
INTRODUCTION
The widespread success of cisplatin (cis-diamino-dichloroplatinum(II)) [1] (cisplatin) for the clinical treatment of various types of cancers in the last 40 years has placed the coordination chemistry of metal-based drugs at the front line in the battle against cancer. The toxic side effects, the low bioavailability and the innate or developed drug resistance of the first FDA-approved platinum drug have only partially been overcome by second and third generation platinum compounds [2]. These issues have stimulated the development of alternative anticancer drugs based on other noble metals [3–6]. Among the first row transition metal complexes, copper coordination compounds are the most promising metal-based anticancer/cytotoxic agents [7]. Copper alteration in cancer has been studied for several decades and aberrant levels of this metal have been found in malignant tissues of both tumour bearing mice and patients suffering from cancer [8–11]. Additionally, high serum concentrations of copper have been correlated to cancer stage, recurrence and/or progression [12–17].
All together these findings suggest that copper might be a diagnostic/prognostic marker [10] and support the idea that the homeostatic machinery of this metal ion could be used as tumour-specific target. Since the 1965 [18], the copper-related tumour progression, has stimulated the development of copper-specific chelating ligands capable to inhibit many malignant processes. These membrane-permeating ligands, that also include diimines and related derivatives, form simple and mixed copper(II) complexes [19–21] and cause an increase of the copper concentration inside cells larger than that resulting from treatment with copper salts alone [22]. Such a behaviour is similar to that shown by metal ionophores which also deliver and release copper to cells [23]. Amongst others, proteasome inhibition [24], generation of reactive oxygen species (ROS) [25], and DNA damage [26] are the mechanisms invoked to account for the anticancer activity of copper complexes.
The family of metal complexes named casiopeinas, i.e. copper-based ternary complexes with diimines and ligands with O, N or O, O donor atoms [27–31], exhibits in vitro anticancer activities greater than that of cisplatin for human cell lines [32]. This class of compounds also displays promising in vivo activity, often at lower concentrations and with lesser side effects than cisplatin [33]. Recently, the different and not mutually exclusive mechanisms of action of these copper coordination compounds with diimines have been reviewed [34]. These compounds have been found to cause tumour cell death by apoptosis [35, 36], both in caspase dependent and independent mechanisms [27, 31, 36], as well as by autophagy [32]. The activities of these metal complexes seem to be linked to: i) generation of ROS [31, 37] with DNA oxidation and degradation [38, 39], with concomitant depletion of antioxidants such as glutathione (GSH) [40, 41]; ii) mitochondrial toxicity [28, 30, 42] and iii) DNA damage through direct interaction with metal complexes (the interaction may take place through intercalation, coordination of the metal to the negatively charged phosphate groups, insertion into the minor groove, partial substitution of some coordinating groups in DNA) [43–45].
ROS, usually generated as side products [46] of the mitochondrial respiratory chain, when present at high levels may cause cell damage by regulating the expression of various apoptosis regulatory proteins [47]. Neoplastic cells possess higher ROS levels than normal cells [48]; consequently, a further increase of ROS may bring these levels to a lethal threshold [15], while resulting safer to normal cells [49]. Owing to its redox characteristics, copper may be involved in processes generating reactive oxygen species (such as the Fenton reaction and the Haber–Weiss reaction) [50], as well as in selective cytotoxicity against cancer cells. In both cases, copper efficiency depends on the properties of the ligands coordinated to the metal ion; for instance, substituents on the phenanthroline rings can affect differently the nuclease activity of the copper complexes [23, 51, 52]. Though the structure and biological properties of copper diimine complexes are still being investigated by various research groups [53–57], a distinctive feature of this class of ligands has attracted our attention: not only the copper complexes of 1,10-phenanthroline and 1,10-phenanthroline-5.6-dione, but also the bare ligands themselves are more cytotoxic than cisplatin in vitro [4, 58–61]. Surprisingly, the mechanism that lies behind such an activity has not received the attention it would have deserved. Being aware that 2,9-dimethyl-1,10-phenathroline and 1,10-phenanthroline form copper complexes having different structure and biological activities [62], here we report on and compare the cytotoxic activities of the copper(II) complexes with 1,10-phenanthroline-5,6-dione (hereafter named phendione) and its 2,9-dimethyl substituted analogue (hereafter named cuproindione) (Scheme 1) on the undifferentiated neuroblastoma cell line (SH-SY5Y). While metal complexes with phendione have already been investigated [63–69], cuproindione copper(II) complexes are reported here for the first time. The present investigation also shows how ROS production consequent to the cell culture treatment with the two 1,10-phenanthroline derivatives alters the metallostasis network (i.e., copper transporters and chaperones) [70] and the redox status of the cells.
Scheme 1: Structures of phendione (A) and cuproindione (B) compounds.
RESULTS AND DISCUSSION
Synthesis and characterization of the compounds
The complex [Cu(cuproindione)2](ClO4)2·2H2O was prepared, isolated and characterized (see Material and Methods) by adapting the procedure used for the preparation of the complexes with phendione [71]. The infrared data highlight the differences between the phendione and the cuproindione copper(II) complexes. The spectrum of [Cu(cuproindione)2](ClO4)2·2H2O displays a strong band at 1699 cm–1 that is assigned to the v(C=O) stretching of coordinated cuproindione [71]. For free cuproindione this band falls at ca 1694 cm-1 (data not shown). The band at about 1590 cm–1 is attributed to v(C=C) of the aromatic ring [72]. The IR spectra of phendione are fairly similar to those obtained for cuproindione, and display smaller shifts for both the v(C=O) stretching and the v(C=O) stretching of the aromatic ring [73]. These bands are detected at ca 1700 cm-1 and 1685 cm-1 (for coordinated and free phendione, respectively) and at 1576 cm-1 for v(C=C) stretching.
In order to gain information on the complexes that form in solution, we followed the changes resulting from addition of copper(II) ion into a solution containing either phendione or cuproindione by UV-vis spectrophotometry (Figure 1). Phendione shows two main absorption bands, centred at 254 and 300 nm. The addition of increasing amounts of copper chloride (from 0.1 to 1.2 equiv.) results in a decrease of the intensity of the band at 254 nm and an increase of the band centred at 300 nm that is also accompanied by a bathochromic shift; The spectrum also shows a quasi-isosbestic point around 284 nm (Figure 1A). Following the addition of 0.5 equivalents of CuCl2, the bands centred at 254 and 300 nm remain nearly constant and increases, respectively. Furthermore, the quasi-isosbestic point is no longer observed and a better-defined absorption band is detected at 314 nm. Slightly different results were obtained for the analogous experiment performed using Cu(ClO4)2 (Figure 1B). Copper(II) perchlorate addition also causes a progressive reduction of the intensity of the band at 254 nm and an increase of the bands around 300 nm, (also accompanied by a bathochromic shift) as well as the appearance of an isosbestic point centred at 295 nm rather than at 284 nm, as observed for CuCl2. For cuproindione, the addition of CuCl2 (from 0.1 to 1.2 equiv.) causes a gradual decrease of the intensity of the bands at 243 and 262 nm and an increase of the shoulder at 309.5 nm (Figure 1C). There is a quasi-isosbestic point centred at 302 nm. There are no significant spectral changes after addition of 0.5 equivalents of CuCl2. A similar result was obtained when using Cu(ClO4)2 (Figure 1D).
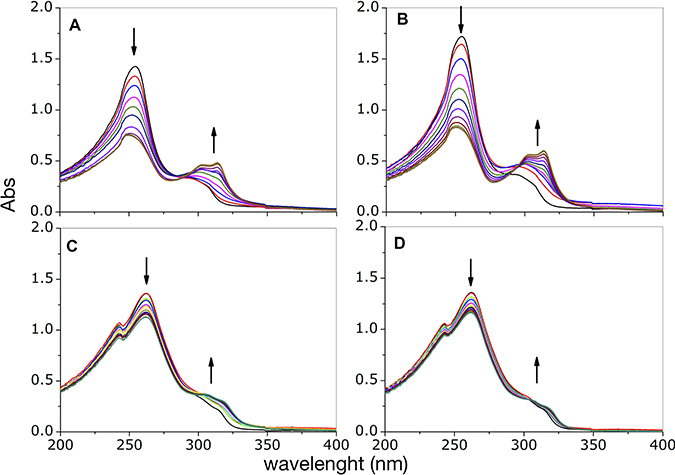
Figure 1: UV-visible curves of phendione (A, B) and cuproindione (C, D) treated with increasing equivalents (0.2- 1.2 range) of CuCl2 (A, C) or Cu(ClO4)2 (B, D).
Figure 2 shows the species that form over the entire range of concentrations explored in the UV-vis titration experiments.
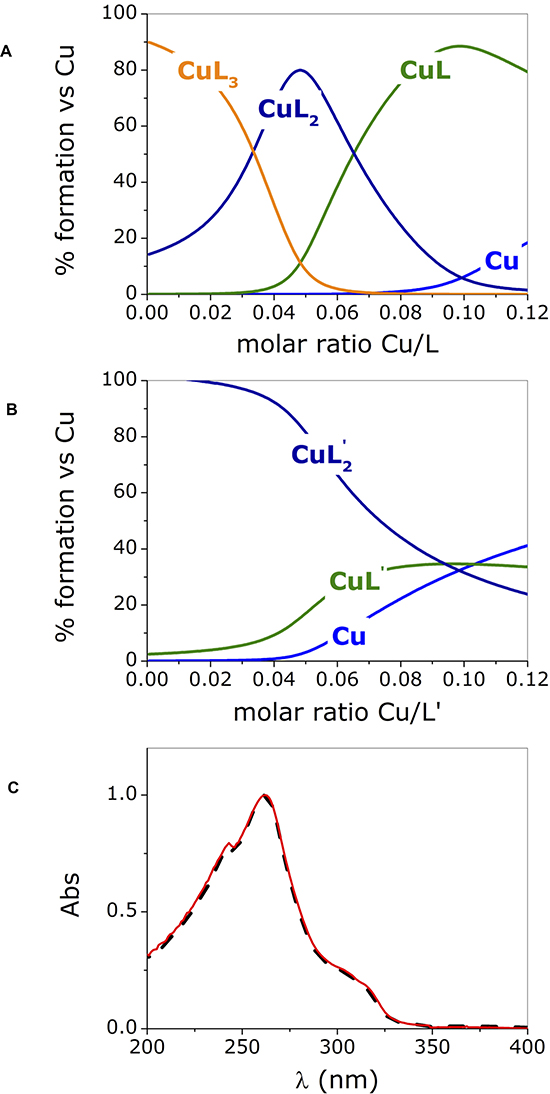
Figure 2: Species distribution computed for phendione (L) (A) and cuproindione (L’) (B) reproducing the conditions employed in the UV-visible experiments (charges are omitted for clarity); (C) UV-spectra of [Cu(cuproindione)2](ClO4)2·2H2O (solid red line) and a solution of cuproindione having a cuproindione/Cu2+ equal to 1:0.5 (dash black line). Titrate concentration= 6x10-5 M; titrant concentration= 1.8x10-3 M.
The distribution diagrams (Figure 2A, 2B) were obtained by using the equilibrium constants reported by Martell et al. [74] for 1,10-phenanthroline (phen) and 2,9-dimethyl-1,10-phenanthroline (2,9-phen), which both lack the carbonyl groups, under the assumption that the carbonyl groups will have similar effects on the two ligands. The assumption is based on a recently published paper showing that the stability constants of copper- phendione complexes is not significantly altered by the presence the carbonyl groups [61]. Figure 2A and 2B clearly show that the species formed in the titration with copper(II) of phendione and cuproindione, under identical experimental conditions are different. In the case of phendione, at least two species coexist (Figure 2A) over the entire range of molar ratios explored. As expected, in the initial part of the titration (i.e. in the presence of large excess of ligand) [Cu(phendione)3]2+ (CuL3) prevails over [Cu(phendione)2]2+ (CuL2); Beyond 0.4 equivalents of Cu2+, the percentage of the latter species rapidly decreases to give way to CuL2 that coexists with the mono-complex (CuL) for a large interval. At a 1:1 molar ratio, CuL is by far the dominant species accompanied by negligible amounts of CuL2 and free copper(II) ion. The UV-vis titrations (Figure 1A and 1B) reflect the simultaneous presence of overlapping species. The picture is less complex for cuproindione (Figure 2B), due to the formation of two species only, namely [Cu(cuproindione)2]2+ (CuL’2) and [Cu(cuproindione)]2+ (CuL’). However, as the 1:1 ratio is approached, the percentage of CuL’2 drops and roughly equivalent amounts (~30%) of CuL’2, CuL’ and free copper(II) ion coexist in these conditions, owing to the smaller formation constants of cuproindione compared to phendione.
The UV-vis spectrum obtained by dissolving the synthesized complex, ([Cu(cuproindione)2] (ClO4)2·2H2O), mirrors the spectrum recorded for a solution with a Cu/L’ (cuproindione/Cu2+ 1:0.5) (Figure 2C). However, the spectrum of both solutions does not result from the presence of a single specie but rather reflects the complex picture resulting from the simultaneous formation of multiple species (Figure 2B). This clearly shows that a solid metal complex with a given stoichiometry does not necessarily survive in aqueous solution with the same stoichiometry, as more than one species can originate, whose concentration ratio depends on their affinity constants.
In order to ascertain the set of donor atoms involved in Cu2+ coordination, EPR measurements were also carried out. The metal to ligand ratios were chosen based on the calculated species distribution (Figure 2), to minimize the overlapping of different complex species. As to phendione, the EPR parameters for the CuL system (g∥ = 2.325 and A∥ = 158 × 10–4 cm-1) indicate the involvement of two nitrogen and two oxygen atoms in a tetragonal arrangement, thus confirming the presence of the [Cu(phendione)]2+ species. The parameters are similar to those reported for analogous complex species formed with 1,10-phenanthroline and its derivatives, in which the metal has the same coordination environment [44]. The EPR spectra obtained by increasing the ligand to metal ratio are characterized by a smaller g value (g∥ = 2.260) and a larger hyperfine coupling constant (A∥ = 175 × 10–4 cm-1), which indicate the substitution of oxygen by nitrogen atoms in the metal coordination environment. These EPR parameters suggest the presence of a CuN4 chromophore, that indicates the formation of a species, [Cu(phendione)2]2+, having nitrogen atoms in a nearly planar disposition. Noteworthy, these parameters closely resemble those reported for the bis-complex species formed by 1,10-phenanthroline [75].
As to cuproindione, the EPR parameters obtained for [Cu(cuproindione]2+ (g∥ =2.323 and A∥ =154 × 10–4 cm-1), are comparable to those obtained for the [Cu(phendione)]2+ species, pointing to the presence of a similar CuN2O2 chromophore. As the metal/ligand ratio increases, the EPR spectra reveal the simultaneous presence of different complex species. At 1:2.5 metal/ligand molar ratio, the parameters of the main species (g∥ = 2.293, A∥ = 162 × 10–4 cm-1) are analogous to those observed for the copper(II) complex species formed by 2,9-dimethyl-1,10-phenanthroline [44, 76]. Such parameters indicate a stronger ligand field compared to [Cu(cuproindione]2+, suggesting the presence of more nitrogen atoms around the metal ions and thus the formation of Cu(cuproindione)2]2+ having a tetragonal geometry more distorted than that of the analogous species formed by phendione.
Cytotoxicity of phendione and cuproindione: the chelators transport copper inside the cell and induce cytotoxicity
Dose-response experiments were performed to compare the activity of cuproindione and phendione on an undifferentiated neuroblastoma cell line (SH-SY5Y). Cells were treated with each compound for 48 hrs exploring the 0.01–10 μM concentration range. The IC50 values of cell viability (MTT assay), obtained by a linear regression of the dose response curves of each compound are reported in Table 1.
Table 1: Cytotoxicity (as IC50 values, in 10-6 M) of phendione (L), cuproindione (L’), [CuCl(phendione)2]ClO4·3/2H2O (1), [Cu(phendione)3](ClO4)2×4H2O (2), [Cu(cuproindione)2] ClO4)2·2H2O (3) and cisplatin (4, [77])
L |
L’ |
1 |
2 |
3 |
L+ BCS |
L’+ BCS |
4 |
|---|---|---|---|---|---|---|---|
1.5 ± 0.1 |
0.78 ± 0.09 |
0.41 ±0.02 |
0.38 ± 0.03 |
0.39 ± 0.03 |
1.56 ± 0.02 |
4.0 ± 0.1 |
2 |
The two ligands (L and L’) show IC50 values lower than that reported for cisplatin, with cuproindione being more cytotoxic than phendione. Moreover, the copper(II) complexes exhibit cytotoxicity values five times greater than that of cisplatin, similarly to what previously reported for [Cu(phendione)3](ClO4)2 and [Ag(Phendione)2] (ClO4), which induce a dose-dependent decrease of DNA synthesis [59]. Analogous results by Igdaloff et al. [58] for metal-free phendione inhibiting the growth of mouse-derived lymphoma cells, were related to the inhibition of DNA and RNA synthesis, as confirmed by Deegan et al. in DNA synthesis studies dealing with human kidney adenocarcinoma and human hepatocellular carcinoma cells [59]. However, other hypothesis have been raised and the anti-proliferative activity of phendione has been attributed to an unspecified cellular microenvironment [61].
Since metal salts are essential components of cell culture media where copper(II) concentration ranges from 0.94 μM to 1.9 μM [78], cells were also pre-treated for 3 hours with BCS (bathocuproine disulfonate), a copper chelator unable to cross the cell membrane [79].
Figure 3 shows that the ability of BCS to remove Cu2+ from the medium affects the anti-proliferative ability of the two ligands differently. Indeed, the toxicity of cuproindione decreases approximately by a factor of four under copper deprivation conditions, whereas the toxicity of phendione is unaffected by BCS. The large effect of BCS upon the toxicity of cuproindione can be explained by the very similar stability constants of copper(II) complexes with BCS and 2,9-phen, (used to mimic the copper(II) affinity of cuproindione). Thus, the large amount of BCS (not permeable to cell membrane) is likely responsible for sequestering Cu2+ outside the cell. By contrast, the negligible effect of BCS upon the toxicity of phendione can be explained by the different affinity constants of Cu2+ complexes with BCS and phen (used to simulate the copper(II) affinity of phendione). The affinity constant of the Cu2+ complex with phen is four orders of magnitude larger than that with BCS. Thus, BCS cannot compete with phendione for copper(II) coordination, notwithstanding its larger concentration. These findings highlight the role of Cu2+ present in the culture medium and may account for the different cytotoxicity of the two phenanthroline derivatives, whose activities can be attributed to their copper complexes. Although both ligands have similar copper chelating features, the distribution diagrams, computed under the assumption that the complexation characteristics of phen and 2,9-phen roughly parallel those of phendione and cuproindione, respectively, highlight the different percentages of complex species that form in the two cases (Supplementary Figure 1).
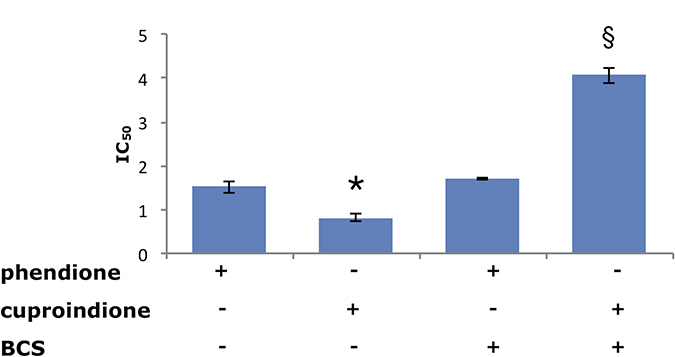
Figure 3: Viability of SH-SY5Y cells assessed by IC50 values (μM) following a 48-hrs treatment with phendione or cuproindione, either in the basal culture medium or after pre-incubation (3 hrs) with 50 μM BCS. Values are expressed as mean ± SEM over at least three independent experiments. (*=p ≤ 0.05 level vs phendione, §=p ≤ 0.05 level vs cuproindione; One-way Anova).
The distribution diagrams obtained by using the Cu2+ concentration reported for the medium by Huang et al. [78] show that for both ligands the mono- and bis-complex are the dominant species over the range of ligand concentrations investigated (0.01 μM - 10 μM) in the biological assays. At small ligand to metal (L/Cu or L’/Cu) ratios, CuL (or CuL’) is the main species; however, CuL reaches percentages as large as 90% (Supplementary Figure 1A) only in the case of phendione thanks to the higher affinity of phen for Cu2+ compared to 2,9-phen (Supplementary Figure 1A and 1E). As the L/Cu (or L’/Cu) ratio increases (Supplementary Figure 1B–1D and 1F–1H), CuL2 and CuL’2 override the formation of the CuL (or CuL’) species. CuL3 forms only in the case of phendione and in the presence of excess of ligand. However, even when the ratio is pushed up to reproduce roughly the conditions of the toxicity found in the MTT assay, CuL2 and CuL’2 are still the main species in both systems and reach roughly the same percentage (Supplementary Figure 1D and 1H). CuL2 stimulates hydroxyl radical formation from molecular oxygen by redox cycling and thus might promote oxidative stress [59]. The reaction pathway proceeds via the formation of a cuprous complex in solution ([Cu(phen)2]+), characterized by a non-intercalative binding of the tetrahedral metal complex in the DNA minor groove [80]. The [Cu(2,9-phen)2]+ stability constant (log β = 19.1) is significantly larger than that of [Cu(phen)2]+ (log β = 15.8) [81], due to the more favourable disposition of the methyl groups of 2,9 phen in the tetrahedral copper(I) bis-chelate complex.
The transcription inhibitor ability of [Cu(2,9-phen)2]+ contributes to its cytotoxicity on eukaryotic and prokaryotic cells [82]; altogether, these evidences can justify the copper-dependent different anti-proliferative activity of cuproindione and phendione (Table 1). The greater stability of the cuprous complex may explain, at least in part, the larger anti-proliferative effect of cuproindione with respect to phendione.
Phendione and cuproindione induce nuclease activity, oxidative stress, and apoptosis and affect mitochondria by p53 activation
Several factors have been invoked to explain the different nuclease activity of the mono and bis-complexes of copper (II)/copper(I) with 1,10-phenanthroline and its derivatives [51, 62, 83–86]. These include: a) stoichiometry; b) metal complex geometry (planar vs tetrahedral) [61, 87, 88]; c) redox ability (phen vs 2,9-phen complex species) [62, 89, 90]; d) metal ion (Cu+ vs Cu2+) affinity for the ligand; e) phen and 2,9-phen complex species interaction with reductants within the cell (ternary complex formation with thiols by mono-complex species vs. thiol oxidation by bis-complex species) [91–94]; f) copper complex species interaction with DNA (intercalation vs minor groove binding) [31, 43–45].
Previous studies have focused on copper only, working in a cell-free environment and without differentiating between cupric and cuprous complexes, which have both different stability constants and redox potentials. This may account for some misleading reports on the cleavage efficiency of mono- and bis-complex of copper with phen and 2,9-phen [24, 95, 96].
The biochemical assays carried out in the present work may help unravelling some of the above still unresolved questions. To ascertain whether also in living cells a DNA damage could be one of the toxic effect of phenanthroline derivatives, cells were treated with the compounds under investigation and specific markers (e.g. Poly [ADP-ribose] polymerase 1 (PARP-1)) were analysed. PARP-1 is a well-known apoptotic marker and undergoes proteolytic cleavage by activated caspase 3 [97]. PARP-1 is very sensitive to cleavage as it controls genomic stability [98]. Supplementary Figure 2 shows the assays carried out by Western Blot (WB) analysis for PARP-1 expression (at metal-to-ligand ratios lower than 1, with ligand concentration lower than IC50) and for PARP-1 cleavage (at IC50 values of the two ligands where the cuprous bis-complexes are the main species). The expression of PARP-1 (Supplementary Figure 2), upon treatment with non- toxic concentrations of the ligands (until 0.1 μM), increases with respect to control and reaches similar values with both phendione and cuproindione complexes. Since the induction of PARP-1 expression may be indicative of an interaction between these copper-binding phenanthroline derivatives and DNA, with a consequent endonuclease cleavage [68], our results suggest that the species that forms under these conditions (i.e., CuL or CuL’) interacts with DNA by intercalation and cause a lengthening, stiffening, and unwinding of the helix [99] that is sensed by PARP-1. The lack of toxicity and oxidative cleavage may be ascribed to the formation of ternary complexes of copper(I)-phendione or cuproindione with thiols (likely glutathione), as observed in cell-free experiments employing copper-phendione complexes that showed a delayed oxidative nuclease effect [96]. Following treatment with IC50 concentrations of ligands, the cleaved PARP-1/full PARP ratios (Supplementary Figure 2B), increases by about 120% and 150% (compared to control) for phendione and cuproindione, respectively. For both compounds, the pre-treatment with BCS nullifies the enhanced PARP-1 cleavage compared to the control. We also tested the PARP-1 24 kDa cleaved fragment and obtained roughly the same result (data not shown).
The different oxidative nuclease efficiency of phendione and cuproindione may relate both to the different amount of cuprous species (resulting from the larger affinity constant value of the metal-bis ligand complex species of 2,9-phen compared to phen, log β = 19.1 vs 15.8) and to the higher redox potential of the cupric complexes with 2,9-phen relative to phen (+594 vs +174 mV) [81]. These two parameters affect the oxidative efficiency of the two systems toward GSH, the most relevant reductant in the cytosol (GSH/GSSG redox potential < −300 mV [100]).
In agreement with such hypothesis, we found a decrease of the GSH/GSSG ratio in the cell that is more pronounced for cuproindione than for phendione (Figure 4). This supports the effective role played by the methyl groups of cuproindione and evidences the prevailing role of Cu+ complexes over Cu2+ species. Such a trend has also been reported for the analogous complexes lacking the carbonyl groups [24].
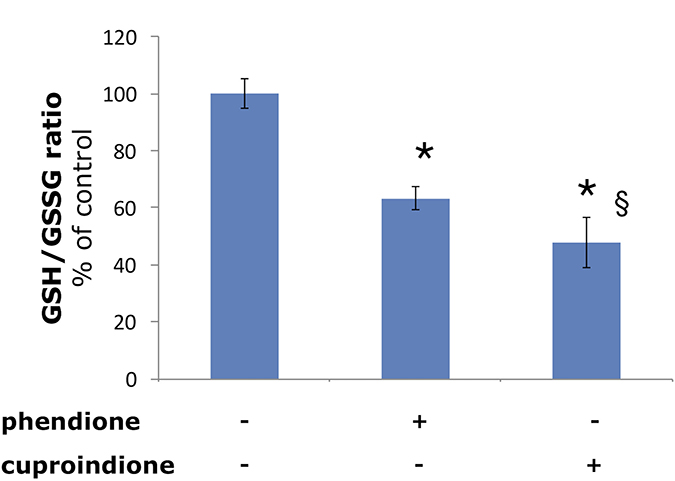
Figure 4: Effect of phendione or cuproindione treatment on GSH redox state in SH-SY5Y cells treated with a phendione or cuproindione 3 × IC50 concentration for 90 minutes. Results are expressed as mean ± SEM from triplicate experiments and normalized with respect to the control (untreated) cells. (*p ≤ 0.05 level vs control; §p ≤ 0.05 level vs phendione; One-way Anova).
The mechanism of apoptosis is a multifaceted process under the control of different pathways and cell life check control. Since copper levels are significantly elevated in a number of malignancies, cancer cells would be more subject to redox cycling between copper species and thus generate larger amounts of ROS, responsible for DNA breakage [101]. In addition to DNA damage and specific cell death signalling, mitochondrial activity disturbances may be sensed by cells as critical steps to the decision for suicide or survival.
Redox cycling between Cu2+ and Cu+ can be efficiently mediated by different compounds exerting cancer cell toxicity owing to a copper related mechanism [102]. The same mechanism might be responsible for mitochondria destabilization and apoptosis activation. Mitochondrial destabilization can easily be followed by monitoring the membrane potential (Δψm), that is rapidly affected by many toxic compounds and oxidative stress as well as by excitotoxic conditions [103].
Figure 5 shows the changes of the mitochondrial membrane potential as well as mitochondrial ROS production following treatment with phendione or cuproindione. The mitochondrial membrane potential was measured through a specific mitochondrial fluorophore, JC-1, which undergoes a fluorescence emission change if depolarization occurs. Figure 5A, 5B show that JC-1 accumulates in the mitochondria of healthy cells under the form of aggregates (red-orange emission), whereas it remains in the cytoplasm in its monomeric form (green fluorescence) in cells treated for 90 min with phendione or cuproindione, as a result of the collapse of Δψm. Noteworthy, the amount of O2•− detected in treated cells (30 min) by monitoring MitoSOX emission is significantly higher for cuproindione (Figure 5C). This suggests a different efficiency of the two compounds, with cuproindione having a stronger effect on the permeability of the mitochondrial membrane. The alteration of Δψm through the generation of ROS can both provide evidence of the disruption of the outer mitochondrial membrane [104] and indicate an important pathway of induced apoptosis, as that attributable to p53 [105].
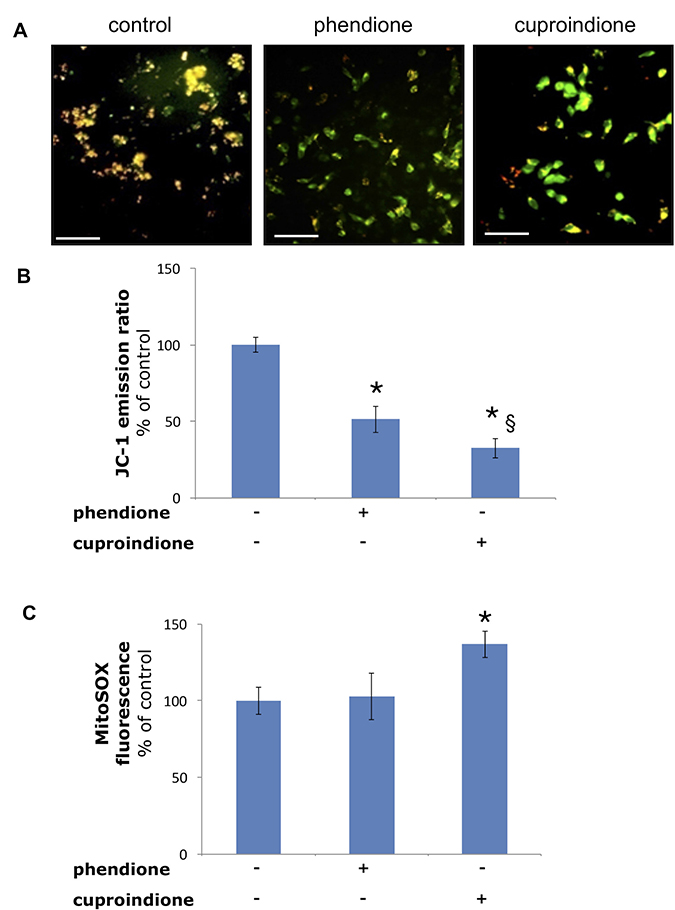
Figure 5: Mitochondria polarization of the cells treated with a phendione or cuproindione 3 × IC50 concentration for 90 minutes. (A) fluorescence micrographs; (B) calculated ratios of JC-1 fluorescence emission at 595/530. Mitochondrial O2•− production of the cells treated with a phendione or cuproindione 3 × IC50 concentration for 30 minutes: (C) MitoSOX emission (ex 510, em 580). Results are expressed as mean ± SEM over three independent experiments with eight replicas for each condition (*= p ≤ 0.05 level vs control, § = p ≤ 0.05 level vs phendione; One-way Anova). Scale bar = 50 μm.
The tumour suppressor protein p53 is a zinc-binding transcription factor [106], often inactivated in cancer cells [107]. DNA damage causes p53 accumulation in the nucleus, binding to a DNA specific sequence, and the transactivation of several target genes [108] involved in cell cycle [109] and apoptosis [110]. DNA strand breaks, induced by oxidative stress [111], is one of the most potent signals leading to p53 induction. Consistently with their diverse oxidative nuclease ability, phendione and cuproindione up regulate differently p53 expression (Figure 6). Noteworthy, once again copper relevance emerges because pre-treatment with BCS only increases the expression of p53. The p53 protein itself is redox-sensitive and contains several cysteines located within the DNA-binding domain [112]. Three of these cysteines and a histidine are involved in the tetrahedral coordination to zinc, leading to a protein arrangement that interacts with the minor groove of target DNA. Several other cysteines are located in the regions that bind within the major groove of target DNA [113]. A murine in vitro assay showed that physiological concentrations of copper(II) ion perturb the conformation of wild-type p53 and inhibit its sequence-specific DNA-binding, while BCS protects against the effect of Cu2+ and prevents the metal ion cellular uptake [114]. The effects of ionophores (e.g. pyrrolidinedithiocarbamate, PDTC) and chelating ligands (e.g. phen) on p53 activity further support the above results and highlight the differences between copper(II) ionophores and chelating ligands [115]. PDTC increases intracellular copper concentration [57, 116], affecting the p53 pathway. The ionophore inhibits the nuclear translocation of p53, induces a “wild-type” to “mutant” transformation, and down-regulates the DNA-binding activity of p53. These multiple effects result from an increase in the oxidation state of p53 cysteine thiols. In turn, the increased oxidation may result from the capacity of PDTC to bind extracellular copper and release it within the cell [117].
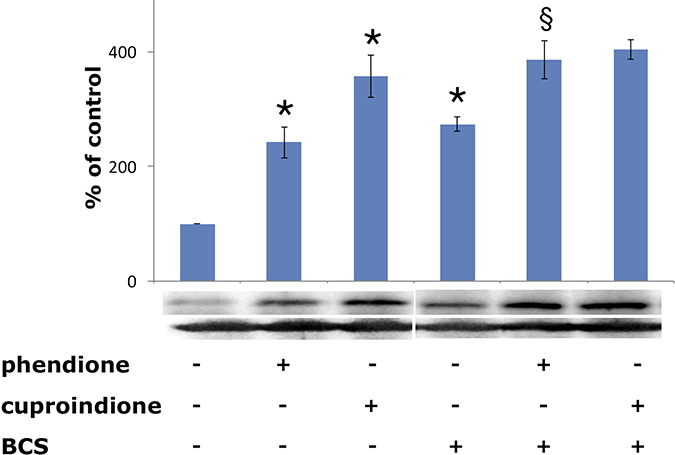
Figure 6: Expression of p53 in SH-SY5Y cells after a 48-hr treatment with phendione and cuproindione IC50 concentration in the absence and in the presence of 50 μM BCS. Results are the mean ± SEM over at least three independent experiments. (*p ≤ 0.05 level vs control, §p ≤ 0.05 level vs phendione; One-way Anova).
Like phendione and cuproindione, phen also favours copper internalization in the cell; however, in the presence of phen, the process takes place through a complex species that upregulates p53 DNA-binding activity via a DNA damage-dependent pathway [57]. Though having similar effects on intracellular copper levels, PDTC and phen have opposite effects on the DNA-binding activity of p53. Furthermore, our findings on phenathroline derivatives further indicate that the effects of phen and PDTC cannot solely result from their ability to increase intracellular copper levels but also depend upon the redox activity of the metal species formed by the two ligands within the cell.
A marked connection between p53 and B-cell lymphoma-2 (Bcl-2) protein family has been reported [118]. The mitochondrial apoptotic pathway is strictly regulated by selective interactions between anti-apoptotic and pro-apoptotic proteins belonging to the Bcl-2 family [119]. This family regulates the intrinsic apoptotic pathway by controlling the mitochondrial outer membrane integrity [120], preventing the p53-induced Δψm alteration [105], and halting apoptosis through an anti-oxidative effect [121, 122]. On the other hand, Bcl-2 may contribute to inhibit caspase-dependent cell death by modulating the activity of the copper-dependent cytochrome c oxidase, the terminal complex of mitochondrial respiratory chain [123]; consequently, copper homeostasis derangements [124] or the Bcl-2 down-regulation, represent critical mitochondrial targets in fighting tumour cells. Bax, one of pro-apoptotic Bcl-2 family protein, can be activated by p53 [125] during apoptosis with the translocation of Bax from the cytosol to mitochondria [126]. Both in vitro [127] and in vivo [128] assays indicate that the pro-apoptotic effects of Bax may be elicited through an intrinsic pore-forming activity [129] accompanied by the release of cytochrome c [130].
Supplementary Figure 3 shows that short-time treatments (90 min) with phendione and cuproindione induce Bax translocation to the mitochondrial membrane (Supplementary Figure 3A) as well as cytochrome C release (Supplementary Figure 3B). Cuproindione is more effective than phendione, consistently with p53 and JC-1 findings. By contrast, long-time treatments (48 hrs) with the two compounds down regulate Bcl-2 expression (Supplementary Figure 3C). The pre-treatment of the medium with BCS nullifies the effect of cuproindione that forms weaker complexes than phendione, highlighting the relevance of the larger stability of copper-phendione complexes in the competition with BCS.
The ratio between Bcl-2 and Bax could determine the fate of cells undergoing apoptosis. The tight control of apoptosis is of critical importance for cancer cells to overcome their highly stressing condition and to counteract the constitutive expression of pro-apoptotic proteins. Noteworthy, the ratio between Bcl-2 and Bax has paved the way to target cell death in cancer and monitor the responses to therapies [131]. The effects observed following cell cultures treatments may well fit in this pro-apoptotic anticancer strategy, and highlight the participation of the copper homeostatic machinery in the apoptotic control of cell life, thereby indicating a new possible way to induce tumour cells to death.
Oxidative stress affects metallostasis
Cellular copper homeostasis is ensured by some copper binding proteins [132] that control concentrations, binding interactions and location of single metal species that determine copper metallome [70, 133]. Prominent modulators of copper homeostasis are: i) import (high-affinity copper transporter 1, CTR1 [134]) and export (ATPase) Cu+ transporters across membranes; ii) trafficking components, including small molecules (GSH) and chaperones (CCS, Atox1, COX17/Sco1-Sco2, COX17) that escort Cu+ inside cell [135]; iii) insertase agents, CCS and Sco1/Sco2 (that insert Cu+ into apo-SOD1 and cytochrome c oxidase, respectively), Atox1 (that transfers Cu+ to ATP7A and ATP7B); iv) storage molecules (metallothionines); and v) metal transcription factors (Atox1, p53, MTF-1, Sp1). These metallostasis [136] regulators can partially remedy copper ion dyshomeostasis, that is responsible for a broad range of human diseases, including tumours [137, 138].
Protein expression experiments were performed to evaluate how copper bound to phendione and cuproindione affects the copper homeostasis network of an undifferentiated neuroblastoma cell line (Figure 7). Figure 7A shows a significant CTR1 up-regulation in cells treated with BCS that sequestrates copper present in the medium and blocks the metal ion outside the cell. Like BCS, phendione and cuproindione also bind copper and markedly increase the expression of the protein devoted to the transferring of the metal ion inside the cell.
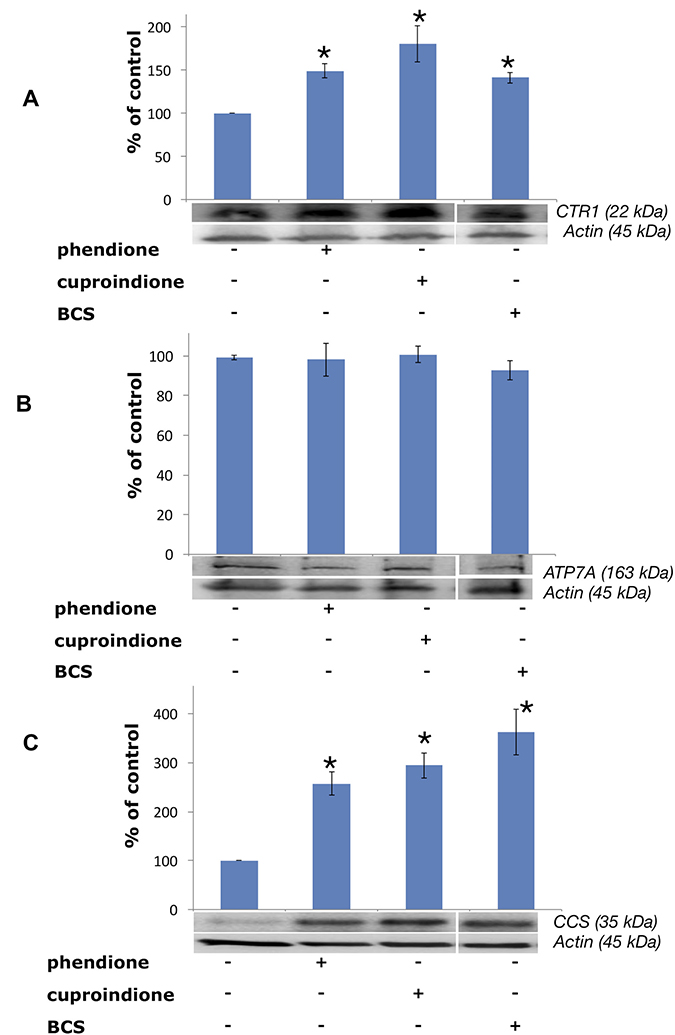
Figure 7: Expression of CTR1 (A), ATP7A (B) and CCS (C) in SH-SY5Y cells after treatment with a phendione (IC50 concentration), cuproindione (IC50 concentration) and BCS (50 μM) for 48 hrs. Results are expressed as mean ± SEM over at least three independent experiments. (*p ≤ 0.05 level with respect to control, One-way Anova).
Noteworthy, CTR1 levels depend upon both the transporter localization and abundance [139]. The plasma membrane is the main location designed to respond to copper cellular demand, while the influx protein is mainly located in the membrane of intracellular vesicles in the presence of elevated exogenous copper [139, 140]. The strong binding of the metal ion to both ligands may account for the observed up-regulation.
Treating cells with membrane-impermeable copper chelators, including ATTM (ammonium tetrathiomolybdate), D-P (D-penicillamine) and BCS causes CTR1 up-regulation [141]. Conversely, these copper chelators do not alter the expression density and pattern of copper efflux ATPases in the same cell lines [142]. In line with such findings, our results show that the expression of ATP7A does not change when neuroblastoma cells are treated with BCS (Figure 7B). Similarly, the membrane permeable ligands reported here do not perturb the expression of ATPA7. The intracellular location of ATPases is controlled by cytoplasm copper concentration; in cultured cells exposed to physiological copper concentrations both ATPases are located in the trans Golgi network, to supply the metal ion to secreted cuproenzymes. In the presence of large copper concentrations, ATPases re-localise to either the plasma membrane (ATP7A) [143] or a vesicular compartment (ATP7B) [144].
The resulting metal efflux and compartmentalization, together with ATPases overexpression, have been associated with drug resistance in different tumors [145].
CCS is a 70 kDA Cu+ protein,203 that is required for the conversion of apo-SOD to holo-SOD, that, in turn, involves the conversion of specific thiols to disulphide [146], process that takes place in the presence of an oxygen-activated enzyme [147]. Figure 7C shows that CCS expression is upregulated by BCS as well as by the two ligands investigated in the present work, i.e., the chaperone behaves as if the cytosol were depleted of metal ion.
To ensure chaperone speciation and redox homeostasis, copper is transferred from a given protein to a different protein having a higher affinity [148], while the chaperone redox status is preserved by GSH (the most abundant antioxidant) that, together with its partners, is believed to maintain the redox potential in tissues, cells and individual compartments [100, 149].
The thiols of cysteines characterize the Cu+ binding to all components involved in metallostasis; in addition, oxidative stress, due to increased levels of Cu+ released within the cell by copper salts, can induce disulphide bond formation [150].
As a result, GSH is transformed into GSSG, while copper chaperone integrity is lost thus perturbing redox and chaperon speciation homeostasis [151].
Atox1 is another chaperon protein with specific properties [152]. It is a 68-amino acid protein where two of its three thiols bind Cu+; it also functions as a copper-dependent transcription factor [153] that mediates copper-induced cell proliferation. Atox1-deficient cells accumulate high levels of intracellular copper; metabolic studies indicate that this deficit results from an impaired cellular copper efflux [154].
Western Blot analyses of Atox1 by polyclonal or monoclonal antibodies are shown in Figure 8. Following a 48 hrs treatment with either phendione or cuproindione, oligomeric Atox 1 forms (i.e., the group of bands at higher MW, Figure 8A) increased and this was paralleled by a decrease of monomeric Atox1 (at a MW of approximately 8 kDa, Figure 8B). The in vitro incubation of recombinant Atox1 samples with copper only (Figure 8C) and with copper/H2O2 (Figure 8D) for 20 hrs results in the formation of dimers/tetramers and higher molecular weight species, respectively.
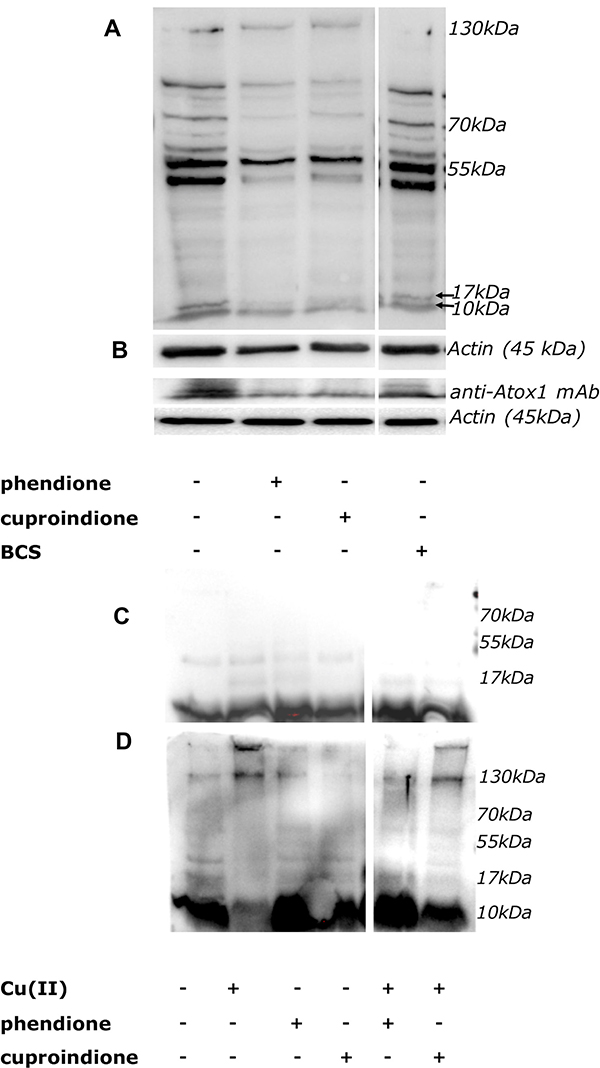
Figure 8: (A, B): Expression of Atox1 detected by polyclonal (A) or monoclonal (B) antibody in SH-SY5Y cells after 48 hrs of treatment. From the left to the right: control cells, cells treated with IC50 concentration of phendione or cuproindione or 50 μM BCS. (C, D): In vitro incubation of the Atox1 purified protein in the absence (C) or presence of H2O2 (D) for 20 hrs. From the left to the right: Atox1; Atox1 + Cu(II) (ratio 1:1); Atox1 + phendione (ratio 1:1); Atox1 + cuproindione (ratio 1:1); Atox1 + phendione + Cu(II) (ratio 1:1:1); Atox1 + cuproindione + Cu(II) (ratio 1:1:1).
Atox1 is mainly present in a reduced form that is ensured by the markedly lower GSH steady-state redox potential [148]. The redox potential of GSH is mainly maintained by NADPH via GSH reductase [155]. In addition, NADPH in association with thioredoxin reductases reduces the oxidized thioredoxin (Trx-S-S) to its active dithiolic form [156] that is required under low GSH conditions [157]. Our experiments show that phendione and cuproindione copper complexes markedly decrease GSH levels (Supplementary Figure 4). Noteworthy, this is consistent with some reports showing that both free phendione and its complexes with some transition metal ions exhibit electro catalytic oxidations [66, 67]. All together these results justify the different redox forms of Atox1 found by WB assays. We might further hypothesize that under the pro-oxidant effect due to phenantroline treatments, ATOX1 oligomerization could affect its own function as copper efflux agent.
Evidence indicates that an increase of p53 due to oxidative stress caused by platinum compound results in the accumulation of nuclear copper in colorectal cells [158]. More recently, it has been demonstrated that in the same colorectal cell line treated with platinum compound [159], p53 can influence nuclear copper transport by affecting the regulation of Atox1 expression.
Phendione and cuproindione induced oxidative stress affects the metallostasis network in mitochondria
Cellular copper homeostasis is accomplished by a highly complex and interconnected network of molecular interactions that balance: i) metal cytosol and sub-cellular uptake, ii) trafficking, iii) storage, iv) speciation and v) signaling [160].
In order to test whether the investigated compounds increase also nuclear copper in addition to p53, we monitored the subcellular changes of copper levels by laser scanning confocal microscopy (LSM) using CS1 [161, 162], a cell permeable chemosensor that specifically discriminates monovalent copper. Copper sub-cellular localization is illustrated in Figure 9. Compared to untreated control cells (Figure 9A), the cells treated with phendione (Figure 9B) and cuproindione (Figure 9C) show a cytosolic increased green fluorescence, consistent with Cu+ uptake. The presence of bright spots is likely due to the aggregation of the lipophilic CS1 probe in the aqueous intracellular environment [163]. The image analyses and the detection of CS1 emission at sub-cellular resolution (Supplementary Figure 5), indicate that the overall Cu+ content in cells treated with either phendione (Supplementary Figure 5B) or cuproindione (Supplementary Figure 5C) statistically increases both in the nuclei and in the mitochondria with respect to untreated control cells (Supplementary Figure 5A). We may conclude that the two compounds investigated cause Cu+ translocation into nuclear and mitochondrial compartments; in this context, cuproindione is more effective than phendione.
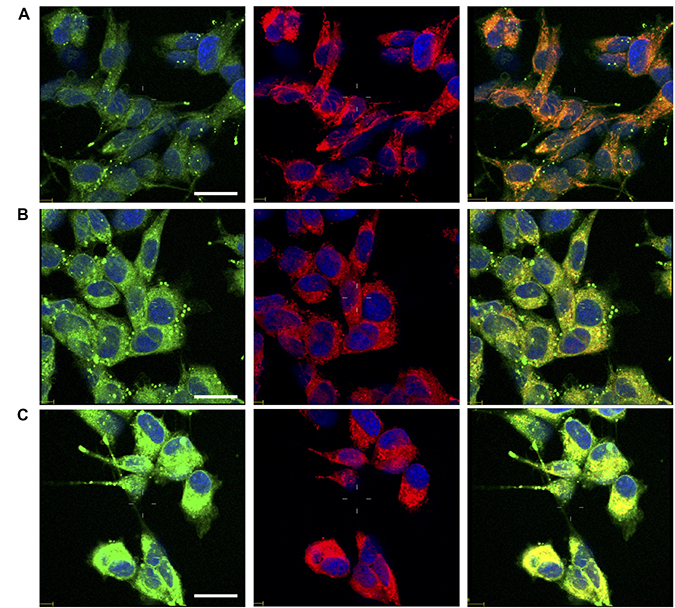
Figure 9: Confocal micrographs of untreated cells (A) and cells treated for 90 min with 4.4 mM phendione (B) or 2.2 mM cuproindione (C). After treatments cells were stained with copper sensor CS1 (green), Mitotracker Deep Red (red) and Hoechst33342 (blue). From the left to the right, merged channels for: copper + nuclei; mitochondria + nuclei; copper + mitochondria + nuclei. Scale bar= 30 μm.
Mitochondria participate in a number of processes critical to cellular homeostasis, including the homeostatic maintenance of many metal ions like copper [164, 165] that is involved in the metal-assembly pathways of cytochrome c oxidase (CCO) and superoxide dismutase-1 (SOD1), the only two copper enzymes present within the mitochondria. Alterations of the metallostasis network in the cytosol, that drives the mitochondria enzyme metallation, causes fatal diseases characterized by CCO deficiency [166, 167]. Though the incorporation of metal ion in both SOD1 and CCO relies on the cysteine thiol redox status of the metallochaperones, the mitochondrial assembly of the copper sites in CCO involves a series of accessory proteins, including (but not limited to) COX17, Sco1 and Sco2; by contrast SOD1 needs CCS only. Sco1, Sco2 and COA6 [168], form a metallochaperone set to deliver and insert copper into the CuA site of CCO. This metallation process requires a dedicated upstream copper donor within the intermembrane space (IMS) of mitochondria; COX17 [169], that was previously hypothesized to shuttle copper between the cytosol and mitochondria based on its dual localization [170], is able to fulfil this role [171].
Although there are reports that have contributed to understand how Cu+ is trafficked to and within mitochondria [172], the IMS copper translocation and transfer to COX17 is still to unveil [173]; recently, a new mitochondrial copper transporter required for cytochrome c oxidase biogenesis has been identified [174]. There is no doubt, however, that COX17 transfers copper to both Sco1 and Sco2 [175]. This 69-residue cysteine-rich protein, containing three pairs of cysteines, has three oxidation states, namely an oxidized Cox173S–S with three disulphide bonds, a reduced Cox170S–S with no disulphide bonds, and an intermediate state Cox172S–S with two disulphide bonds. Cox172S–S transfers Cu(I) and two electrons to the oxidized form of apo-Sco1 (containing a disulphide bond), but only to reduced Sco2 [176].
Copper is delivered to the CuA site only by Sco1. Sco2 acts as an oxidoreductase maintaining the cysteines of CuA under reduced conditions, thereby aiding copper delivery by Sco1 [177]. According to an alternative hypothesis, a ternary complex (Sco2-CCO-Sco1) would form and then Sco2 would sequentially transfer copper to CCO; Sco1 would then do the same process [178]. Following copper transfer, Sco2 acts as an oxidoreductase regulating the redox status of the cysteines in (oxidized) Sco1. This favours copper insertion into Sco1 by COX17 and activates the next cycle.
Following treatment with BCS, COX17 is markedly up-regulated (Figure 10), as expected due to the decreased copper availability to the chaperone resulting from copper complexation to BCS; by contrast, COX17 expression decreases in cells treated with phendione and cuproindione (Figure 10A). To the best of our knowledge, this is the first time that a cytotoxic copper complex has been shown to affect the expression of this CCO chaperone, which raises some questions as to the causes that determine such an effect. The considerations that follow may help answering these questions.
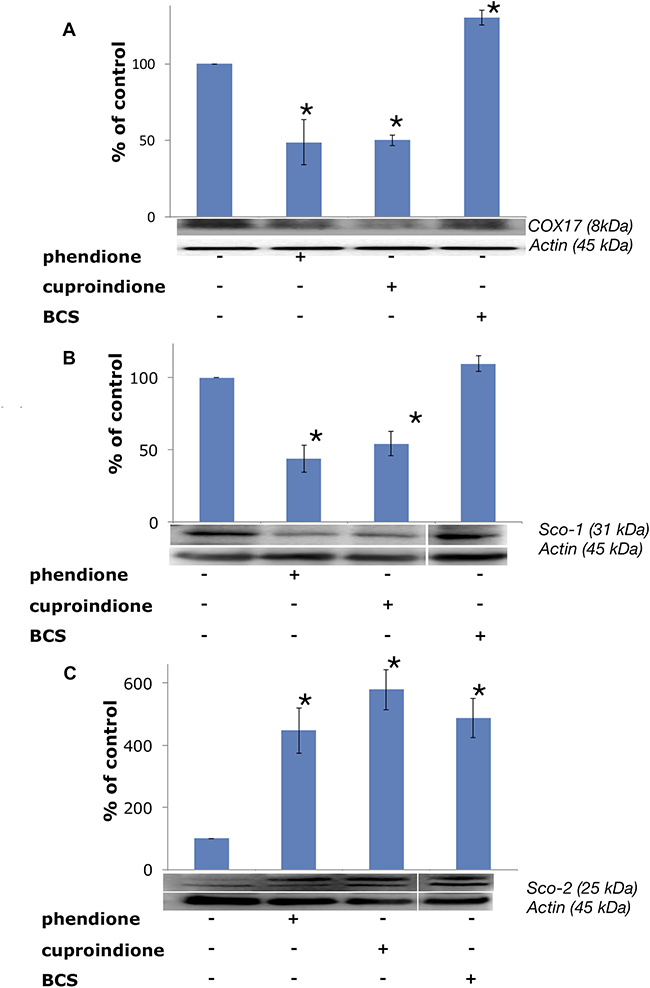
Figure 10: Expression of mitochondrial metallochaperones (A) Sco-1, (B) COX17 and (C) Sco-2 in SH-SY5Y cells after treatment with a phendione, cuproindione IC50 concentration and 50 μM BCS for 48 hrs. Results are expressed as mean ± SEM over at least three independent experiments. (*p ≤ 0.05 level vs control, One-way Anova).
In the first place, the over-expression of COX17, found in some cancer cells, seems to favour proliferation and to maintain CCO efficiency, thus suggesting the use of the chaperone for tumour treatment [179]. Secondly, COX17 is also a newly identified auxiliary factor involved in the control of the architecture of the MICOS complex, a protein complex that is in contact with the outer membrane of mitochondria and is critical to establish and maintain the inner membrane structure. In any case, the COX17-MICOS interaction is promoted by copper ions [180]. Moreover, COX17 forms two independent assemblies, one with SCO1 for the biogenesis of cytochrome c oxidase [181], and one with the MICOS complex for mitochondrial membrane organization [182]. Copper regulation is common to both assemblies. The oxidative stress induced by the two ligands investigated in the present study suggests a link between the decreased expression of the copper chaperone and the Bax translocation to the mitochondrial membrane, whose architecture is altered by pore formation. This process is accompanied by the contemporary release of cytochrome C that results in the altered assembly and Sco1 biogenesis assisted by CCO.
The analysis of different colon carcinoma cell lines (Caco-2, HT116, HT29) and cancer cell lines of different tissue origin (MCF7, PC3) showed a transcript-level upregulation of Sco1 [183, 184], suggesting that also this metallochaperone can be a drug target for cancer. The results for Sco1 expression (Figure 10B) are similar to those obtained for COX17, indicating that the down-regulation of this chaperone could contribute to the inhibition of cell proliferation. A link between the oxidative stress induced by our ligands and Sco1 is supported by the reported finding that production of ROS partly induces Fas-mediated cell death of glioma cells by Sco1 down-regulation [185].
Unlike Sco1, Sco2 expression is up-regulated by the cellular treatments with phendione and cuproindione (Figure 10C). This is consistent with the indication that one of the factors for a poor prognosis of patients with breast cancer results also from a low SCO2 expression [186]; noteworthy, exogenous addition of the SCO2 gene to hypoxic cancer cells induces apoptosis and causes a significant regression of tumour xenografts [187]. The complex role played by the two ligands is proved by the involvement of p53 (activated by the oxidative stress) in Sco2 transcription, while the reduced oxidative phosphorylation in p53-null cells is rescued by bringing Sco2 expression back to physiological levels [188]. Our results further stress the dual role of p53 that may serve as a tumour suppressor (activation of Bax) as well as a regulator of mitochondrial aerobic respiration via the modulation of Sco2 synthesis.
MATERIALS AND METHODS
Compounds synthesis and characterization
1,10-phenanthroline-5,6-dione and 2,9-dimethyl-1,10-phenanthroline-5,6-dione (Scheme 1) were prepared according to procedures previously reported [189, 190].
Synthesis of [CuCl(phendione)2] ClO4× 3/2H2O. This complex was prepared as described in the literature [191] with some modifications. A solution of CuCl2 (0.016 gr, 0.118 mmol) dissolved in 5 mL of ethanol was added to a solution containing phendione (0.050 g, 0.237 mmol) in 20 mL of ethanol at 60° C. The mixture was stirred at 60° C for 5 h. After removing any undissolved materials by filtration, LiClO4 (0.253 gr, 2.36 mmol) was added to the filtrate. The obtained green fine precipitated was filtered off, washed with ethanol and then air-dried. Yield: 40%. Anal. Calc. for [CuCl(phendione)2]ClO4·3/2H2O (C24H12Cl2CuN4O8·3/2H2O): C, 44.63; H, 2.34; N, 8.67%. Anal. Found: C, 44.76; H, 2.34; N, 8.58%. ESI-MS: calculated for [CuCl(phendione)2]+([C24H12ClCuN4 O4]+): 517.9. Found: m/z (% relative to the base peak) [M+]: 517.9 (100). UV-Vis in NaNO3 100 mM: λmax, nm (A): 253 (1.003), 301 (0.370). IR (KBr): 3419.2; 1702.2; 1577.0; 1430.3; 1302.3; 1090.3; 730.3; 936.3; 624.6 cm-1.
Synthesis of [Cu(phendione)3](ClO4)2× 4H2O. This complex was prepared as described in the literature [192] with some modifications. Phendione (0.050 gr, 0.237 mmol) suspended in 5 mL of ethanol was added to 1.5 mL of pale blue solution containing Cu(ClO4)2× 6H2O (0.029 gr, 0.079 mmol) in ethanol and the resulting green suspension was stirred at r.t. for 30 minutes. The precipitated green solid was filtered off, washed with ethanol and ether, and then air-dried. Yield: 74%. Anal. Calc. for [Cu(phendione)3](ClO4)2·4H2O (C36H26Cl2CuN6O18): C, 44.80; H, 2.72; N, 8.71%. Anal. Found: C, 44.34; H, 2.32; N, 8.44%. ESI-MS: calculated for [Cu(phendione)3 + (ClO4)3 + 2H2O]- ([C36H22Cl3CuN6O20]-): 1027.9. Found: m/z (% relative to the base peak): 1027.6 (20). Calculated for [Cu(phendione)2 + (ClO4)3]- ([C24H12Cl3CuN4O16]-): 781.9. Found: m/z (% relative to the base peak): 781.8 (85). Calculated for [Cu(Phendione)2(ClO4)]+([C24H12ClCuN4O8]+): 581.0. Found: m/z (% relative to the base peak) 581.9 (24). UV-VIS in NaNO3 100 mM: λmax, nm (A): 253 (1.003), 301 (0.346). IR (KBr): 3418.5; 1699.4; 1576.3; 1429.1; 1302.1; 1088.9; 730.3; 625.7 cm-1.
Synthesis of [Cu(cuproindione)2](ClO4)2·2H2O. Cuproindione (0.050 gr, 0.209 mmol) dissolved in 4.5 mL of ethanol was added to 1 mL of a pale blue solution of Cu(ClO4)2·6H2O (0.029 gr, 0.079 mmol) in ethanol; the resulting yellow suspension was stirred at r.t. for 30 minutes. The yellow precipitate was filtered off, washed with ethanol and ether and then air-dried. Yield: 70%. Anal. Calc. for [Cu(cuproindione)2](ClO4)2·2H2O (C28H24Cl2CuN4O14): C, 43.40; H, 3.12; N, 7.23%. Anal. Found: C, 43.14; H, 3.35; N, 6.90%. ESI-MS: calculated for [Cu(cuproindione)2-H]+ ([C28H20CuN4O4]+): 539.1. Found: m/z (% relative to the base peak) [M-H] +: 539.3 (100). UV-VIS in NaNO3 100 mM: λmax, nm (A): 243 (0.778), 262 (0.998), 303 (0.248), 315 (0.178). IR (KBr): 3436.2; 1698.6; 1590.9; 1315.4; 1090.0; 625.4; 222.8 cm-1. The powder samples were stored at −20° C in the darkness. Stock solutions of the compounds were prepared in DMSO (Sigma-Aldrich, St. Louis, MO) at the concentration of 10–1 M. Ultrapure milliQ water (18.2 mΩ·cm at 25° C, Millipore) was used throughout. All other chemicals and reagents were of high purity grade and obtained from standard commercial sources. Electrospray ionization mass spectrometry (ESI-MS) was performed with a dual electrospray interface and a quadrupole time-of-flight mass spectrometer (Agilent 6530 Series Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) LC-MS). Elemental analyses were carried out with a Eurovector EA 3000 CHN instrument. IR Spectra were recorded by a Perkin–Elmer Spectrum One and Perkin–Elmer 1600 Series FT-IR spectrophotometers using KBr as solid support for pellets.
Copper-complexes characterization
UV-visible spectroscopy (UV-vis)
UV-vis spectra were recorded with a Varian-Cary 100 UV-Vis Spectrophotometer (Agilent). UV-vis titration experiments were carried out by addition of 1.82 mM aqueous solutions of copper chloride and copper perchlorate (Sigma Aldrich, St. Louis, MO) (from 0.1 equiv. to 1.2 equiv.) to the water solution of phendione or cuproindione (0.065 mM) into the measuring cell containing a known volume (2.8 mL) of the ligand solution.
Electron paramagnetic resonance (EPR)
Isotopically pure aqueous solution of 63Cu, in the (8·10–4 –1·10–3) M range of concentration, were obtained from a 5 × 10–2 M stock solution of 63Cu(NO3)2 (Sigma Aldrich, St. Louis, MO). EPR spectra were carried out by using a Bruker Elexsys E500 CW-EPR spectrometer driven by a PC running the XEpr program on Linux and equipped with a Super-X microwave bridge, operating at 9.3–9.5 GHz, and a SHQE cavity. The spectra were recorded at 150 K by means of a variable temperature apparatus (ER4131VT). EPR magnetic parameters were obtained directly from the experimental EPR spectra and were calculated from the 2nd and 3rd line to avoid second order effects. The spectra were recorded as an average of 5 scans, microwave frequency 9.344–9.378 GHz; modulation frequency 100 kHz; modulation amplitude 0.2–0.6 mT; time constant 164–327 ms; sweep time 2.8 min; microwave power 20–40 mW; receiver gain 50–60 dB. The solutions were prepared at 1:1 and 1:2 metal-to-ligand ratios in the concentration range 1–1.5 mM. The pH of aqueous solution was adjusted by adding NaOH. 10% of methanol was added to aqueous solutions to enhance spectral resolution at low temperatures.
Distribution diagrams were obtained by using the 2009 version of the Hyperquad Simulation and Speciation program (HySS) [193]. titrations are simulated by specifying a set of titration conditions and calculating the concentrations of each complex species as the titration proceeds. The results are summarized in tabular form and shown graphically.
Cellular experiments
Cell culture maintenance and treatments
Dulbecco’s modified eagle medium (DMEM), Ham’s F-12 medium (F12), streptomycin, L-glutamine, fetal bovine serum (FBS) were provided by Lonza (Verviers, Belgium). Human neuroblastoma (SH-SY5Y line) cells were cultivated not longer than 20 passages in full medium, i.e., DMEM/F12 supplemented with 10% FBS, 2 mM L-glutamine and 100 μg ml−1 streptomycin. The cell culture was grown in tissue-culture treated Corning® flasks (Sigma-Aldrich, St. Louis, MO) in humidified atmosphere (5% CO2) at 37° C (Heraeus Hera Cell 150C incubator). For the cellular treatments, the day before the experiment cells were seeded at a density of 2·105 cells/mL in full medium on Corning® tissue-culture treated culture dishes (Sigma-Aldrich, St. Louis, MO). Immediately before use, stocks of phendione and cuproindione compounds were diluted (100x) in ultrapure H2O and then added to the cells in the culture medium at the desired final concentration (always less than 0.01% v/v of DMSO). For the cell experiments in copper-deprived medium, cells were pre-incubated with 50 μM of extracellular copper chelator BCS for three hrs and, maintaining the same medium, further incubated with the compounds.
Cytotoxicity assays
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide and the hydrated disodium salt of bathocuproinedisulfonic acid (BCS) were purchased from Sigma-Aldrich (St. Louis, MO). The effect of phendione and cuproindione on cell viability was tested at 60–70% of cell confluence by incubation with the compounds with concentrations ranging from 0.01 to 10 × 10–6 M for 48 hrs.
In order to calculate the concentration of each compound that produces a 50% cell mortality (IC50), the viable cells were quantified by the reaction with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT method, as previously described [194]). After 90 min, the reaction was stopped by adding DMSO, and absorbance was measured at 569 nm (Multiskan Ascent 96/384 Plate Reader). Results were expressed as% of viable cells over the concentration of each compound. The experiments were repeated at least 5 times in triplicate and results expressed as mean ± SEM. The statistical analysis was performed with a one-way Analysis of Variance (ANOVA test, by using the Microcal Origin software, version 8.6).
Western blot (WB) analysis
Tris-HCl buffer, ethylenediaminetetraacetic acid (EDTA), Triton X-100, EGTA, nonyl phenoxy polyethoxylethanol (NP40), phenylmethylsulfonylfluoride (PMSF) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). For the determination of protein amount by WB, cells were incubated at 37° C (in 5% CO2 atmosphere) with the compounds. Treatments with IC50 concentrations (48 hrs) or 3 × IC50 concentrations (90 min) were used for full protein extract or sub-cellular fractions, respectively.
Cells lysates were prepared by cells treatment with RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 1% Triton X-100, 0.5 mM EGTA, 1% NP40) containing 2 mM PMSF, an inhibitor of the protease cocktail. Immediately after the addition of the buffer, cells were collected by the scratch method and transferred to Eppendorf tubes (1.5 mL of size, purchased from Sigma-Aldrich, St. Louis, MO) for an incubation on ice for 30 min. After a centrifugation step (10 min at 14,000 r.p.m.) the supernatants were collected and the protein concentration was measured by Bradford’s method using BSA as the standard curve [195]. To prepare sub-cellular fractions (mitochondria and cytoplasm) cell were homogenated in sucrose buffer (0.32 M sucrose, 1 mM EDTA, 10 mM Tris, 1 mM PMSF, pH 7.4) instead of RIPA buffer, and separated by centrifugation according to standard protocols (Abcam).
SDS-PAGE with precast gel (4–20%, BioRad mini-PROTEAN) or with 15% Tricine gel [196] was used to separate proteins lysates or human Atox1 protein oligomers, respectively. Nitrocellulose membranes (Sigma-Aldrich, St. Louis, MO) were used to transfer proteins from the gel. Membranes were incubated with blocking buffer (0.1% Tween20 in tris-buffered saline added with either 5% BSA or 5% non-fat milk, depending on the primary antibody) at room temperature for 1 h, and then incubated with primary antibodies overnight at 4° C. After that, 1 h treatment with horseradish peroxidase-conjugated secondary antibodies was performed.
Primary antibodies used were as follows. From Santa Cruz Biotechnology (Santa Cruz, CA): against PARP-1 (F-2) (code: sc-8007, 1:500 dilution), PARP-1 (B-10) (code: sc-74470, 1:500 dilution), CCS (code: sc-20141, 1:500 dilution), Bcl-2 (code: sc-7382, 1:500 dilution), p53 (code: sc-6243, 1:500 dilution), Bax (code: sc-526, 1:1000 dilution). From Abcam (MA, USA): against GAPDH (code: ab8245, 1:2000 dilution) and CTR1 (code: ab129067, 1:3000 dilution). From Aviva Systems Biology (San Diego, USA): against ATP7A (code: ARP33797, 1:2000 dilution). From Abnova Corporation (Taiwan): polyclonal antibody against Atox1 (code: 15530, 1:2000 dilution) and monoclonal antibody against Atox1 (code: H00000475M01, 1:2000 dilution). From Cell Signaling Technologies Inc. (MA, USA): against Cytochrom C (code: 11940, 1:1000 dilution) and β-Actin (code: 4970, 1:2000 dilution). The secondary antibodies used were from EMD Millipore Bioscience (MA, USA): goat anti-mouse and anti-rabbit IgG horseradish peroxidase-conjugated (AP181P and AP307P, respectively, 1:3000 dilution). Measurements were performed by a ChemiDoc MP Imaging System (BioRad) using enhanced Western Lighting Chemiluminescence Reagent Plus (PerkinElmer (MA, USA). Experiments were repeated independently at least three times and results analysed by one-way ANOVA. The data reported throughout the paper are the mean values ± SEM and to the most representative membrane images.
Mitochondrial membrane potential measurements
5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) was purchased from Molecular Probes (ThermoFisher Scientific, MA, USA) [197]. In order to determine the mitochondrial membrane potential, cells were treated with 3 × IC50 concentration of the compounds for 90 min, further incubated with 2 μg/mL JC-1 (in the dark at 37° C) for 30 min, and washed twice with PBS to remove the unbound dye. A fluorescence plate reader (Varioskan® Flash Spectral Scanning Multimode Readers, Thermo Scientific (excitation wavelength = 488 nm) was used to monitor the fluorescence intensities for JC-1 molecules, either in monomer (emission wavelength = 530 nm) and in aggregated (emission wavelength = 595 nm) forms, respectively. Experiments were carried out simultaneously in by using eight wells for each compound; at least three independent experiments were performed. Results were analysed by one-way ANOVA expressed as fluorescence ratio (I = 595/530 nm) mean values ± SEM. For the detection of mitochondrial O2•−, after the treatment with phendione or cuproindione (at 3 × IC50 concentrations) for 30 minutes, cells were stained with superoxide probe MitoSOX (ThermoFisher Scientific, MA, USA). Hoechst 33342 solution was used for normalizing data to the actual cell number for each well. The fluorescence intensities were recorded at excitation/emission wavelengths of 510/580 nm for MitoSOX and 361/497 for Hoechst 33342 solution, respectively. In addition, in this case, experiments were carried out simultaneously in by using eight wells for each compound; at least three independent experiments were performed. Results were analysed by one-way ANOVA and expressed as mean values ± SEM.
Glutathione (GSH) levels determination
The changes of total intracellular GSH in treated with respect to untreated cells was assessed by using Glutathione Detection Assay Kit (Fluorometric, ab65322, Abcam, MA, USA). Cells were incubated with phendione or cuproindione at IC50 concentration for 24 hrs, and lysates obtained by addition of the lysis buffer provided in the kit. After an incubation on ice for 30 min, the lysates were centrifuged (10 min at 14,000 r.p.m.), the supernatants collected and processed according to the kit protocol, to eventually measure fluorescence (excitation/emission wavelengths = 380/461 nm).
Glutathione redox state determination
Cells were incubated with phendione or cuproindione at 3 × IC50 concentration for 90 min, then trypsinized and counted to obtain a cell suspension of 8·105 cells/mL. In order to evaluate the ratio of GSH/GSSG we used the detection assay kit from Abcam (ab138881, Abcam, MA, USA). Lysates were obtained by adding PBS with 0.5% of NP-40. Cells were homogenized by pipetting and centrifuged (10 min at 14,000 r.p.m.). Supernatants were treated by deproteinizing Sample Preparation Kit–TCA (ab204708, Abcam, MA, USA) and were then collected and processed according to the kit protocol (excitation/emission wavelengths = 490/520 nm).
Scanning confocal microscopy (LSM) analyses
For confocal microscope imaging, cells were seeded on glass bottom dishes (WillCo-dish®, Willco Wells, B.V.) with 12 mm of glass diameter at a density of 1.5 × 104 cells per dish, with complete medium for 24 hrs until cellular adhesion was attained. Cells were incubated with phendione and cuproindione (at 3 × IC50 concentrations) for 90 min in complete medium without FBS. Fifteen minutes before stopping incubation, Hoechst 33342, MitoTracker™ Deep Red FM (Thermo Fisher) and Coppersensor-1 (CS1) (1 μM, Cu(I) selective probe were added, for the staining of nuclei, mitochondria, and intracellular copper, respectively [161, 162, 198]. Cells were then fixed with high purity 2% paraformaldehyde in PBS (pH = 7.3). Images were acquired with an Olympus FV1000 confocal microscope equipped with the following UV/visible lasers: diode laser (50 mW, λex = 405 nm), Argon laser (20 mW, jλex=488 nm) and HeNe(R) laser (1 mW, λex = 633 nm) for the imaging of nuclei (blue channel), monovalent copper by CS1 (green channel) and mitochondria (red channel), respectively. An oil immersion objective (60xO PLAPO) was used; the detector gain was fixed at a constant value, and the emitted light was detected in sequential mode. All images were taken randomly throughout the area of the cell culture wells. The image analysis was carried out using Huygens Essential software (by Scientific Volume Imaging B.V., The Netherlands). The statistical analysis was performed with a one-way ANOVA.
Atox1 oligomerisation
Atox 1 protein was expressed and purified according to previously reported procedures [199]. For oligomerisation, Atox1 samples (2.2·10–5 M) were incubated in 0.01 PBS with phendione or cuproindione (1:1 mole ratio) both with and without CuSO4 (1:1:1 mole ratio) for 20 hrs. Parallel experiments were run in the presence of H2O2 (2.5-fold excess with respect to the cystein residues of the Atox1) [200]. Atox1 oligomeric species were separated on 15% Tricine gel in non-reducing conditions, transferred to nitrocellulose membranes and then incubated with primary antibodies (polyclonal anti-Atox1 antibody (code: 15530, 1:2000 dilution) overnight at 4° C (For a more detailed description see the WB analysis section reported above).
CONCLUSIONS
Phendione and cuproindione bind and transport inside the cell the copper ion present in the culture medium, giving rise to oxidative stress. Assuming that the two carbonyl groups present in phendione and cuproindione do not alter the copper affinity constants of the parent ligands (2,9-dimethyl-1,10-phenanthroline-5,6-dione and 2,9-dimethyl-1,10-phenanthroline), speciation results indicate that the bis-chelate complex species are mainly responsible for the cellular response. In three cases (increase of IC50 in SH-SY5Y cells (Figure 3), increase of cleaved/full PARP-1 ratio (Supplementary Figure 2), and downregulation of anti-apoptotic Bcl-2 (Supplementary Figure 3), the addition of BCS nullifies the cuproindione-induced effect, reflecting the different copper affinity of the two investigated ligands.
The binding of the metal ion to cuproindione and phendione changes copper chaperone speciation and alters copper redox homeostasis, through the oxidation of thiol groups. These copper complex species can also cause DNA-strand break by direct interaction with DNA (non-intercalative binding to the minor groove of DNA[80]). We found several possibly interconnected effects, i.e., the decrease of the GSH/GSSG ratio (Figure 4), the depolarization of mitochondrial membrane and production of ROS (Figure 5), the decrease of monomeric Atox1 (Figure 8) and the decrease of GSH levels (Supplementary Figure 4). Moreover, phendione and cuproindione up regulate differently p53 expression, depending on their different oxidative nuclease ability, (Figure 6). In addition, the p53 transcription factor is not only the key driver of mitochondria apoptosis but also participates in the mitochondria metallostasis both by altering the intracellular location of metal ion and by controlling the synthesis of Sco-2 that, in turn, increases ROS levels.
Usually, copper homeostasis has been investigated focusing on a single component of the metallostasis network; instead, our investigation involves several of the factors controlling the metallostasis. This is the only approach that allows for an exhaustive understanding of how the communication within the dynamic metallome drives the final decisions made by the cells.
Author contributions
IN has taken care of cell culture experiments. CS has performed confocal microscopy measurements. AC, NM and GN have dealt with complex synthesis and characterization. GA has taken care of species distribution and calculation. DL performed EPR experiments. GA, CS, NM, GN, DL, VN and ER have contributed to writing the manuscript. The study was mainly planned and designed by ER and VN.
ACKNOWLEDGMENTS
Authors acknowledge Dr. G. Trusso Sfrazzetto (Department of Chemistry of University of Catania) for the synthesis of the CS1 probe.
CONFLICTS OF INTEREST
There are no conflicts to declare.
FUNDING
This research was partially supported by MIUR under Grant PRIN 2015 (2015MP34H3 and 20152EKS4Y projects) and University of Catania (Piano della Ricerca di Ateneo 2016–2018).
REFERENCES
1. Zhang CX, Lippard SJ. New metal complexes as potential therapeutics. Curr Opin Chem Biol. 2003; 7:481–9.
2. Arnesano F, Natile G. Mechanistic insight into the cellular uptake and processing of cisplatin 30 years after its approval by FDA. Coordination Chemistry Reviews. 2009; 253:2070–81. https://doi.org/10.1016/j.ccr.2009.01.028.
3. Bednarski PJ, Mackay FS, Sadler PJ. Photoactivatable platinum complexes. Anticancer Agents Med Chem. 2007; 7:75–93.
4. Devereux M, O Shea D, Kellett A, McCann M, Walsh M, Egan D, Deegan C, Kędziora K, Rosair G, Müller-Bunz H. Synthesis, X-ray crystal structures and biomimetic and anticancer activities of novel copper(II)benzoate complexes incorporating 2-(4′-thiazolyl)benzimidazole (thiabendazole), 2-(2-pyridyl)benzimidazole and 1,10-phenanthroline as chelating nitrogen donor ligands. Journal of Inorganic Biochemistry. 2007; 101:881–92. https://doi.org/10.1016/j.jinorgbio.2007.02.002.
5. Komeda S, Casini A. Next-generation anticancer metallodrugs. Curr Top Med Chem. 2012; 12:219–35.
6. Nagy EM, Sitran S, Montopoli M, Favaro M, Marchiò L, Caparrotta L, Fregona D. Zinc(II) complexes with dithiocarbamato derivatives: Structural characterisation and biological assays on cancerous cell lines. Journal of Inorganic Biochemistry. 2012; 117:131–9. https://doi.org/10.1016/j.jinorgbio.2012.09.004.
7. Santini C, Pellei M, Gandin V, Porchia M, Tisato F, Marzano C. Advances in Copper Complexes as Anticancer Agents. Chemical Reviews. 2014; 114:815–62. https://doi.org/10.1021/cr400135x.
8. Narayanan VS, Fitch CA, Levenson CW. Tumor suppressor protein p53 mRNA and subcellular localization are altered by changes in cellular copper in human Hep G2 cells. J Nutr. 2001; 131:1427–32.
9. Rizk SL, Sky-Peck HH. Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer Res. 1984; 44:5390–4.
10. Schwartz MK. Role of trace elements in cancer. Cancer Res. 1975; 35:3481–7.
11. Zowczak M, Iskra M, Torliński L, Cofta S. Analysis of Serum Copper and Zinc Concentrations in Cancer Patients. Biological Trace Element Research. 2001; 82:001–8. https://doi.org/10.1385/bter:82:1-3:001.
12. Delves HT, Alexander FW, Lay H. Copper and zinc concentration in the plasma of leukaemic children. Br J Haematol. 1973; 24:525–31.
13. Diez M, Cerdan FJ, Arroyo M, Balibrea JL. Use of the copper/zinc ratio in the diagnosis of lung cancer. Cancer. 1989; 63:726–30.
14. Gupta SK, Shukla VK, Vaidya MP, Roy SK, Gupta S. Serum and tissue trace elements in colorectal cancer. J Surg Oncol. 1993; 52:172–5.
15. Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treatment Reviews. 2009; 35:32–46. https://doi.org/10.1016/j.ctrv.2008.07.004.
16. Kaiafa GD, Saouli Z, Diamantidis MD, Kontoninas Z, Voulgaridou V, Raptaki M, Arampatzi S, Chatzidimitriou M, Perifanis V. Copper levels in patients with hematological malignancies. European Journal of Internal Medicine. 2012; 23:738–41. https://doi.org/10.1016/j.ejim.2012.07.009.
17. Yoshida D, Ikeda Y, Nakazawa S. Quantitative analysis of copper, zinc and copper/zinc ratio in selected human brain tumors. J Neurooncol. 1993; 16:109–15.
18. Dwyer FP, Mayhew E, Roe EM, Shulman A. Inhibition of Landschuetz Ascites Tumour Growth by Metal Chelates Derived from 3,4,7,8-Tetramethyl-1,10-Phenanthroline. Br J Cancer. 1965; 19:195–9.
19. Maheswari PU, van der Ster M, Smulders S, Barends S, van Wezel GP, Massera C, Roy S, den Dulk H, Gamez P, Reedijk J. Structure, Cytotoxicity, and DNA-Cleavage Properties of the Complex [CuII(pbt)Br2]. Inorganic Chemistry. 2008; 47:3719–27. https://doi.org/10.1021/ic702306f.
20. Chen D, Milacic V, Frezza M, Dou QP. Metal complexes, their cellular targets and potential for cancer therapy. Curr Pharm Des. 2009; 15:777–91.
21. Tardito S, Marchio L. Copper compounds in anticancer strategies. Curr Med Chem. 2009; 16:1325–48.
22. Cai X, Pan N, Zou G. Copper-1,10-Phenanthroline-Induced Apoptosis in Liver Carcinoma Bel-7402 Cells Associates with Copper Overload, Reactive Oxygen Species Production, Glutathione Depletion and Oxidative DNA Damage. Biometals. 2006; 20:1–11. https://doi.org/10.1007/s10534-006-9008-0.
23. Marzano C, Pellei M, Tisato F, Santini C. Copper complexes as anticancer agents. Anticancer Agents Med Chem. 2009; 9:185–211.
24. Xiao Y, Chen DI, Zhang X, Cui Q, Fan Y, Bi C, Dou QP. Molecular study on copper-mediated tumor proteasome inhibition and cell death. Int J Oncol. 2010; 37:81–7.
25. Fatfat M, Merhi RA, Rahal O, Stoyanovsky DA, Zaki A, Haidar H, Kagan VE, Gali-Muhtasib H, Machaca K. Copper chelation selectively kills colon cancer cells through redox cycling and generation of reactive oxygen species. BMC Cancer. 2014; 14:527. https://doi.org/10.1186/1471-2407-14-527.
26. Liu C, Zhou J, Li Q, Wang L, Liao Z, Xu H. DNA damage by copper(II) complexes: coordination-structural dependence of reactivities. J Inorg Biochem. 1999; 75:233–40.
27. Gracia-Mora I, Ruiz-Ramírez L, Gómez-Ruiz C, Tinoco-Méndez M, Márquez-Quiñones A, Lira LR, Marín-Hernández Á, Macías-Rosales L, Bravo-Gómez ME. Knigth's Move in the Periodic Table, From Copper to Platinum, Novel Antitumor Mixed Chelate Copper Compounds, Casiopeinas, Evaluated by an in Vitro Human and Murine Cancer Cell Line Panel. Met Based Drugs. 2001; 8:19–28. https://doi.org/10.1155/mbd.2001.19.
28. Hernández-Esquivel L, Marín-Hernández A, Pavón N, Carvajal K, Moreno-Sánchez R. Cardiotoxicity of copper-based antineoplastic drugs casiopeinas is related to inhibition of energy metabolism. Toxicology and Applied Pharmacology. 2006; 212:79–88. https://doi.org/10.1016/j.taap.2005.06.023.
29. Leal-García M, García-Ortuño L, Ruiz-Azuara L, Gracia-Mora I, Luna-delVillar J, Sumano H. Assessment of Acute Respiratory and Cardiovascular Toxicity of Casiopeinas in Anaesthetized Dogs. Basic & Clinical Pharmacology & Toxicology. 2007; 101:151–8. https://doi.org/10.1111/j.1742-7843.2007.00038.x.
30. Marin-Hernandez A, Gracia-Mora I, Ruiz-Ramirez L, Moreno-Sanchez R. Toxic effects of copper-based antineoplastic drugs (Casiopeinas) on mitochondrial functions. Biochem Pharmacol. 2003; 65:1979–89.
31. Rivero-Müller A, De Vizcaya-Ruiz A, Plant N, Ruiz L, Dobrota M. Mixed chelate copper complex, Casiopeina IIgly®, binds and degrades nucleic acids: A mechanism of cytotoxicity. Chemico-Biological Interactions. 2007; 165:189–99. https://doi.org/10.1016/j.cbi.2006.12.002.
32. De Vizcaya-Ruiz A, Rivero-Muller A, Ruiz-Ramirez L, Kass GE, Kelland LR, Orr RM, Dobrota M. Induction of apoptosis by a novel copper-based anticancer compound, casiopeina II, in L1210 murine leukaemia and CH1 human ovarian carcinoma cells. Toxicol In Vitro. 2000; 14:1–5.
33. Carvallo-Chaigneau F, Trejo-Solís C, Gómez-Ruiz C, Rodríguez-Aguilera E, Macías-Rosales L, Cortés-Barberena E, Cedillo-Peláez C, Gracia-Mora I, Ruiz-Azuara L, Madrid-Marina V, Constantino-Casas F. Casiopeina III-ia induces apoptosis in HCT-15 cells in vitro through caspase-dependent mechanisms and has antitumor effect in vivo. Biometals. 2007; 21:17–28. https://doi.org/10.1007/s10534-007-9089-4.
34. Serment-Guerrero J, Bravo-Gomez ME, Lara-Rivera E, Ruiz-Azuara L. Genotoxic assessment of the copper chelated compounds Casiopeinas: Clues about their mechanisms of action. Journal of Inorganic Biochemistry. 2017; 166:68–75. https://doi.org/10.1016/j.jinorgbio.2016.11.007.
35. Bravo-Gómez ME, García-Ramos JC, Gracia-Mora I, Ruiz-Azuara L. Antiproliferative activity and QSAR study of copper(II) mixed chelate [Cu(N–N)(acetylacetonato)]NO3 and [Cu(N–N)(glycinato)]NO3 complexes, (Casiopeínas®). Journal of Inorganic Biochemistry. 2009; 103:299–309. https://doi.org/10.1016/j.jinorgbio.2008.10.006.
36. Ruiz-Azuara L, Bravo-Gomez ME. Copper compounds in cancer chemotherapy. Curr Med Chem. 2010; 17:3606–15.
37. Alemón-Medina R, Muñoz-Sánchez JL, Ruiz-Azuara L, Gracia-Mora I. Casiopeína IIgly induced cytotoxicity to HeLa cells depletes the levels of reduced glutathione and is prevented by dimethyl sulfoxide. Toxicology in Vitro. 2008; 22:710–5. https://doi.org/10.1016/j.tiv.2007.11.011.
38. Serment-Guerrero J, Cano-Sanchez P, Reyes-Perez E, Velazquez-Garcia F, Bravo-Gomez ME, Ruiz-Azuara L. Genotoxicity of the copper antineoplastic coordination complexes casiopeinas®. Toxicology in Vitro. 2011; 25:1376–84. https://doi.org/10.1016/j.tiv.2011.05.008.
39. Alemón-Medina R, Breña-Valle M, Muñoz-Sánchez JL, Gracia-Mora MI, Ruiz-Azuara L. Induction of oxidative damage by copper-based antineoplastic drugs (Casiopeínas®). Cancer Chemotherapy and Pharmacology. 2007; 60:219–28. https://doi.org/10.1007/s00280-006-0364-9.
40. Alemón-Medina R, Bravo-Gómez ME, Gracia-Mora MI, Ruiz-Azuara L. Comparison between the antiproliferative effect and intracellular glutathione depletion induced by Casiopeína IIgly and cisplatin in murine melanoma B16 cells. Toxicology in Vitro. 2011; 25:868–73. https://doi.org/10.1016/j.tiv.2011.02.007.
41. Kachadourian R, Brechbuhl HM, Ruiz-Azuara L, Gracia-Mora I, Day BJ. Casiopeína IIgly-induced oxidative stress and mitochondrial dysfunction in human lung cancer A549 and H157 cells. Toxicology. 2010; 268:176–83. https://doi.org/10.1016/j.tox.2009.12.010.
42. López-Rodríguez A, Cárabez-Trejo A, Rosas-Sánchez F, Mejía C, Ruiz-Azuara L, Miledi R, Martínez-Torres A. The metal-coordinated Casiopeína IIIEa induces the petite-like phenotype in Saccharomyces cerevisiae. Biometals. 2011; 24:1189–96. https://doi.org/10.1007/s10534-011-9477-7.
43. Huang R, Wallqvist A, Covell DG. Anticancer metal compounds in NCI's tumor-screening database: putative mode of action. Biochemical Pharmacology. 2005; 69:1009–39. https://doi.org/10.1016/j.bcp.2005.01.001.
44. Chikira M, Tomizawa Y, Fukita D, Sugizaki T, Sugawara N, Yamazaki T, Sasano A, Shindo H, Palaniandavar M, Antholine WE. DNA-fiber EPR study of the orientation of Cu(II) complexes of 1,10-phenanthroline and its derivatives bound to DNA: mono(phenanthroline)-copper(II) and its ternary complexes with amino acids. J Inorg Biochem. 2002; 89:163–73.
45. Chakravarty AR, Anreddy PAN, Santra BK, Thomas AM. Copper complexes as chemical nucleases. Journal of Chemical Sciences. 2002; 114:391–401. https://doi.org/10.1007/bf02703829.
46. Roy Chowdhury S, Banerji V. Targeting Mitochondrial Bioenergetics as a Therapeutic Strategy for Chronic Lymphocytic Leukemia. Oxidative Medicine and Cellular Longevity. 2018; 2018:1–10. https://doi.org/10.1155/2018/2426712.
47. Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell. 2006; 10:241–52. https://doi.org/10.1016/j.ccr.2006.08.009.
48. Fruehauf JP, Meyskens FL. Reactive Oxygen Species: A Breath of Life or Death? Clinical Cancer Research. 2007; 13:789–94. https://doi.org/10.1158/1078-0432.ccr-06-2082.
49. Schumacker PT. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell. 2006; 10:175–6. https://doi.org/10.1016/j.ccr.2006.08.015.
50. Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996; 145:532–41.
51. Sigman DS, Graham DR, D'Aurora V, Stern AM. Oxygen-dependent cleavage of DNA by the 1,10-phenanthroline. cuprous complex. Inhibition of Escherichia coli DNA polymerase I. J Biol Chem. 1979; 254:12269–72.
52. Sigman DS. Nuclease activity of 1,10-phenanthroline-copper ion. Accounts of Chemical Research. 2002; 19:180–6. https://doi.org/10.1021/ar00126a004.
53. Laws K, Bineva-Todd G, Eskandari A, Lu C, O'Reilly N, Suntharalingam K. A Copper(II) Phenanthroline Metallopeptide That Targets and Disrupts Mitochondrial Function in Breast Cancer Stem Cells. Angew Chem Int Ed Engl. 2018; 57:287–91. https://doi.org/10.1002/anie.201710910.
54. Robertazzi A, Vargiu AV, Magistrato A, Ruggerone P, Carloni P, de Hoog P, Reedijk J. Copper−1,10-Phenanthroline Complexes Binding to DNA: Structural Predictions from Molecular Simulations. J Phys Chem B. 2009; 113:10881–90. https://doi.org/10.1021/jp901210g.
55. Thati B, Noble A, Creaven BS, Walsh M, Kavanagh K, Egan DA. Apoptotic cell death: A possible key event in mediating the in vitro anti-proliferative effect of a novel copper(II) complex, [Cu(4-Mecdoa)(phen)2] (phen=phenanthroline, 4-Mecdoa=4-methylcoumarin-6,7-dioxactetate), in human malignant cancer cells. European Journal of Pharmacology. 2007; 569:16–28. https://doi.org/10.1016/j.ejphar.2007.04.064.
56. Tsang SY, Tam SC, Bremner I, Burkitt MJ. Research communication copper-1,10-phenanthroline induces internucleosomal DNA fragmentation in HepG2 cells, resulting from direct oxidation by the hydroxyl radical. Biochem J. 1996; 317:13–6.
57. Verhaegh GW, Richard MJ, Hainaut P. Regulation of p53 by metal ions and by antioxidants: dithiocarbamate down-regulates p53 DNA-binding activity by increasing the intracellular level of copper. Mol Cell Biol. 1997; 17:5699–706.
58. Igdaloff D, Santi DV, Eckert TS, Bruice TC. Effects of 1,7- and 1,10-phenanthroline dione on tissue culture cells. Biochem Pharmacol. 1983; 32:172–4.
59. Deegan C, Coyle B, McCann M, Devereux M, Egan DA. In vitro anti-tumour effect of 1,10-phenanthroline-5,6-dione (phendione), [Cu(phendione)3](ClO4)2·4H2O and [Ag(phendione)2]ClO4 using human epithelial cell lines. Chemico-Biological Interactions. 2006; 164:115–25. https://doi.org/10.1016/j.cbi.2006.08.025.
60. Roy S, Hagen KD, Maheswari PU, Lutz M, Spek AL, Reedijk J, van Wezel GP. Phenanthroline Derivatives with Improved Selectivity as DNA-Targeting Anticancer or Antimicrobial Drugs. ChemMedChem. 2008; 3:1427–34. https://doi.org/10.1002/cmdc.200800097.
61. Pivetta T, Trudu F, Valletta E, Isaia F, Castellano C, Demartin F, Tuveri R, Vascellari S, Pani A. Novel copper(II) complexes as new promising antitumour agents. A crystal structure of [Cu(1,10-phenanthroline-5,6-dione) 2 (OH 2)(OClO 3)](ClO 4). Journal of Inorganic Biochemistry. 2014; 141:103–13. https://doi.org/10.1016/j.jinorgbio.2014.08.011.
62. Sigman DS, Mazumder A, Perrin DM. Chemical nucleases. Chemical Reviews. 1993; 93:2295–316. https://doi.org/10.1021/cr00022a011.
63. Calucci L, Pampaloni G, Pinzino C, Prescimone A. Transition metal derivatives of 1,10-phenanthroline-5,6-dione: Controlled growth of coordination polynuclear derivatives. Inorganica Chimica Acta. 2006; 359:3911–20. https://doi.org/10.1016/j.ica.2006.04.040.
64. Farhangian H, Eslami Moghadam M, Divsalar A, Rahiminezhad A. Anticancer activity of novel amino acid derivative of palladium complex with phendione ligand against of human colon cancer cell line. Journal of Biological Inorganic Chemistry. 2017; 22:1055–64. https://doi.org/10.1007/s00775-017-1483-y.
65. Ghosh S, Barve AC, Kumbhar AA, Kumbhar AS, Puranik VG, Datar PA, Sonawane UB, Joshi RR. Synthesis, characterization, X-ray structure and DNA photocleavage by cis-dichloro bis(diimine) Co(III) complexes. Journal of Inorganic Biochemistry. 2006; 100:331–43. https://doi.org/10.1016/j.jinorgbio.2005.11.022.
66. Goss CA, Abruna HD. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorganic Chemistry. 1985; 24:4263–7. https://doi.org/10.1021/ic00219a012.
67. Hilt G, Jarbawi T, Heineman WR, Steckhan E. An Analytical Study of the Redox Behavior of 1,10-Phenanthroline-5,6-dione, its Transition-Metal Complexes, and itsN-Monomethylated Derivative with regard to their Efficiency as Mediators of NAD(P)+ Regeneration. Chemistry. 1997; 3:79–88. https://doi.org/10.1002/chem.19970030114.
68. Luis DV, Silva J, Tomaz AI, de Almeida RF, Larguinho M, Baptista PV, Martins LM, Silva TF, Borralho PM, Rodrigues CM, Rodrigues AS, Pombeiro AJ, Fernandes AR. Insights into the mechanisms underlying the antiproliferative potential of a Co(II) coordination compound bearing 1,10-phenanthroline-5,6-dione: DNA and protein interaction studies. J Biol Inorg Chem. 2014; 19:787–803. https://doi.org/10.1007/s00775-014-1110-0.
69. Wu Q, Maskus M, Pariente F, Tobalina F, Fernández VM, Lorenzo E, Abruña HD. Electrocatalytic Oxidation of NADH at Glassy Carbon Electrodes Modified with Transition Metal Complexes Containing 1,10-Phenanthroline-5,6-dione Ligands. Analytical Chemistry. 1996; 68:3688–96. https://doi.org/10.1021/ac960395y.
70. La Mendola D, Giacomelli C, Rizzarelli E. Intracellular Bioinorganic Chemistry and Cross Talk Among Different -Omics. Curr Top Med Chem. 2016; 16:3103–30.
71. Saravani H, Rezvani AR, Mansouri G, Salehi Rad AR, Khavasi HR, Hadadzadeh H. Crystal structure, magnetic and electrochemical properties of five-coordinate copper (II) complexes with 1,10-phenanthroline-5,6-dione. Inorganica Chimica Acta. 2007; 360:2829–34. https://doi.org/10.1016/j.ica.2007.01.010.
72. Abdus Subhan M, Saifur Rahman M, Alam K, Mahmud Hasan M. Spectroscopic analysis, DNA binding and antimicrobial activities of metal complexes with phendione and its derivative. Spectrochim Acta A Mol Biomol Spectrosc. 2014; 118:944–50. https://doi.org/10.1016/j.saa.2013.09.110.
73. Stephenson MD, Hardie MJ. Coordination and hydrogen bonded network structures of Cu(ii) with mixed ligands: a hybrid hydrogen bonded material, an infinite sandwich arrangement, and a 3-D net. Dalton Transactions. 2006: 28:3407–17. https://doi.org/10.1039/b600357e.
74. Smith RM, Martell AE. Critical Stability Constants. (Plenum Press, New York: Springer US). 1975.
75. Hirohama T, Kuranuki Y, Ebina E, Sugizaki T, Arii H, Chikira M, Tamil Selvi P, Palaniandavar M. Copper(II) complexes of 1,10-phenanthroline-derived ligands: Studies on DNA binding properties and nuclease activity. Journal of Inorganic Biochemistry. 2005; 99:1205–19. https://doi.org/10.1016/j.jinorgbio.2005.02.020.
76. Zoroddu MA, Zanetti S, Pogni R, Basosi R. An electron spin resonance study and antimicrobial activity of copper(II)-phenanthroline complexes. J Inorg Biochem. 1996; 63:291–300.
77. Tardito S, Isella C, Medico E, Marchiò L, Bevilacqua E, Hatzoglou M, Bussolati O, Franchi-Gazzola R. The Thioxotriazole Copper(II) Complex A0 Induces Endoplasmic Reticulum Stress and Paraptotic Death in Human Cancer Cells. J Biol Chem. 2009; 284:24306–19. https://doi.org/10.1074/jbc.M109.026583.
78. Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer?s A? peptides. Journal of Biological Inorganic Chemistry. 2004; 9:954–60. https://doi.org/10.1007/s00775-004-0602-8.
79. Ashino T, Sudhahar V, Urao N, Oshikawa J, Chen GF, Wang H, Huo Y, Finney L, Vogt S, McKinney RD, Maryon EB, Kaplan JH, Ushio-Fukai M, et al. Unexpected Role of the Copper Transporter ATP7A in PDGF-Induced Vascular Smooth Muscle Cell Migration. Circulation Research. 2010; 107:787–99. https://doi.org/10.1161/circresaha.110.225334.
80. Thederahn TB, Kuwabara MD, Larsen TA, Sigman DS. Nuclease activity of 1,10-phenanthroline-copper: kinetic mechanism. Journal of the American Chemical Society. 1989; 111:4941–6. https://doi.org/10.1021/ja00195a057.
81. James BR, Williams RJ. 383. The oxidation–reduction potentials of some copper complexes. J Chem Soc. 1961; 0:2007–19. https://doi.org/10.1039/jr9610002007.
82. Feig AL, Thederahn T, Sigman DS. Mutagenicity of the nuclease activity of 1,10-phenanthroline-copper ion. Biochem Biophys Res Commun. 1988; 155:338–43.
83. Shang Z, Ebright YW, Iler N, Pendergrast PS, Echelard Y, McMahon AP, Ebright RH, Abate C. DNA affinity cleaving analysis of homeodomain-DNA interaction: identification of homeodomain consensus sites in genomic DNA. Proc Natl Acad Sci U S A. 1994; 91:118–22.
84. Pan CQ, Landgraf R, Sigman DS. DNA-binding proteins as site-specific nucleases. Mol Microbiol. 1994; 12:335–42.
85. Pan CQ, Johnson RC, Sigman DS. Identification of New Fis Binding Sites by DNA Scission with Fis-1,10-phenanthroline−Copper(I) Chimeras†. Biochemistry. 1996; 35:4326–33. https://doi.org/10.1021/bi952040z.
86. Xiao G, Cole DL, Gunsalus RP, Sigman DS, Chen CH. Site-specific DNA cleavage of synthetic NarL sites by an engineered Escherichia coli NarL protein-1,10-phenanthroline cleaving agent. Protein Science. 2008; 11:2427–36. https://doi.org/10.1110/ps.0212502.
87. Chikira M, Ng C, Palaniandavar M. Interaction of DNA with Simple and Mixed Ligand Copper(II) Complexes of 1,10-Phenanthrolines as Studied by DNA-Fiber EPR Spectroscopy. International Journal of Molecular Sciences. 2015; 16:22754–80. https://doi.org/10.3390/ijms160922754.
88. Sigman DS, Bruice TW, Mazumder A, Sutton CL. Targeted chemical nucleases. Accounts of Chemical Research. 2002; 26:98–104. https://doi.org/10.1021/ar00027a004.
89. Czapski G, Goldstein S. When do metal complexes protect the biological system from superoxide toxicity and when do they enhance it? Free Radic Res Commun. 1986; 1:157–61.
90. Kucková L, Jomová K, Švorcová A, Valko M, Segľa P, Moncoľ J, Kožíšek J. Synthesis, Crystal Structure, Spectroscopic Properties and Potential Biological Activities of Salicylate–Neocuproine Ternary Copper(II) Complexes. Molecules. 2015; 20:2115–37. https://doi.org/10.3390/molecules20022115.
91. Cadoni E, Valletta E, Caddeo G, Isaia F, Cabiddu MG, Vascellari S, Pivetta T. Competitive reactions among glutathione, cisplatin and copper-phenanthroline complexes. Journal of Inorganic Biochemistry. 2017; 173:126–33. https://doi.org/10.1016/j.jinorgbio.2017.05.004.
92. Carrasco-Pozo C, Aliaga ME, Olea-Azar C, Speisky H. Double edge redox-implications for the interaction between endogenous thiols and copper ions: In vitro studies. Bioorganic & Medicinal Chemistry. 2008; 16:9795–803. https://doi.org/10.1016/j.bmc.2008.09.068.
93. Gilbert BC, Silvester S, Walton PH. Spectroscopic, kinetic and mechanistic studies of the influence of ligand and substrate concentration on the activation by peroxides of CuI–thiolate and other CuI complexes. Journal of the Chemical Society, Perkin Transactions 2. 1999: 1115-22. https://doi.org/10.1039/a901179j.
94. Veal JM, Merchant K, Rill RL. The influence of reducing agent and 1,10-phenanthroline concentration on DNA cleavage by phenanthroline + copper. Nucleic Acids Res. 1991; 19:3383–8.
95. Lu LP, Zhu ML, Yang P. Crystal structure and nuclease activity of mono(1,10-phenanthroline) copper complex. J Inorg Biochem. 2003; 95:31–6.
96. Bales BC. Mechanistic studies on DNA damage by minor groove binding copper-phenanthroline conjugates. Nucleic Acids Research. 2005; 33:5371–9. https://doi.org/10.1093/nar/gki856.
97. Spina-Purrello V, Patti D, Giuffrida-Stella AM, Nicoletti VG. Parp and Cell Death or Protection in Rat Primary Astroglial Cell Cultures Under LPS/IFNγ Induced Proinflammatory Conditions. Neurochemical Research. 2008; 33:2583–92. https://doi.org/10.1007/s11064-008-9835-1.
98. Nicoletti VG, Stella AM. Role of PARP under stress conditions: cell death or protection? Neurochem Res. 2003; 28:187–94.
99. Kemp S, Wheate NJ, Wang S, Collins JG, Ralph SF, Day AI, Higgins VJ, Aldrich-Wright JR. Encapsulation of platinum(II)-based DNA intercalators within cucurbit[6,7,8]urils. Journal of Biological Inorganic Chemistry. 2007; 12:969–79. https://doi.org/10.1007/s00775-007-0269-z.
100. Calabrese G, Morgan B, Riemer J. Mitochondrial Glutathione: Regulation and Functions. Antioxidants & Redox Signaling. 2017; 27:1162–77. https://doi.org/10.1089/ars.2017.7121.
101. Khan HY, Zubair H, Ullah MF, Ahmad A, Hadi SM. A prooxidant mechanism for the anticancer and chemopreventive properties of plant polyphenols. Curr Drug Targets. 2012; 13:1738–49.
102. Rehman SU, Zubair H, Sarwar T, Husain MA, Ishqi HM, Nehar S, Tabish M. Redox cycling of Cu(II) by 6-mercaptopurine leads to ROS generation and DNA breakage: possible mechanism of anticancer activity. Tumour Biol. 2014; 36:1237–44. https://doi.org/10.1007/s13277-014-2743-x.
103. Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB. Mitochondrial membrane potential. Anal Biochem. 2018; 552:50–59. https://doi.org/10.1016/j.ab.2017.07.009.
104. Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997; 91:627–37.
105. Li PF. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. The EMBO Journal. 1999; 18:6027–36. https://doi.org/10.1093/emboj/18.21.6027.
106. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harbor Perspectives in Medicine. 2016; 6:a026070. https://doi.org/10.1101/cshperspect.a026070.
107. Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000; 77:81–137.
108. Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death and Differentiation. 2017; 25:133–43. https://doi.org/10.1038/cdd.2017.174.
109. Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death and Differentiation. 2017; 25:114–32. https://doi.org/10.1038/cdd.2017.172.
110. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death and Differentiation. 2017; 25:104–13. https://doi.org/10.1038/cdd.2017.169.
111. Nelson WG, Kastan MB. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol. 1994; 14:1815–23.
112. Meplan C, Richard MJ, Hainaut P. Redox signalling and transition metals in the control of the p53 pathway. Biochem Pharmacol. 2000; 59:25–33.
113. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994; 265:346–55.
114. Hainaut P, Rolley N, Davies M, Milner J. Modulation by copper of p53 conformation and sequence-specific DNA binding: role for Cu(II)/Cu(I) redox mechanism. Oncogene. 1995; 10:27–32.
115. Ding WQ, Lind SE. Metal ionophores - an emerging class of anticancer drugs. IUBMB Life. 2009; 61:1013–8. https://doi.org/10.1002/iub.253.
116. Wild AC, Mulcahy RT. Pyrrolidine dithiocarbamate up-regulates the expression of the genes encoding the catalytic and regulatory subunits of gamma-glutamylcysteine synthetase and increases intracellular glutathione levels. Biochem J. 1999; 338:659–65.
117. Wu HH, Thomas JA, Momand J. p53 protein oxidation in cultured cells in response to pyrrolidine dithiocarbamate: a novel method for relating the amount of p53 oxidation in vivo to the regulation of p53-responsive genes. Biochem J. 2000; 351:87–93.
118. Hemann MT, Lowe SW. The p53–Bcl-2 connection. Cell Death and Differentiation. 2006; 13:1256–9. https://doi.org/10.1038/sj.cdd.4401962.
119. Rautureau GJ, Day CL, Hinds MG. Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis. International Journal of Molecular Sciences. 2010; 11:1808–24. https://doi.org/10.3390/ijms11041808.
120. Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Molecular Cell. 2011; 44:517–31. https://doi.org/10.1016/j.molcel.2011.10.001.
121. Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993; 75:241–51.
122. Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993; 262:1274–7.
123. Chen ZX, Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2009; 17:408–20. https://doi.org/10.1038/cdd.2009.132.
124. Lee K, Hart MR, Briehl MM, Mazar AP, Tome ME. The copper chelator ATN-224 induces caspase-independent cell death in diffuse large B cell lymphoma. International Journal of Oncology. 2014; 45:439–47. https://doi.org/10.3892/ijo.2014.2396.
125. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009; 458:1127–30. https://doi.org/10.1038/nature07986.
126. Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A. 1997; 94:3668–72.
127. Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A. 1998; 95:4997–5002.
128. Rossé T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998; 391:496–9. https://doi.org/10.1038/35160.
129. Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997; 277:370–2.
130. Chandra D, Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD. Necroptosis Interfaces with MOMP and the MPTP in Mediating Cell Death. PLoS One. 2015; 10:e0130520. https://doi.org/10.1371/journal.pone.0130520.
131. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. Relative Mitochondrial Priming of Myeloblasts and Normal HSCs Determines Chemotherapeutic Success in AML. Cell. 2012; 151:344–55. https://doi.org/10.1016/j.cell.2012.08.038.
132. Kaplan JH, Lutsenko S. Copper Transport in Mammalian Cells: Special Care for a Metal with Special Needs. J Biol Chem. 2009; 284:25461–5. https://doi.org/10.1074/jbc.R109.031286.
133. Fraústo da Silva JJR, Williams RJP. The Trinity of Life: The Genome, the Proteome, and the Mineral Chemical Elements. Journal of Chemical Education. 2004; 81:738. https://doi.org/10.1021/ed081p738.
134. Zhou B, Gitschier J. hCTR1: a human gene for copper uptake identified by complementation in yeast. Proc Natl Acad Sci U S A. 1997; 94:7481–6.
135. Maryon EB, Molloy SA, Kaplan JH. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am J Physiol Cell Physiol. 2013; 304:C768-C79. https://doi.org/10.1152/ajpcell.00417.2012.
136. Milardi D, Rizzarelli E. (2011). Neurodegeneration metallostasis and proteostasis. (Cambridge: Royal Society of Chemistry).
137. Jong NN, McKeage MJ. Emerging roles of metal solute carriers in cancer mechanisms and treatment. Biopharmaceutics & Drug Disposition. 2014; 35:450–62. https://doi.org/10.1002/bdd.1903.
138. Robinson NJ, Winge DR. Copper Metallochaperones. Annual Review of Biochemistry. 2010; 79:537–62. https://doi.org/10.1146/annurev-biochem-030409-143539.
139. Nose Y, Wood LK, Kim BE, Prohaska JR, Fry RS, Spears JW, Thiele DJ. Ctr1 Is an Apical Copper Transporter in Mammalian Intestinal Epithelial Cells in Vivo That Is Controlled at the Level of Protein Stability. J Biol Chem. 2010; 285:32385–92. https://doi.org/10.1074/jbc.M110.143826.
140. Molloy SA, Kaplan JH. Copper-dependent Recycling of hCTR1, the Human High Affinity Copper Transporter. J Biol Chem. 2009; 284:29704–13. https://doi.org/10.1074/jbc.M109.000166.
141. Cui H, Zhang AJ, McKeage MJ, Nott LM, Geraghty D, Guven N, Liu JJ. Copper transporter 1 in human colorectal cancer cell lines: Effects of endogenous and modified expression on oxaliplatin cytotoxicity. Journal of Inorganic Biochemistry. 2017; 177:249–58. https://doi.org/10.1016/j.jinorgbio.2017.04.022.
142. Banci L, Bertini I, Cantini F, Ciofi-Baffoni S. Cellular copper distribution: a mechanistic systems biology approach. Cellular and Molecular Life Sciences. 2010; 67:2563–89. https://doi.org/10.1007/s00018-010-0330-x.
143. Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J. 1996; 15:6084–95.
144. Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD. Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem. 1997; 272:21461–6.
145. Abada PB, Larson CA, Manorek G, Adams P, Howell SB. Sec61 Controls Sensitivity to Platinum-Containing Chemotherapeutic Agents through Modulation of the Copper-Transporting ATPase ATP7A. Molecular Pharmacology. 2012; 82:510–20. https://doi.org/10.1124/mol.112.079822.
146. Furukawa Y, Torres AS, O'Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. The EMBO Journal. 2004; 23:2872–81. https://doi.org/10.1038/sj.emboj.7600276.
147. M Fetherolf M, Boyd SD, Winkler DD, Winge DR. Oxygen-dependent activation of Cu,Zn-superoxide dismutase-1. Metallomics. 2017; 9:1047–59. https://doi.org/10.1039/c6mt00298f.
148. Hatori Y, Lutsenko S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants. 2016; 5:25. https://doi.org/10.3390/antiox5030025.
149. Boysen G. The Glutathione Conundrum: Stoichiometric Disconnect between Its Formation and Oxidative Stress. Chemical Research in Toxicology. 2017; 30:1113–6. https://doi.org/10.1021/acs.chemrestox.7b00018.
150. Ba LA, Doering M, Burkholz T, Jacob C. Metal trafficking: from maintaining the metal homeostasis to future drug design. Metallomics. 2009; 1:292. https://doi.org/10.1039/b904533c.
151. Hatori Y, Inouye S, Akagi R. Thiol-based copper handling by the copper chaperone Atox1. IUBMB Life. 2017; 69:246–54. https://doi.org/10.1002/iub.1620.
152. Klomp LW, Lin SJ, Yuan DS, Klausner RD, Culotta VC, Gitlin JD. Identification and functional expression of HAH1, a novel human gene involved in copper homeostasis. J Biol Chem. 1997; 272:9221–6.
153. Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, Fukai T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. 2008; 283:9157–67. https://doi.org/10.1074/jbc.M709463200.
154. Hamza I, Faisst A, Prohaska J, Chen J, Gruss P, Gitlin JD. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis. Proc Natl Acad Sci U S A. 2001; 98:6848–52. https://doi.org/10.1073/pnas.111058498.
155. Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008; 1780:1273–90. https://doi.org/10.1016/j.bbagen.2008.01.011.
156. Holmgren A. Antioxidant Function of Thioredoxin and Glutaredoxin Systems. Antioxidants & Redox Signaling. 2000; 2:811–20. https://doi.org/10.1089/ars.2000.2.4-811.
157. Hatori Y, Clasen S, Hasan NM, Barry AN, Lutsenko S. Functional Partnership of the Copper Export Machinery and Glutathione Balance in Human Cells. J Biol Chem. 2012; 287:26678–87. https://doi.org/10.1074/jbc.M112.381178.
158. Eiblmaier M, Meyer LA, Anderson CJ. The role of p53 in the trafficking of copper-64 to tumor cell nuclei. Cancer Biol Ther. 2008; 7:63–9.
159. Beaino W, Guo Y, Chang AJ, Anderson CJ. Roles of Atox1 and p53 in the trafficking of copper-64 to tumor cell nuclei: implications for cancer therapy. Journal of Biological Inorganic Chemistry. 2014; 19:427–38. https://doi.org/10.1007/s00775-013-1087-0.
160. Lutsenko S. Human copper homeostasis: a network of interconnected pathways. Current Opinion in Chemical Biology. 2010; 14:211–7. https://doi.org/10.1016/j.cbpa.2010.01.003.
161. Rampazzo E, Bonacchi S, Genovese D, Juris R, Sgarzi M, Montalti M, Prodi L, Zaccheroni N, Tomaselli G, Gentile S, Satriano C, Rizzarelli E. A versatile strategy for signal amplification based on core/shell silica nanoparticles. Chemistry. 2011; 17:13429–32. https://doi.org/10.1002/chem.201101851.
162. Zeng L, Miller EW, Pralle A, Isacoff EY, Chang CJ. A selective turn-on fluorescent sensor for imaging copper in living cells. J Am Chem Soc. 2006; 128:10–1. https://doi.org/10.1021/ja055064u.
163. Trusso Sfrazzetto G, Satriano C, Tomaselli GA, Rizzarelli E. Synthetic fluorescent probes to map metallostasis and intracellular fate of zinc and copper. Coordination Chemistry Reviews. 2016; 311:125–67. https://doi.org/10.1016/j.ccr.2015.11.012.
164. Leary SC. Redox Regulation of SCO Protein Function: Controlling Copper at a Mitochondrial Crossroad. Antioxidants & Redox Signaling. 2010; 13:1403–16. https://doi.org/10.1089/ars.2010.3116.
165. Leary SC, Winge DR, Cobine PA. “Pulling the plug” on cellular copper: The role of mitochondria in copper export. Biochim Biophys Acta. 2009; 1793:146–53. https://doi.org/10.1016/j.bbamcr.2008.05.002.
166. Baertling F, A M van den Brand M, Hertecant JL, Al-Shamsi A, P van den Heuvel L, Distelmaier F, Mayatepek E, Smeitink JA, Nijtmans LG, Rodenburg RJ. Mutations inCOA6cause CytochromecOxidase Deficiency and Neonatal Hypertrophic Cardiomyopathy. Human Mutation. 2015; 36:34–8. https://doi.org/10.1002/humu.22715.
167. Leary SC, Antonicka H, Sasarman F, Weraarpachai W, Cobine PA, Pan M, Brown GK, Brown R, Majewski J, Ha KC, Rahman S, Shoubridge EA. Novel Mutations inSCO1as a Cause of Fatal Infantile Encephalopathy and Lactic Acidosis. Human Mutation. 2013; 34:1366–70. https://doi.org/10.1002/humu.22385.
168. Stroud DA, Maher MJ, Lindau C, Vögtle FN, Frazier AE, Surgenor E, Mountford H, Singh AP, Bonas M, Oeljeklaus S, Warscheid B, Meisinger C, Thorburn DR, et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Human Molecular Genetics. 2015; 24:5404–15. https://doi.org/10.1093/hmg/ddv265.
169. Glerum DM, Shtanko A, Tzagoloff A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J Biol Chem. 1996; 271:14504–9.
170. Beers J, Glerum DM, Tzagoloff A. Purification, characterization, and localization of yeast Cox17p, a mitochondrial copper shuttle. J Biol Chem. 1997; 272:33191–6.
171. Heaton DN, George GN, Garrison G, Winge DR. The mitochondrial copper metallochaperone Cox17 exists as an oligomeric, polycopper complex. Biochemistry. 2001; 40:743–51.
172. Vest KE, Leary SC, Winge DR, Cobine PA. Copper Import into the Mitochondrial Matrix in Saccharomyces cerevisiae Is Mediated by Pic2, a Mitochondrial Carrier Family Protein. J Biol Chem. 2013; 288:23884–92. https://doi.org/10.1074/jbc.M113.470674.
173. Winge DR. Filling the mitochondrial copper pool. J Biol Chem. 2018; 293:1897–8. https://doi.org/10.1074/jbc.H118.001457.
174. Boulet A, Vest KE, Maynard MK, Gammon MG, Russell AC, Mathews AT, Cole SE, Zhu X, Phillips CB, Kwong JQ, Dodani SC, Leary SC, Cobine PA. The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochromecoxidase biogenesis. J Biol Chem. 2018; 293:1887–96. https://doi.org/10.1074/jbc.RA117.000265.
175. Jett KA, Leary SC. Building the CuAsite of cytochromecoxidase: A complicated, redox-dependent process driven by a surprisingly large complement of accessory proteins. J Biol Chem. 2018; 293:4644–52. https://doi.org/10.1074/jbc.R117.816132.
176. Banci L, Bertini I, Ciofi-Baffoni S, Hadjiloi T, Martinelli M, Palumaa P. Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer. Proc Natl Acad Sci U S A. 2008; 105:6803–8. https://doi.org/10.1073/pnas.0800019105.
177. Morgada MN, Abriata LA, Cefaro C, Gajda K, Banci L, Vila AJ. Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuAin human cytochrome oxidase. Proc Natl Acad Sci U S A. 2015; 112:11771–6. https://doi.org/10.1073/pnas.1505056112.
178. Leary SC, Sasarman F, Nishimura T, Shoubridge EA. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Human Molecular Genetics. 2009; 18:2230–40. https://doi.org/10.1093/hmg/ddp158.
179. Suzuki C, Daigo Y, Kikuchi T, Katagiri T, Nakamura Y. Identification of COX17 as a therapeutic target for non-small cell lung cancer. Cancer Res. 2003; 63:7038–41.
180. Chojnacka M, Gornicka A, Oeljeklaus S, Warscheid B, Chacinska A. Cox17 Protein Is an Auxiliary Factor Involved in the Control of the Mitochondrial Contact Site and Cristae Organizing System. J Biol Chem. 2015; 290:15304–12. https://doi.org/10.1074/jbc.M115.645069.
181. Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific Copper Transfer from the Cox17 Metallochaperone to Both Sco1 and Cox11 in the Assembly of Yeast Cytochrome c Oxidase. J Biol Chem. 2004; 279:35334–40. https://doi.org/10.1074/jbc.M404747200.
182. Rampelt H, Zerbes RM, van der Laan M, Pfanner N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim Biophys Acta. 2017; 1864:737–46. https://doi.org/10.1016/j.bbamcr.2016.05.020.
183. Barresi V, Trovato-Salinaro A, Spampinato G, Musso N, Castorina S, Rizzarelli E, Condorelli DF. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation ofSLC31A1,SCO1, andCOX11in colorectal cancer. FEBS Open Bio. 2016; 6:794–806. https://doi.org/10.1002/2211-5463.12060.
184. Carden T, Singh B, Mooga V, Bajpai P, Singh KK. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J Biol Chem. 2017; 292:20694–706. https://doi.org/10.1074/jbc.M117.797001.
185. Jayanthi S, Lewis BD, Cadet JL. Fas-induced apoptosis of glioma cells is associated with down-regulation of the hSCO1 protein, a subunit of complex IV. Brain Res Mol Brain Res. 2001; 91:131–6.
186. Won KY, Lim SJ, Kim GY, Kim YW, Han SA, Song JY, Lee DK. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Human Pathology. 2012; 43:221–8. https://doi.org/10.1016/j.humpath.2011.04.021.
187. Madan E, Gogna R, Kuppusamy P, Bhatt M, Mahdi AA, Pati U. SCO2 Induces p53-Mediated Apoptosis by Thr845 Phosphorylation of ASK-1 and Dissociation of the ASK-1-Trx Complex. Molecular and Cellular Biology. 2013; 33:1285–302. https://doi.org/10.1128/mcb.06798-11.
188. Matoba S. p53 Regulates Mitochondrial Respiration. Science. 2006; 312:1650–3. https://doi.org/10.1126/science.1126863.
189. Calderazzo F, Marchetti F, Pampaloni G, Passarelli V. Co-ordination properties of 1,10-phenanthroline-5,6-dione towards group 4 and 5 metals in low and high oxidation states†. Journal of the Chemical Society, Dalton Transactions. 1999: 0:43 89–96. https://doi.org/10.1039/a906016b.
190. Margiotta N, Bertolasi V, Capitelli F, Maresca L, Moliterni AG, Vizza F, Natile G. Influence of steric and electronic factors in the stabilization of five-coordinate ethylene complexes of platinum(II): X-ray crystal structure of [PtCl2(2,9-dimethyl-1,10-phenanthroline-5,6-dione)]. Inorganica Chimica Acta. 2004; 357:149–58. https://doi.org/10.1016/s0020-1693(03)00382-7.
191. Yamada Y, Sakurai H, Miyashita Y, Fujisawa K, Okamoto KI. Crystal structures, electronic absorption and reflectance spectral behaviors, and electrochemical properties of five-coordinated chlorocopper(II) complexes with 5,6-disubstituted-1,10-phenanthroline. Polyhedron. 2002; 21:2143–7. https://doi.org/10.1016/s0277-5387(02)01157-9.
192. McCann M, Coyle B, McKay S, McCormack P, Kavanagh K, Devereux M, McKee V, Kinsella P, O'Connor R, Clynes M. Synthesis and X-ray crystal structure of [Ag(phendio)2] ClO4 (phendio = 1,10-phenanthroline-5,6-dione) and its effects on fungal and mammalian cells. Biometals. 2004; 17:635–45. https://doi.org/10.1007/s10534-004-1229-5.
193. http://www.hyperquad.co.uk/hyss.htm.
194. Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods. 1988; 20:309–21.
195. Kruger NJ. The Bradford Method for Protein Quantitation. Methods Mol Biol. 2002; 0:15–22. https://doi.org/10.1385/1-59259-169-8:15.
196. Schagger H. Tricine-SDS-PAGE. Nat Protoc. 2006; 1:16–22. https://doi.org/10.1038/nprot.2006.4.
197. Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995; 260:406–17.
198. Giacomelli C, Trincavelli ML, Satriano C, Hansson Ö, La Mendola D, Rizzarelli E, Martini C. Copper (II) ions modulate Angiogenin activity in human endothelial cells. The International Journal of Biochemistry & Cell Biology. 2015; 60:185–96. https://doi.org/10.1016/j.biocel.2015.01.005.
199. Belviso BD, Galliani A, Lasorsa A, Mirabelli V, Caliandro R, Arnesano F, Natile G. Oxaliplatin Binding to Human Copper Chaperone Atox1 and Protein Dimerization. Inorganic Chemistry. 2016; 55:6563–73. https://doi.org/10.1021/acs.inorgchem.6b00750.
200. Naletova IN, Muronetz VI, Schmalhausen EV. Unfolded, oxidized, and thermoinactivated forms of glyceraldehyde-3-phosphate dehydrogenase interact with the chaperonin GroEL in different ways. Biochim Biophys Acta. 2006; 1764:831–8. https://doi.org/10.1016/j.bbapap.2006.02.002.