2004; 22: 247-306. https://doi.org/10.1146/annurev.immunol.22.012703.104753.
10. Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013; 13:559–71. https://doi.org/10.1038/nrc3563.
11. Khatri A, Wang J, Pendergast AM. Multifunctional ABL kinases in health and disease. J Cell Sci. 2016; 129: 9-16. https://doi.org/10.1242/jcs.175521.
12. Choi JR, Koh SB, Kim HR, Lee H, Kang DR. Radon Exposure-induced Genetic Variations in Lung Cancers among Never Smokers. J Korean Med Sci. 2018; 33: e207. https://doi.org/10.3346/jkms.2018.33.e207.
13. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007; 131: 1190-203. https://doi.org/10.1016/j.cell.2007.11.025.
14. Gu JJ, Rouse C, Xu X, Wang J, Onaitis MW, Pendergast AM. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight. 2016; 1: e89647. https://doi.org/10.1172/jci.insight.89647.
15. Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010; 463: 501-6. https://doi.org/10.1038/nature08675.
16. Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J, Hogan BL, Onaitis MW. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci U S A. 2012; 109: 4910-5. https://doi.org/10.1073/pnas.1112499109.
17. Barletta JA, Yeap BY, Chirieac LR. Prognostic significance of grading in lung adenocarcinoma. Cancer. 2010; 116: 659-69. https://doi.org/10.1002/cncr.24831.
18. Sun Z, Aubry MC, Deschamps C, Marks RS, Okuno SH, Williams BA, Sugimura H, Pankratz VS, Yang P. Histologic grade is an independent prognostic factor for survival in non-small cell lung cancer: an analysis of 5018 hospital- and 712 population-based cases. J Thorac Cardiovasc Surg. 2006; 131: 1014-20. https://doi.org/10.1016/j.jtcvs.2005.12.057.
19. Lange AW, Sridharan A, Xu Y, Stripp BR, Perl AK, Whitsett JA. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J Mol Cell Biol. 2015; 7: 35-47. https://doi.org/10.1093/jmcb/mju046.
20. Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, Li J, Huang H, Cai T, Ji H, Yang C, Tang N. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016; 16: 1810-9. https://doi.org/10.1016/j.celrep.2016.07.020.
21. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007; 17: 2054-60. https://doi.org/10.1016/j.cub.2007.10.039.
22. Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010; 24: 862-74. https://doi.org/10.1101/gad.1909210.
23. Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003; 114: 445-56.
24. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, Chinnaiyan A, Israel MA, Goldstein LS, Abujarour R, Ding S, Guan KL. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010; 24: 1106-18. https://doi.org/10.1101/gad.1903310.
25. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 2011; 144: 782-95. https://doi.org/10.1016/j.cell.2011.02.031.
26. Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009; 16: 425-38. https://doi.org/10.1016/j.ccr.2009.09.026.
27. Kim MH, Kim YK, Shin DH, Lee HJ, Shin N, Kim A, Lee JH, Choi KU, Kim JY, Lee CH, Sol MY. Yes associated protein is a poor prognostic factor in well-differentiated lung adenocarcinoma. Int J Clin Exp Pathol. 2015; 8: 15933-9.
28. Sun Z, Xu R, Li X, Ren W, Ou C, Wang Q, Zhang H, Zhang X, Ma J, Wang H, Li G. Prognostic Value of Yes-Associated Protein 1 (YAP1) in Various Cancers: A Meta-Analysis. PLoS One. 2015; 10: e0135119. https://doi.org/10.1371/journal.pone.0135119.
29. Fukui T, Shaykhiev R, Agosto-Perez F, Mezey JG, Downey RJ, Travis WD, Crystal RG. Lung adenocarcinoma subtypes based on expression of human airway basal cell genes. Eur Respir J. 2013; 42: 1332-44. https://doi.org/10.1183/09031936.00144012.
30. Khatri A, Kraft BD, Tata PR, Randell SH, Piantadosi CA, Pendergast AM. ABL kinase inhibition promotes lung regeneration through expansion of an SCGB1A1+ SPC+ cell population following bacterial pneumonia. Proc Natl Acad Sci U S A. 2019; 116: 1603-12. https://doi.org/10.1073/pnas.1816030116.
31. Szymaniak AD, Mahoney JE, Cardoso WV, Varelas X. Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev Cell. 2015; 34: 283-96. https://doi.org/10.1016/j.devcel.2015.06.020.
32. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016; 29: 783-803. https://doi.org/10.1016/j.ccell.2016.05.005.
33. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015; 15: 73-9. https://doi.org/10.1038/nrc3876.
34. Cheng H, Zhang Z, Rodriguez-Barrueco R, Borczuk A, Liu H, Yu J, Silva JM, Cheng SK, Perez-Soler R, Halmos B. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget. 2016; 7: 28976-88. https://doi.org/10.18632/oncotarget.6721.
35. Ma K, Xu Q, Wang S, Zhang W, Liu M, Liang S, Zhu H, Xu N. Nuclear accumulation of Yes-Associated Protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer Lett. 2016; 375: 84-91. https://doi.org/10.1016/j.canlet.2016.02.045.
36. Ghiso E, Migliore C, Ciciriello V, Morando E, Petrelli A, Corso S, De Luca E, Gatti G, Volante M, Giordano S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia. 2017; 19: 1012-21. https://doi.org/10.1016/j.neo.2017.10.003.
37. Singleton KR, Crawford L, Tsui E, Manchester HE, Maertens O, Liu X, Liberti MV, Magpusao AN, Stein EM, Tingley JP, Frederick DT, Boland GM, Flaherty KT, et al. Melanoma Therapeutic Strategies that Select against Resistance by Exploiting MYC-Driven Evolutionary Convergence. Cell Rep. 2017; 21: 2796-812. https://doi.org/10.1016/j.celrep.2017.11.022.
38. Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol. 2016; 29: 26-33. https://doi.org/10.1016/j.coph.2016.05.002.
39. Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol. 2003; 23: 4013-25.