
INTRODUCTION
Mucosal melanoma is a rare subtype of melanoma that originates from melanocytes in the epithelial lining of the conjunctiva, respiratory, alimentary, and genitourinary tracts. Although a majority of mucosal melanomas arise from the mucosa of the nasal cavity, oral cavity, anorectum, vulvovaginal, they can arise in any part of mucosal membranes. Mucosal melanomas comprise approximately 1% of all melanomas in European populations, but up to 25% in Asian populations [1]. Because mucosal melanoma occurs at sites that are not easily amenable to clinical inspection, patients with mucosal melanoma frequently present with advanced-stage disease often with regional and/or distant metastases, and their prognosis is generally poor [2, 3]. In contrast to cutaneous melanomas that are typically associated with exogenous (ultraviolet light exposure) and endogenous (genetic predisposition) risk factors [4], there are no clear risk factors for mucosal melanomas, and the molecular pathogenesis of mucosal melanoma is not well defined.
Several studies have shown that mucosal melanomas have distinct molecular profiles [5–11]. Molecular drivers such as BRAF p.V600 mutations that are amenable to therapeutic intervention (with a combination of BRAF and MEK inhibitors) are uncommon in mucosal melanomas (<10% of cases) as compared to cutaneous melanoma, where approximately 40% are BRAF mutant; of these 74% are the V600E genotype and 22% are V600K [5–9]. Mutations in NRAS only occur in 10–20% of mucosal melanomas [5, 6], while mutations in GNAQ and GNA11 that are commonly detected in uveal melanoma, occur in approximately 9.5% of mucosal melanomas [10, 11]. Other activating oncogenic events, including the gain-of-function mutations of KIT, are present in approximately 15% of mucosal melanomas [10, 11] but are rare in cutaneous melanomas [12–14]. Unfortunately, the oncogenic drivers of mucosal melanoma remain poorly defined, nor it is known whether they vary in prevalence among melanomas from different mucosal sites of the body.
Recently, we performed whole-genome sequencing (WGS) on a large cohort (n =183) of cutaneous (n = 140), acral (n = 35) and mucosal melanomas (n = 8), and SF3B1 (splicing factor 3B subunit 1) was identified as significantly mutated gene in mucosal melanoma [15]. SF3B1 mutations are the most common spliceosomal component gene mutation implicated in the pathogenesis of cancer and act by causing aberrant RNA splicing events [16–19]. Among the different subtypes of melanoma, deleterious somatic variants in SF3B1 were identified in 20% of uveal melanomas [17, 20, 21]. Several studies have identified mutations in SF3B1 in subsets of solid tumors, as well as in myelodysplastic syndrome and chronic lymphocytic leukemia (CLL), in which they occurred in almost 15% of the reported cases [18, 22].
Common molecular drivers and mutations affecting spliceosomal components such as SF3B1 have been reported to be associated with disease outcome in some cancer types, but not in mucosal melanoma [20, 23–26]. In this study, we sought to determine the prevalence of genetic alterations in SF3B1 and of common oncogenic driver genes in mucosal melanomas, and investigate their impact on clinicopathologic characteristics and patient outcomes. To do this, we performed a novel dual-strand amplicon-based targeted sequencing covering all the previously defined significantly mutated melanoma genes [15] in a cohort of 27 mucosal melanomas arising from a variety of anatomical locations including vulvovaginal, anorectal, nasopharyngeal, conjunctival and oropharyngeal sites.
RESULTS
Mucosal melanoma patient characteristics
There were 27 patients included in this study (Table 1 and Supplementary Table 1); 22 females (81%) and 5 males (19%), with a median age at diagnosis of 65.5 years (range 29 to 109 years). The primary melanomas were located in the vulva/vagina (n = 15, 55%), anorectal region (n = 5, 18.5%), nasopharynx (n = 5, 18.5%), conjunctiva (n = 1, 4%) and palate (n = 1, 4%). Fourteen (52%) patients had T4 disease, three (11%) had T3 disease, and the remaining patients had T0, T1 or T2 disease. Tumor thickness was >1 mm in 22 (82%) patients. Median mitotic rate was 15 mitoses/mm2 and ulceration was present in 16 of 22 patients (72%) with known ulceration status.
Table 1: Clinicopathological characteristics of patients with mucosal melanoma (n = 27)
Characteristics |
N (%)a |
|---|---|
Age (median, IQR) |
61 years, 51–78 years |
Gender |
|
Female |
22 (81%) |
Male |
5 (19%) |
Site |
|
Vulva |
12 (44%) |
Vagina |
3 (11%) |
Rectum |
3 (11%) |
Anus |
2 (7%) |
Conjunctiva |
1 (4%) |
Nasal cavity |
4 (15%) |
Nasal Sinus |
1 (4%) |
Palate |
1 (4%) |
T classification |
|
0 |
1 (4%) |
1 |
1 (4%) |
2 |
5 (19%) |
3 |
3 (11%) |
4 |
14 (52%) |
Data unavailable |
3 (11%) |
N classification |
|
N0 |
16 (59%) |
N+ |
7 (26%) |
Data unavailable |
4 (15%) |
Mitotic rate (number of mitoses/mm2) |
|
< 10 |
7 (26%) |
≥ 10 |
14 (52%) |
Data unavailable |
6 (22%) |
Ulceration |
|
Absent |
6 (22%) |
Present |
16 (59%) |
Data unavailable |
5 (19%) |
Tumor thickness (mm) |
|
<1 |
1 (4%) |
1–4 |
8 (30%) |
≥ 4 |
14 (52%) |
Data unavailable |
4 (15%) |
aUnless otherwise indicated.
IQR indicates interquartile range.
SF3B1 and KIT mutations are oncogenic driver mutations in mucosal melanoma
A 45 gene targeted NGS panel was designed to include all significantly mutated genes (SMG) identified in cutaneous, mucosal or acral melanomas in our previous publications [15, 27]. The panel includes all SMG identified in the Hayward et al or TCGA SKCM datasets that were detected using the MutSig, OncodriveFML or IntOGen driver detection tools for coding mutations and OncodriveFML for non-coding genes, as outlined in the prior studies [15, 27, 28]. The targeted NGS panel identified a total of 1435 variants that passed the variant caller filters with a median coverage depth of 2,700X (1,000–22,113X). The NGS panel identified SF3B1 (6 of 27: 22%) as the most commonly mutated gene, followed by KIT (3 of 27: 11%) (Figure 1A). Other less frequently mutated genes included BRAF (7%), NRAS (7%), ARID2, CTNNB1, DICER1, MAP2K1, NF1, PTEN, SETD2 and TP53 (all 4%, Figure 1A). Both SF3B1 (50%) and KIT (67%) mutations were most frequently mutated in tumors of female genital origin and anorectal region (33.3% for SF3B1 and KIT) compared to a single SF3B1 mutant conjunctival melanoma in the upper body sites (Figure 1B). All five SF3B1 mutations (5 of 6: 83%) that occurred in the known hotspot p.R625H/L originated in the anorectal or female genital region, while the conjunctival primary harbored a SF3B1 p.C1123Y mutation (Figure 1B). The KIT mutations were all missense, and of these, two were the hotspot mutation p.L576P in exon 11, and the other a p.T670I mutation in exon 14. KIT mutations in exon 11 and 14 are known to occur in thymic cancer and cutaneous melanoma, and gastrointestinal stromal tumors, respectively. The details of the oncogenic classification (oncodriveMUT), nucleotide and amino acid changes are summarized in Supplementary Table 2.
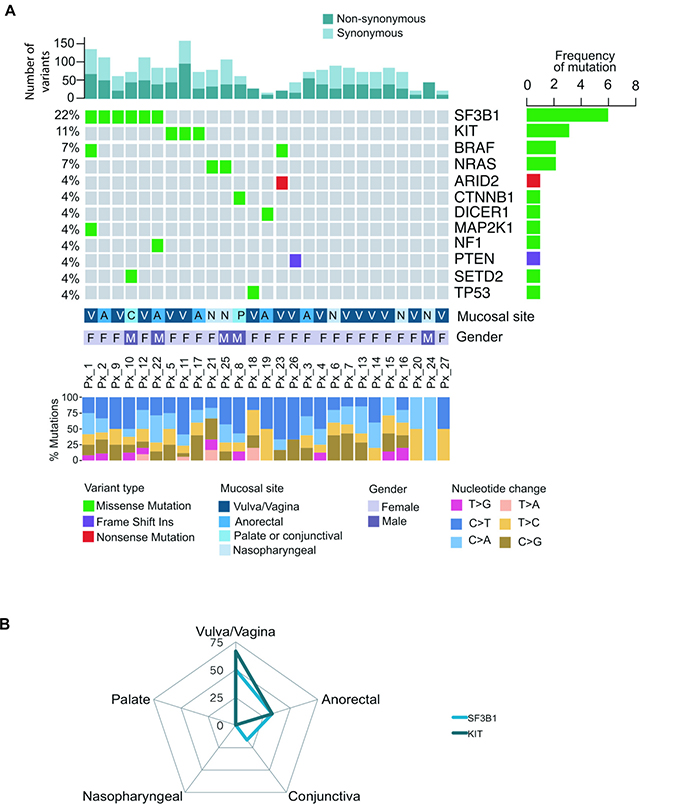
Figure 1: Mutational landscape of 27 melanoma patients across different mucosal sites. (A) The oncoplot showing the distribution of different mutational types across 19 genes. Each column represents a mucosal melanoma sample (individual patient). Number of driver events for each patient is shown at the top of the panel. The frequency of mutation for each gene is shown in the right panel. The mucosal site, gender and percentage of nucleotide changes for individual patients are presented in the lower panel. (B) The radar graph displays the frequency of various gene mutations in different mucosal sites including vulva/vagina, anorectal, nasopharyngeal, conjunctiva and palate. Height of the peak indicates the frequency of individuals with a mutation in the respective mucosal site.
Recurrent hotspot SF3B1 mutations at codon 625 in anorectal and vulva/vaginal melanomas
We previously reported splicing factor SF3B1 as a significantly mutated gene in mucosal melanoma using OncodriveFML (Hayward et al. 2017). Similar to The Cancer Genome Atlas cohort and our previous published work [15, 29], the recurrent SF3B1 mutations occured at codon 625, comprising four p.R625H and one p.R625L alterations (Figure 2A). These recurrent SF3B1 mutations were only identified in anorectal and vulva/vagina mucosal melanomas and not in melanoma samples from the conjunctiva, nasopharynx or palate (representative histological images in Figure 2B and 2C). The specific mutations found in each patient are described in Supplementary Table 2.
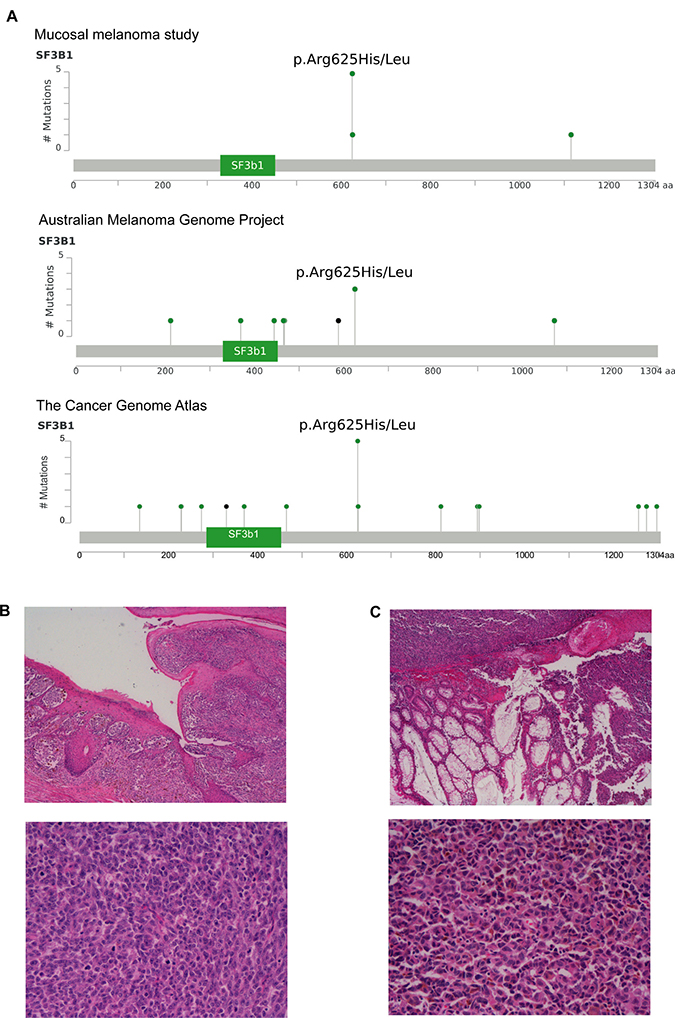
Figure 2: Recurrent hotspot SF3B1 R625 mutations and co-occurrence mutation events in mucosal melanoma. (A) Lollipop plots showing amino acid changes (p.Arg625His/Leu) at codon 625 on SF3B1 protein structure in the present mucosal melanoma study, Australian Melanoma Genome Project and The Cancer Genome Atlas (SKCM-TCGA). The y-axis shows the number of mutations and the x-axis represent the amino acid residues of SF3B1 protein. Representative images of two cases (H & E x 4–upper panel, H & E x 20–lower panel) of mucosal melanoma with SF3B1 mutation obtained from vulva (B) and (C) rectal sites.
Association of mutations with clinical and survival outcomes
The clinicopathological characteristics of tumors with SF3B1 and non-SF3B1 mutations are detailed and compared in Table 2. Representative histological images of SF3B1 mutant tumors are presented in Figures 2B and 2C. Of all the patients with SF3B1 mutations, two (33.3%) had T2 disease, one (16.7%) had T3 disease, and the remaining three (50%) had T4 disease. The depth of invasion of SF3B1 mutant tumors ranged from 1.9 to 12 mm. Melanomas with SF3B1 mutations had a similar mitotic rate when compared to non-SF3B1 mutated cases (15.6±4.4 vs 15.5±2.54 mitoses/mm2) but were more often ulcerated (SF3B1: 6 out of 6 cases vs non-SF3B1: 10 of 16 cases with ulceration data), and were comprised of heterogeneous cell types. We tested whether SF3B1 mutated and non-SF3B1 cases had different prognosis. Median overall survival (OS) was 34.9 months in patients harboring SF3B1 mutations compared to 79.7 months in patients with non-SF3B1 mutations (HR: 2.44, 95% CI: 0.54 to 11, Log-rank test P = 0.117; Figure 3A). Median progression-free survival (PFS) was 16.9 months in the SF3B1 mutant group and 35.7 months in non-SF3B1 patients (HR: 0.474, 95% CI: 0.139 to 1.62, Log-rank test P = 0.0963; Figure 3B).
Table 2: Association of SF3B1 mutations with clinicopathological features
Characteristics |
SF3B1 mutations identified (n = 6) N (%)a |
Non-SF3B1 mutations identified (n = 21)a N (%)a |
P-value |
|---|---|---|---|
Age (median, IQR) |
52.5 years, 46.5–71.5 years |
64 years, 54.5–79 years |
0.309 |
Gender |
0.303 |
||
Female |
4 (67%) |
18 (86%) |
|
Male |
2 (33%) |
3 (14%) |
|
Site |
>0.999 |
||
Vulva/vagina |
3 (50%) |
12 (57%) |
|
Other sites (total) |
3 (50%) in total |
9 (43%) in total |
|
Anorectal |
2 (33%) |
3 (14%) |
|
Nasopharyngeal |
0 (0%) |
5 (24%) |
|
Conjunctiva |
1 (17%) |
0 (0%) |
|
Palate |
0 (0%) |
1 (5%) |
|
T classification |
>0.999 |
||
0–2 |
2 (33%) |
5 (24%) |
|
3–4 |
4 (67%) |
13 (62%) |
|
Data unavailable |
0 (0%) |
3 (14%) |
|
N classification |
0.272 |
||
N0 |
5 (83%) |
12 (57%) |
|
N+ |
0 (0%) |
7 (33%) |
|
Data unavailable |
1 (17%) |
2 (10%) |
|
Mitotic rate (number of mitoses/mm2) |
0.624 |
||
< 10 |
1 (17%) |
6 (29%) |
|
≥ 10 |
4 (67%) |
10 (48%) |
|
Data unavailable |
1 (17%) |
5 (24%) |
|
Ulceration |
0.133 |
||
Absent |
0 (0%) |
6 (29%) |
|
Present |
6 (100%) |
10 (48%) |
|
Data unavailable |
0 (0%) |
5 (24%) |
|
Tumor thickness (mm) |
0.655b |
||
<1 |
0 (0%) |
1 (5%) |
|
1–4 |
3 (50%) |
6 (29%) |
|
≥ 4 |
3 (50%) |
10 (48%) |
|
Data unavailable |
0 (0%) |
4 (19%) |
|
Cell morphology |
0.362c |
||
Epithelioid |
3 (50%) |
13 (62%) |
|
Mixed |
2 (33%) |
1 (5%) |
|
Spindle |
1 (17%) |
4 (19%) |
|
Data unavailable |
0 (0%) |
3 (14%) |
aUnless otherwise indicated.
bTissue with <1 mm is excluded from statistical testing.
cStatistical testing was performed by comparing epithelioid and other cell morphology (mixed and spindle).
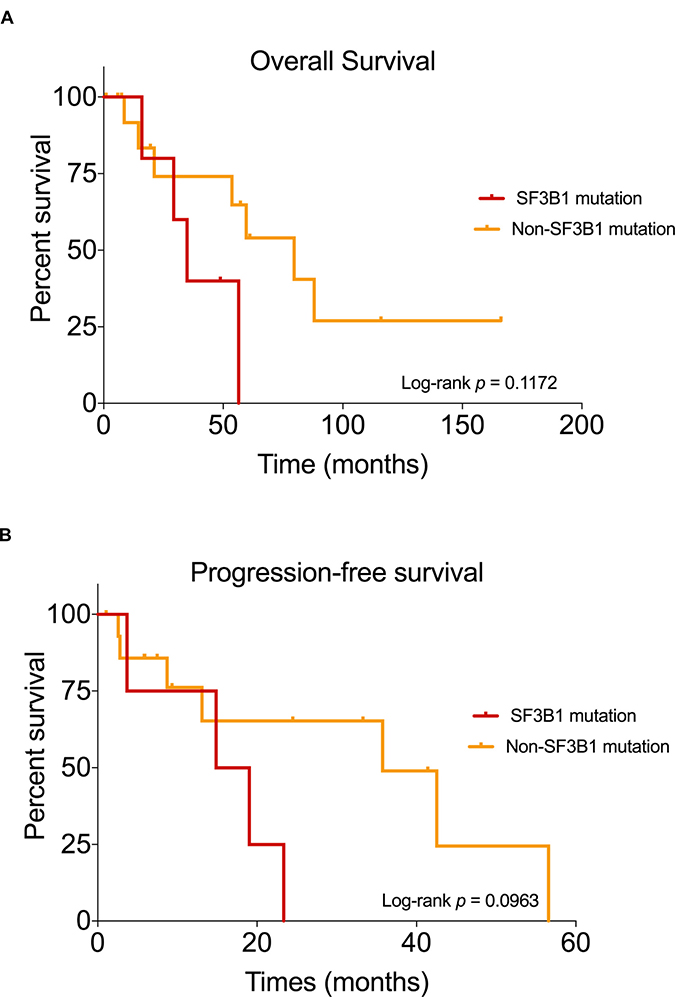
Figure 3: Kaplan–Meier curves showing the survival outcomes of mucosal melanoma patients. Comparison of overall survival (A) and progression-free survival (B) in patients with mutated SF3B1 and non-SF3B1 mutation group.
DISCUSSION
In this study, 27 cases of mucosal melanoma were screened for mutations across 45 key oncogenes identified in our previous whole-genome sequencing study of major melanoma subtypes [15]. We found that SF3B1 and KIT mutations predominantly occurred in melanomas originating in vulval/vaginal sites. Genes that were known to be mutated in cutaneous and uveal melanoma subtypes, including BRAF, NRAS, NF1, GNAQ and GNA11, were rarely mutated in our cohort of patients.
Mutations within exons 12–15 of SF3B1, encoding the C-terminal portion of the protein, have been described in 20% of uveal melanoma, 19% of CLL and 1.8% of breast cancers [20, 22, 30]. In our cohort of 27 mucosal melanoma patients, six (22%) carried a SF3B1 mutation (in exons 14 and 23). Five patients harbored a mutation at exon 14, including p.R625H (n = 4) and p.R625L (n = 1), while one patient carried a p.C1123Y mutation within exon 23 in SF3B1. Of particular interest is the confirmation that the SF3B1 hotspot mutant cases are apparently unique to mucosal melanomas of the lower body sites, hinting to divergent biology with those of upper body sites [24]. Furthermore, somatic mutations in KIT have been reported in 15–20% of mucosal melanomas, and are more commonly observed in anorectal and vulval/vaginal tumors (15–25%) [13, 31–33]. Our study supports these previous reports [13, 31–33], and detected KIT mutations in 11% of mucosal melanomas, particularly in the vulval and vaginal sites. KIT functions as a receptor tyrosine kinase, which transmits signals from the cell membrane into the cell [34, 35]. Once activated, KIT plays an important role in initiating the activation of MAPK/MEK and PI3K/AKT pathways that are critical in cancer development [36]. The aberrant MAPK/MEK and PI3K/AKT pathways impact a variety of cellular activities including cell proliferation and differentiation, which can result in neoplastic growth. Furthermore, besides SF3B1 and KIT mutations, other variants have been described in the context of mucosal melanoma, including amplifications of CCND1, MDM2 and KRAS ([15, 24]), however this was not assessed this cohort and could represent a source of additional driver eventsin this cohort.
Our findings are in line with the growing evidence that the recurrent hotspot mutation at codon 625 of SF3B1 has functional impact in initiating aberrant 3’ splice site selection causing down-regulation of canonical protein expression to promote tumorigenesis [16–18, 20, 24]. The cancer-associated p.R625 mutation resulted in a conformational change in the U2 snRNP complex, such as p14 or U2AF, leading to the binding of alternative branchpoints for RNA splicing that processed these aberrant transcripts into aberrant proteins with altered functions [16, 17, 24, 25]. In melanoma, there has been significant interest in establishing whether the splicing inhibition contributes to cancer development and progression by specific pathologic splicing events. SF3B1 inhibitors have now been studied in patients with locally advanced or metastatic solid tumors. As an example, E7107 was tested in the phase I, open-label and single-arm clinical trial (Study E7107-A001-101; Trial registration ID: NCT00499499) for solid tumors, including colorectal, esophageal and pancreatic carcinomas [37, 38]. Pharmacodynamic analysis revealed that splicing inhibition in the peripheral blood mononuclear cells was achieved, and that it was dose dependant and reversible. Overall treatment was well tolerated, however, an unexpected toxicity of bilateral optic neuritis was detected in the patients, leading to the suspension of the clinical trial [37, 38]. Future design of clinical trials that include the spliceosome inhibitors should consider the risk of toxicity in conjunction with the on- and off-target effect of SF3B1 inhibition. Elucidating the impact of SF3B1 mutation in mucosal melanomas may provide more understanding of its role in tumorigenesis, and facilitate the development of new drugs (i.e. SF3B1 inhibitors) for mucosal melanomas with SF3B1 mutations.
SF3B1 mutations have different prognostic associations in different types of cancers [22, 24, 26, 30, 39, 40]. In uveal melanoma and myelodysplastic syndrome, SF3B1 mutations are associated with a better prognosis, whereas in CLL, SF3B1 mutations are correlated with a worse prognosis [17, 20, 40, 41]. In our study, SF3B1-mutated patients had a worse PFS and OS outcomes, however, our study of these rare tumors was underpowered to detect statistically significant differences. Larger studies are required to address this issue.
In summary, we discovered a SF3B1 C1123Y mutation in a conjunctival mucosal melanoma, and a recurrent SF3B1 R625 mutation, which predominantly occurred in female genital tract mucosal melanomas. We validated previous reports that the commonly known mutations in cutaneous melanoma, including BRAF, NRAS, NF1, GNAQ and GNA11, were rarely mutated in mucosal melanomas. This study provides additional insight into genetic alterations that occur in mucosal melanomas. Collectively, these findings provide a better understanding of the oncogenic drivers of tumor development and may the identification of effective systemic therapies for these rare melanomas.
MATERIALS AND METHODS
Patients and sample selection
This study was approved by the New South Wales Department of Health Human Research Ethics Committee (Protocol no. X15-0454). All human research procedures were performed according to the National Health and Medical Research Council of Australia guidelines. Samples were acquired with patients’ informed consent from the Melanoma Institute Australia Biospecimen bank and Royal Prince Alfred Hospital. Mucosal melanomas were defined as occurring in the mucosa membranes of the oral, conjunctiva, respiratory, gastrointestinal and urogenital tracts. The Melanoma Institute Australia’s research database was searched for primary melanomas originating in the aforementioned sites, and all available archival formalin-fixed biopsies were collected from these patients. The H&E slides of the primary melanoma of all cases were carefully reviewed independently by melanoma pathologists (R.V.R, P.M.F, and R.A.S) to ensure they were from a primary mucosal site. Any melanoma that had arisen at the junction of mucosa and cutaneous skin was excluded.
DNA isolation
Formalin-fixed, paraffin-embedded tumor tissue was prepared in 10 μm sections and deparaffinized through xylene and ethanol according to standard procedures. After air-drying, the tumor tissue was manually macro-dissected, as previously reported [42]. Genomic DNA was isolated using AllPrep DNA/RNA kits (Qiagen) according to the manufacturer’s instructions. Samples were quantified using the Qubit dsDNA high sensitivity assay (Life Technologies).
Amplicon library construction and Illumina sequencing
Amplicon dual-strand (DS) library preparation was performed using the TruSeq Custom Amplion Low Input Library Prep Kit according to the manufacturer’s protocol. Briefly, the amount of DNA was quantitated and diluted to a final concentration of 10−25 ng/μl. The DNA samples were hybridized to the custom amplicons across 45 target genes (Supplementary Table 3) using Illumina’s recommended HYB temperature gradient program from 90°C to 40°C. Subsequently, unbound oligos were washed using Stringent Wash Buffer 1 and 60% ethanol, and was followed by extension and ligation of targeted regions of interest. For individual libraries, the extension-ligation products were ligated to i7 and i5 adapters containing an unique indexed sequence, and followed by library amplification. The amplified paired-end DS libraries were pooled and each strand was sequenced independently using the Illumina MiniSeq™ instrument (Illumina).
Amplicon sequence processing and somatic variant analysis
Sequence data were aligned to the UCSC hg19 assembly using BWA 0.7.13 [43], and SAMtools 1.3 [44] was used to convert aligned sequence read format to binary file (BAM) for each respective sample. These BAMs were marked for duplicate reads using Picard 2.1.1 [45]. Somatic variants were detected using Illumina Amplicon DS Somatic Variant Caller. Briefly, the algorithm analyzed each pool separately, and computed the variant scores assuming that the non-reference calls should follow a Poisson distribution: P = 1–CDF(K -1, λ), where P is the probability that no variant is present given K or more observations, and CDF represents Poisson cumulative distribution function. The probability, P, is subsequently converted to a Q-score and only calls with above Q20 were included for further processing. Variants that were marked as passing quality control contained the following criteria: (i) each variant present in both pools, (ii) a cumulative depth of 1000, and (iii) at least 3% or greater variant frequency. Variants were annotated using human genome build UCSC hg19 with ANNOVAR [46]. Variants were further parsed to OncodriveMut in Cancer Genome Interpreter to identify biological and clinical relevance somatic genes [47]. The mutation results and somatic interactions were further analyzed and visualized using R package maftools [48].
Statistical analysis
The association of mutation status with available clinicopathologic characteristics was performed using Mann–Whitney U test and Fischer exact tests for continuous and categorical variables as appropriate. The Kaplan–Meier survival curves were compared according to the type of mutation group using the log-rank test. All statistical analyses were performed using GraphPad Prism 7.0.
Abbreviations
AKT: Protein kinase B, ARID2: AT-Rich Interaction Domain 2, BRAF: Serine/threonine-protein kinase B-raf, C: Cysteine, CI: Confidence interval, CLL: Chronic lymphocytic leukemia, CTNNB1: Catenin Beta 1, DICER1: Ribonuclease III, DNA: Deoxyribonucleic acid, DS: Dual strand, E: Glutamic acid, GNA11: G Protein Subunit Alpha 11, GNAQ: G Protein Subunit Alpha Q, H: Histidine, hg19: human genome reference version 19, HR: Hazard ratio, K: Lysine, KIT: Proto-Oncogene Receptor Tyrosine Kinase, L: Leucine, MAP2K1/MEK/MAPK: Mitogen-Activated Protein Kinase Kinase 1, NF1:Neurofibromatosis 1, NRAS: neuroblastoma RAS viral oncogene homolog, OS: Overall survival, P: Proline, PFS: Progression-free survival, PI3K: Phosphoinositide 3-kinase, PTEN: Phosphatase And Tensin Homolog, R: Arginine, RNA: Ribonucleic acid, SETD2: SET(Su(var)3-9, Enhancer-of-zeste and Trithorax) Domain Containing 2, SF3B1: Splicing factor 3B subunit 1, T: Threonine, TP53: Tumor protein p53, V: Valine, Y: Tyrosine.
Author contributions
C.Q was the lead author, responsible for all major areas of study design, data analysis, and the majority of manuscript composition. R.V.R, P.M.F, and R.A.S were the melanoma pathologists, responsible for reviewing the mucosal melanoma tissues and the clinical data collection. P.S. performed the experiments including DNA isolation, amplicon library construction and targeted sequencing. I.S. assisted the collection of clinical data. R.P.M.S, K.S. and J.F.T were the melanoma surgeons involved in biospecimen collection. N.K.H was involved in the early stages of the study design. G.V.L., G.J.M. and R.A.S. were the supervisory authors on this project. J.S.W was the corresponding author on this study and involved in manuscript edits and the overall study design. All authors were involved in the manuscript review and edits.
ACKNOWLEDGMENTS
Not applicable.
CONFLICTS OF INTEREST
G.L. receives consultant service fees from Amgen, BMS, Array, Pierre Fabre, Novartis, MSD, and Roche.
FUNDING
This work was supported by Melanoma Institute Australia, the New South Wales Department of Health, NSW Health Pathology, National Health and Medical Research Council of Australia (NHMRC) and Cancer Institute NSW (CINSW). G.V.L. and R.A.S. are supported by NHMRC Fellowships. J.S.W. is supported by NHMRC and CINSW fellowships. J.F.T and G.V.L. are supported by the Melanoma Foundation of The University of Sydney through The University of Sydney Medical Foundation.
REFERENCES
1. Mihajlovic M, Vlajkovic S, Jovanovic P, Stefanovic V. Primary mucosal melanomas: a comprehensive review. Int J Clin Exp Pathol. 2012; 5:739–53.
2. Thompson LD, Wieneke JA, Miettinen M. Sinonasal tract and nasopharyngeal melanomas: a clinicopathologic study of 115 cases with a proposed staging system. Am J Surg Pathol. 2003; 27:594–611.
3. Moreno MA, Roberts DB, Kupferman ME, DeMonte F, El-Naggar AK, Williams M, Rosenthal DS, Hanna EY. Mucosal melanoma of the nose and paranasal sinuses, a contemporary experience from the M. D. Anderson Cancer Center. Cancer. 2010; 116:2215–23. https://doi.org/10.1002/cncr.24976.
4. Postow MA, Hamid O, Carvajal RD. Mucosal melanoma: pathogenesis, clinical behavior, and management. Curr Oncol Rep. 2012; 14:441–8. https://doi.org/10.1007/s11912-012-0244-x.
5. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353:2135–47. https://doi.org/10.1056/NEJMoa050092.
6. Tacastacas JD, Bray J, Cohen YK, Arbesman J, Kim J, Koon HB, Honda K, Cooper KD, Gerstenblith MR. Update on primary mucosal melanoma. J Am Acad Dermatol. 2014; 71:366–75. https://doi.org/10.1016/j.jaad.2014.03.031.
7. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949–54. https://doi.org/10.1038/nature00766.
8. Lyle M, Haydu LE, Menzies AM, Thompson JF, Saw RP, Spillane AJ, Kefford RF, Mann GJ, Cooper WA, Yu B, Scolyer RA, O'Toole SA, Long GV. The molecular profile of metastatic melanoma in Australia. Pathology. 2016; 48:188–93. https://doi.org/10.1016/j.pathol.2015.12.008.
9. Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011; 29:1239–46. https://doi.org/10.1200/JCO.2010.32.4327.
10. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010; 363:2191–9. https://doi.org/10.1056/NEJMoa1000584.
11. Sheng X, Kong Y, Li Y, Zhang Q, Si L, Cui C, Chi Z, Tang B, Mao L, Lian B, Wang X, Yan X, Li S, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016; 65:156–63. https://doi.org/10.1016/j.ejca.2016.06.019.
12. Ashida A, Takata M, Murata H, Kido K, Saida T. Pathological activation of KIT in metastatic tumors of acral and mucosal melanomas. Int J Cancer. 2009; 124:862–8. https://doi.org/10.1002/ijc.24048.
13. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006; 24:4340–6. https://doi.org/10.1200/JCO.2006.06.2984.
14. Tzen CY, Wu YH, Tzen CY. Characterization of KIT mutation in melanoma. Dermatologica Sinica. 2014; 32:7–12.
15. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, Jakrot V, Kazakoff S, Holmes O, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017; 545:175–80. https://doi.org/10.1038/nature22071.
16. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, Corson L, Feala J, Fekkes P, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3' Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015; 13:1033–45. https://doi.org/10.1016/j.celrep.2015.09.053.
17. Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, Stern MH, Marais R. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013; 3:1122–9. https://doi.org/10.1158/2159-8290.CD-13-0330.
18. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011; 365:1384–95. https://doi.org/10.1056/NEJMoa1103283.
19. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012; 486:353–60. https://doi.org/10.1038/nature11143.
20. Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013; 45:133–5. https://doi.org/10.1038/ng.2523.
21. Decatur CL, Ong E, Garg N, Anbunathan H, Bowcock AM, Field MG, Harbour JW. Driver Mutations in Uveal Melanoma: Associations With Gene Expression Profile and Patient Outcomes. JAMA Ophthalmol. 2016; 134:728–33. https://doi.org/10.1001/jamaophthalmol.2016.0903.
22. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, Zhang W, Vartanov AR, Fernandes SM, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011; 365:2497–506. https://doi.org/10.1056/NEJMoa1109016.
23. Kfir N, Lev-Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, Ast G. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015; 11:618–29. https://doi.org/10.1016/j.celrep.2015.03.048.
24. Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE, Applegate AJ, Couts KL, Medina TM, Wells KR, Wisell JA, McCarter MD, Box NF, et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017; 27:189–99. https://doi.org/10.1097/CMR.0000000000000345.
25. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, Dutertre M, Stern MH. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016; 7:10615. https://doi.org/10.1038/ncomms10615.
26. DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3’splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015; 11:e1004105.
27. Wilmott JS, Johansson PA, Newell F, Waddell N, Ferguson P, Quek C, Patch AM, Nones K, Shang P, Pritchard AL, Kazakoff S, Holmes O, Leonard C, et al. Whole genome sequencing of melanomas in adolescent and young adults reveals distinct mutation landscapes and the potential role of germline variants in disease susceptibility. Int J Cancer. 2019; 144:1049–60. https://doi.org/10.1002/ijc.31791.
28. Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 2015; 161:1681–96. https://doi.org/10.1016/j.cell.2015.05.044.
29. Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, Friedlander P, Gonzalez R, Weber JS, Gajewski TF, O'Day SJ, Kim KB, Lawrence D, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013; 31:3182–90. https://doi.org/10.1200/JCO.2012.47.7836.
30. Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B, Natrajan RC. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015; 235:571–80. https://doi.org/10.1002/path.4483.
31. Satzger I, Schaefer T, Kuettler U, Broecker V, Voelker B, Ostertag H, Kapp A, Gutzmer R. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br J Cancer. 2008; 99:2065–9. https://doi.org/10.1038/sj.bjc.6604791.
32. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F 3rd, Azar S, Rubin BP, Muller S, West R, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008; 14:6821–8. https://doi.org/10.1158/1078-0432.CCR-08-0575.
33. Torres-Cabala CA, Wang WL, Trent J, Yang D, Chen S, Galbincea J, Kim KB, Woodman S, Davies M, Plaza JA. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Modern Pathology. 2009; 22:1446.
34. Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. 2012; 92:1619–49. https://doi.org/10.1152/physrev.00046.2011.
35. Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, Corless CL, Li L, Li H, Sheng X, Cui C, Chi Z, Li S, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011; 29:2904–9. https://doi.org/10.1200/JCO.2010.33.9275.
36. Woodman SE, Davies MA. Targeting KIT in melanoma: a paradigm of molecular medicine and targeted therapeutics. Biochem Pharmacol. 2010; 80:568–74. https://doi.org/10.1016/j.bcp.2010.04.032.
37. Eskens FA, Ramos FJ, Burger H, O'Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, Tabernero J. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013; 19:6296–304. https://doi.org/10.1158/1078-0432.CCR-13-0485.
38. Hong D, Kurzrock R, Naing A, Wheler J, Falchook G, Schiffman J, Faulkner N, Pilat M, O’Brien J, LoRusso P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs. 2014; 32:436–44.
39. Inoue D, Abdel-Wahab O. Modeling SF3B1 Mutations in Cancer: Advances, Challenges, and Opportunities. Cancer Cell. 2016; 30:371–3. https://doi.org/10.1016/j.ccell.2016.08.013.
40. Damm F, Thol F, Kosmider O, Kade S, Loffeld P, Dreyfus F, Stamatoullas-Bastard A, Tanguy-Schmidt A, Beyne-Rauzy O, de Botton S, Guerci-Bresler A, Gohring G, Schlegelberger B, et al. SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia. 2012; 26:1137–40. https://doi.org/10.1038/leu.2011.321.
41. Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, Elena C, Galli A, Walldin G, Della Porta MG, Raaschou-Jensen K, Travaglino E, Kallenbach K, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015; 126:233–41. https://doi.org/10.1182/blood-2015-03-633537.
42. Madore J, Vilain RE, Menzies AM, Kakavand H, Wilmott JS, Hyman J, Yearley JH, Kefford RF, Thompson JF, Long GV, Hersey P, Scolyer RA. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res. 2015; 28:245–53. https://doi.org/10.1111/pcmr.12340.
43. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
44. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–9. https://doi.org/10.1093/bioinformatics/btp352.
45. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013; 43:11.10.1–33. https://doi.org/10.1002/0471250953.bi1110s43.
46. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. https://doi.org/10.1093/nar/gkq603.
47. Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, Tusquets I, Albanell J, Rodon J, Tabernero J, de Torres C, Dienstmann R, Gonzalez-Perez A, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018; 10:25. https://doi.org/10.1186/s13073-018-0531-8.
48. Mayakonda A, Koeffler HP. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv. 2016. https://doi.org/10.1101/052662.