
INTRODUCTION
Bromodomain and extra-terminal domain (BET) proteins bind to acetylated lysine residues of histones [1], play important roles in cellular homeostasis [2] and regulate gene transcription [3]. The BET subfamily of proteins includes BRD2, BRD3, BRD4 and BRDT, which normally act as epigenetic readers [4, 5]. BET inhibitors competitively bind to the acetyl lysine recognition pocket, displacing BET proteins from the chromatin and causing transcriptional changes, which leads to cell cycle arrest and apoptosis [6, 7]. Consequently, inhibition of BET proteins has been extensively investigated as therapeutic agents of certain cancers, inflammatory diseases and metabolic dysfunctions [8–10]. As a result, several BET inhibitors including I-BET762, OTX015, TEN-010, and CPI-0610 have been approved for clinical trials to target cancerous cells [11]. A less stable but more commonly studied BET inhibitor, JQ1 ((S)-tert-butyl2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3a][1,4] diazepin-6-yl)acetate), is particularly effective against BRD4 and has been shown to downregulate c-MYC, an oncoprotein involved in cell proliferation and cancer pathogenesis [6]. JQ1 inhibition of BET proteins is known to cause decreased proliferation, cell cycle arrest, and induction of apoptosis in several cancer cell types including pancreatic cancer, leukemia, lymphoma, and triple negative breast cancer [7, 8, 12, 13]. Furthermore, JQ1 has been investigated for treating diseases of the central nervous system since it is capable of crossing the blood brain barrier [14]. JQ1 has been shown to reduce proliferation and induce apoptosis in cells from medulloblastomas and glioblastomas [15, 16]. In addition, it has been tested to treat damaged retinal ganglion cells in a mouse model [17]. JQ1 can also be effective against other neurodegenerative diseases such as Alzheimer’s disease by reducing splenomegaly and neuroinflammation [18].
While anti-cancer and anti-inflammatory properties led to the approval of several inhibitors of BRD4 for drug therapy, their toxicological properties have been poorly investigated. A recent study showed that JQ1 caused memory and other neurological problems in mice, suggesting that BRD4 is important for neurological functions [19]. Another report stated that pharmacological concentrations of JQ1 in mice caused significant weight loss as well as lymphoid and hematopoietic toxicity [20]. In mouse embryonic stem cells, JQ1 induced spontaneous differentiation by downregulating pluripotent genes [21]. We also have previously shown that treatment of JQ1 in mesenchymal stem cells (MSCs) derived from human umbilical cord inhibited growth, caused cell cycle arrest, and interfered with signaling pathways by downregulating expression of WNT [22]. In a clinical study, OTX015, a BET inhibitor that inhibits BRD2, BRD3, and BRD4, was reportedly found to cause thrombocytopenia, anemia, neutropenia, diarrhea, fatigue, and nausea in patients being treated for lymphoma and multiple myeloma [23]. While these limited studies show the effects of JQ1 in pluripotent and multipotent stem cells, the exact mechanism of toxicity is unknown. In this study, we show that JQ1 caused selective toxicity of neuronal derivatives (NDs) of MSCs. These findings have implications in the therapeutic application of BET inhibitors requiring more careful evaluation of their toxicological properties.
RESULTS
Examination of JQ1 treatment on cell morphology and growth
Our previous studies have shown that JQ1 inhibited proliferation of MSCs by causing cell cycle arrest and induction of differentiation [22]. In this study, we investigated the effect of JQ1 on derivatives of MSCs. The results depicted in Figure 1A show that MSCs grown in culture medium (CM) displayed typical fibroblastoid morphology but when treated with JQ1 they had flatter morphology and were larger in size as compared to untreated MSCs. MSCs induced to adipogenic, chondrogenic, and osteogenic derivatives (ADs, CDs, and ODs, respectively) did not show significant morphological changes upon treatment with JQ1 (Figure 1A). MSCs cultured in neural induction medium (NM) differentiated into NDs, exhibited cellular extensions, and had large nuclei, typical of early neural progenitors [24, 25]. However, these neural extensions were lost upon treatment with JQ1. In addition, JQ1 caused cell rounding and loss of adhesion, while the remaining adherent cells exhibited fibroblastoid morphology (Figure 1A). Although JQ1 adversely affected cell proliferation of MSCs, ADs, and NDs (Figure 1B), only NDs showed a significant reduction in cell viability (10%) (Figure 1C). On the other hand, an insignificant decrease in the viability of undifferentiated MSCs in the presence of JQ1 was observed. JQ1 also did not significantly affect the viability of ADs, CDs and ODs (Figure 1C), indicating that MSCs differentiated into the neural lineage were selectively sensitive to JQ1.
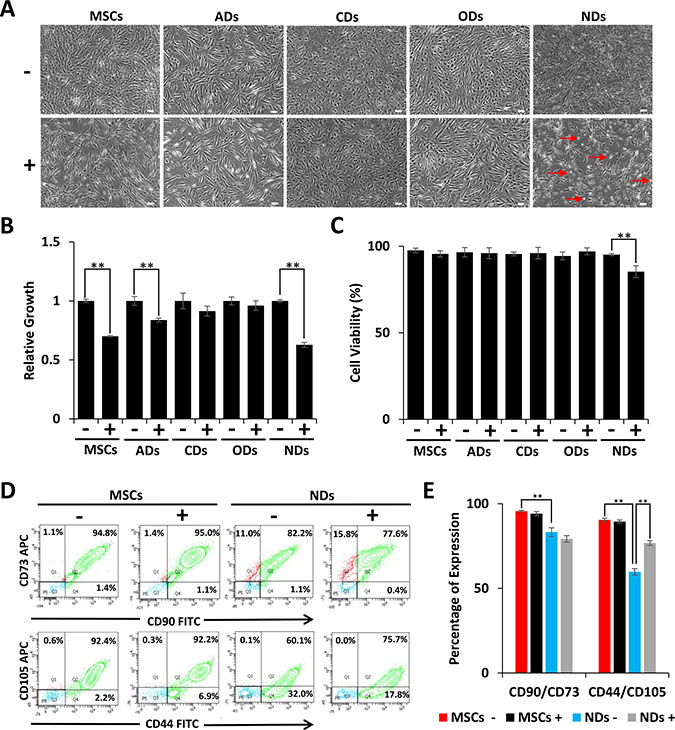
Figure 1: Effect of JQ1 on morphology, viability, and growth of MSCs and derivatives. MSCs were cultured in culture medium (CM) or differentiation media for induction into adipogenic, chondrogenic, osteogenic, and neuronal derivatives (ADs, CDs, ODs, and NDs, respectively). (A) Cell morphology was visualized by phase contrast microscopy. Scale bars represent 100 μm (Magnification: 4X). Arrows in JQ1 treated NDs point to rounded cells. (B) Relative growth of MSCs and their derivatives in the absence or presence of JQ1. (C) Graphical representation of the percentage of viable cells as determined by trypan blue staining. (D) Representative flow cytometric analysis of MSCs and NDs to determine the MSC specific markers, CD90, CD73, CD44, and CD105. (E) Graphical representation of flow cytometric data showing percentage of cells positive for MSC markers. Experiments were performed in triplicate and error bars represent SEM of three independent experiments (n = 3). *p < 0.05 and **p <0.01.
Because the majority of the NDs treated with JQ1 remained viable, we wondered if not all of the MSCs differentiated into NDs. We investigated the expression of MSC surface markers by flow cytometry, and the results depicted in Figure 1D and 1E showed a reduction in CD90/CD73 expression from 95% to 82% in MSCs and NDs, respectively. Furthermore, CD44/CD105 expression decreased from 92% to 60% when MSCs were induced to neural differentiation. This significant loss of MSC markers in NDs suggested that only a specific population of MSCs underwent differentiation.
The expression of MSC markers remained almost the same in undifferentiated MSCs cultured in the absence or presence of JQ1. However, when MSCs were induced to differentiate into NDs and treated with JQ1, CD90/CD73 expression was decreased insignificantly from 82% to 77% but CD44/CD105 expression increased from 60% to 75%. Thus, suggesting that JQ1 was selectively deleterious to differentiated cells.
Effect of JQ1 on the expression of neural markers
The results depicted in Figure 2A show expression of early neurogenic proteins, TUJ1, Nestin, and NeuN, in NDs but not MSCs further confirming that MSCs were induced to the neuronal lineage in NM. Consistent with our previous findings [22], treatment of JQ1 resulted in an increase in TUJ1 expression in MSCs. However, JQ1 caused a significant decrease in the expression of Nestin and NeuN, but not TUJ1 in NDs (Figure 2B and 2C). We then investigated the transcriptional expression of neural markers, TUJ1, Nestin, and PAX6 using quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). The results described in Figure 2D show loss of expression of neural genes in NDs upon treatment with JQ1, suggesting the selective toxicity of differentiated neuronal cells but not the undifferentiated cells (MSCs).
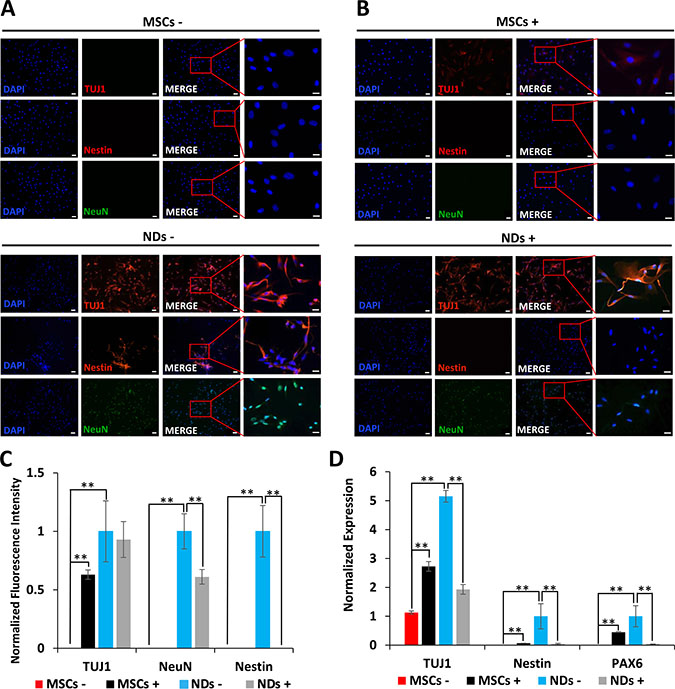
Figure 2: Effect of JQ1 on expression of neural markers. MSCs and NDs were untreated (−) or treated (+) with JQ1 for 48 hours. (A and B) Immunocytochemical analysis of expression of neural proteins TUJ1, Nestin, and NeuN, in MSCs and NDs in the absence or presence of JQ1, respectively. Scale bars represent 50 μm (Magnification: 10X) and 20 μm in high magnification merged inserts (Magnification: 40X), respectively. (C) Quantification of normalized fluorescent intensities of neural proteins in MSCs and NDs treated with and without JQ1 using ImageJ software. (D) Transcriptional analysis of neural genes, TUJ1, Nestin, and PAX6 as determined by qRT-PCR. Experiments were performed in triplicate and error bars represent SEM of three independent experiments (n = 3). *p < 0.05 and **p < 0.01.
Analysis of cell death
The loss of cell viability in NDs exposed to JQ1 was also evaluated using an apoptosis assay. The results shown in Figure 3A and 3B depict representative flow cytometric analysis of Annexin-V and propidium iodide (PI) staining and the average percentage of dead cells, respectively. A significantly higher percentage of dead cells was observed in JQ1 treated NDs (16.7%) as compared to untreated NDs (Figure 3B). The dead cells stained with both Annexin-V and PI were likely to be in the late stages of apoptosis. Based on the fact that the adherent cells had fibroblastoid morphology after JQ1 treatment and expressed MSC markers as shown above, the loss of viability of NDs was confirmed via apoptosis rather than random cell death.
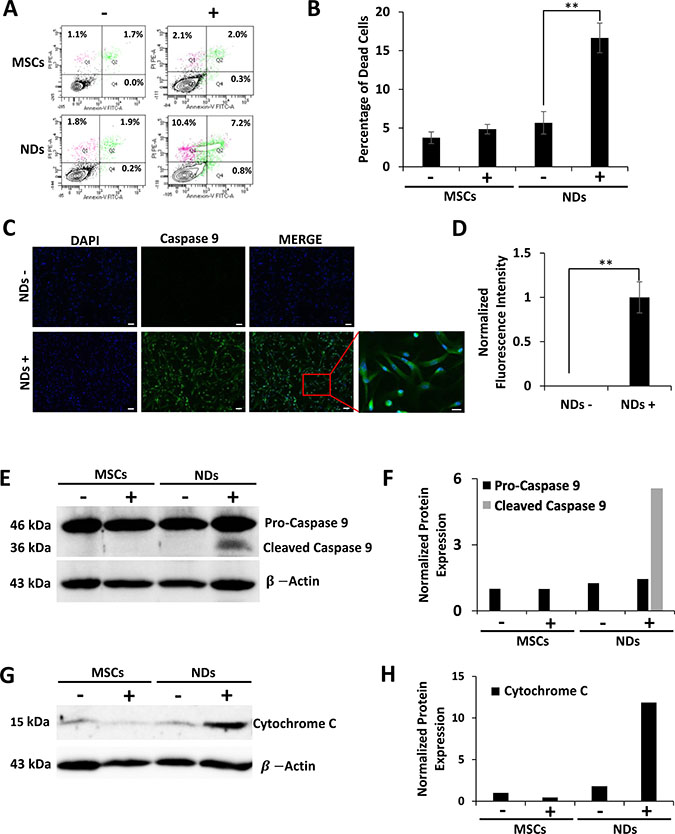
Figure 3: Effect of JQ1 on the expression of Caspase 9 and Cytochrome C. MSCs and NDs untreated (−) and treated (+) with JQ1 for 48 hours and subjected to analysis. (A) Representative flow cytomeric plots of cells stained with Annexin-V/FITC and PI. (B) Graphical representation of the average percentage of dead cells as determined by flow cytometry, error bars represent SEM of three independent experiments (n = 3). (C) Immunocytochemical analysis of Caspase 9 showing protein expression in NDs treated with JQ1. Scale bars represent 50 μm (Magnification: 10X) and 20 μm in high magnification merged insert (Magnification: 40X), respectively. (D) Quantification of normalized fluorescent intensity of Caspase 9 expression in NDs using ImageJ software. *p < 0.05 and **p < 0.01. (E) Western blotting analysis of Caspase 9 protein expression showing cleaved Caspase 9 at 36 kDa in the JQ1 treated NDs. (F) Quantification of Caspase 9 protein expression normalized to β-Actin using ImageJ software. (G) Western blotting analysis showing Cytochrome C protein expression. (H) Quantification of Cytochrome C protein expression normalized to β-Actin using ImageJ software.
To further understand the apoptosis induced in NDs by JQ1, we investigated the expression of proteins involved in cell death. The results of the immunocytochemical analysis given in Figure 3C and quantified in Figure 3D showed that NDs treated with JQ1 had increased fluorescence expression of Caspase 9 as compared to the untreated control. Higher expression of Caspase 9 was confirmed by western blot analysis (Figure 3E and 3F). Furthermore, JQ1 treated NDs showed activation of Caspase 9 as evident by the presence of the 36 kDa cleaved protein. In addition, western blot results shown in Figure 3G and 3H indicated an increase in the expression of Cytochrome C in NDs treated with JQ1 as compared to the untreated control cells.
Further investigation of the action of JQ1 showed upregulation of BRD4 and c-MYC genes in NDs in comparison to MSCs, but downregulation of these genes upon JQ1 treatment (Figure 4A). In MSCs, JQ1 caused upregulation of p53 and p21, suggesting cell cycle arrest as reported previously [22]. In NDs, JQ1 caused a significant downregulation of p53 and p21 but upregulation of BAX, while there was no significant difference in expression of PUMA and NOXA (Figure 4A). Based on these results, we proposed a mechanism of action of JQ1 in MSCs and NDs as depicted in Figure 4B. JQ1 caused p21 mediated cell cycle arrest and induced differentiation in MSCs but resulted in intrinsic apoptosis in NDs. Taken together, these results suggest that JQ1 differentially affected MSCs and NDs.
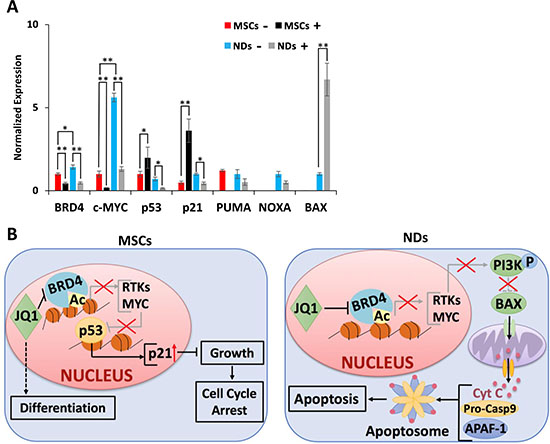
Figure 4: Proposed mechanism of action of JQ1. (A) Transcriptional analysis of BRD4, c-MYC, p53, p21, PUMA, NOXA, and BAX in MSCs and NDs untreated (−) and treated (+) with JQ1 as determined by qRT-PCR. Experiments were performed in triplicate and error bars represent SEM of three independent experiments (n = 3). *p < 0.05 and **p < 0.01. (B) JQ1 caused p21 mediated cell cycle arrest and differentiation in MSCs, and resulted in Caspase 9 mediated intrinsic apoptosis in NDs.
DISCUSSION
Several BET inhibitors have been approved as anti-cancer agents for leukemia, lymphoma, and multiple myeloma [23, 26]. They have been shown to downregulate c-MYC and decrease cell proliferation [6, 27, 28]. BET inhibitors can also cause apoptosis of cancer cells by downregulating receptor tyrosine kinase (RTK) signaling pathways [29]. We have previously shown that JQ1 caused not only cell cycle arrest but differentiation of human umbilical cord MSCs [22].
Studies have shown that JQ1 suppresses differentiation in adipogenic, chondrogenic, osteogenic, and myogenic cells [30–34]. To examine the effect of JQ1 on neural differentiation, MSCs were cultured in neural induction medium, NM. The results indicated that NM induced MSCs to differentiate into NDs, since they expressed PAX6 and Nestin, characteristic of neural stem cells [35, 36], as well as NeuN and TUJ1, markers of neuronal precursors and neurons [37, 38]. A previous report showed that BET inhibition by JQ1 enhanced differentiation of mouse neural progenitor cells into neurons but suppressed cell cycle progression and gliogenesis [39]. In contrast, our results showed that JQ1 caused the loss of cellular extensions in NDs, followed by rounding of cells. These results are similar to previously reported morphological changes observed with other JQ1 sensitive cells, including pancreatic stellate cells and human foreskin fibroblasts [40, 41].
Our preliminary investigation of MSC derivatives showed that JQ1 did not have a significant effect on the morphology of ADs, CDs, and ODs. While JQ1 adversely affected the proliferation of both MSCs, ADs and NDs, only NDs showed a significant decrease in cell viability following exposure to JQ1. This is consistent with our previous study whereby JQ1 inhibited proliferation by cell cycle arrest but did not cause cell death in MSCs [22].
Induction of neural differentiation of MSCs yielded early neuronal derivatives, while a majority of the cells remained undifferentiated as they expressed MSC surface markers. In comparison to MSCs, NDs exhibited only a 15.5% and 31% decrease in CD90 and CD105 expression, respectively. Interestingly, the percentage of cells expressing CD105 was higher in NDs following JQ1 treatment. Evidently, JQ1 was selectively toxic to neuronal cells but not to undifferentiated cells, which may explain the increase in the expression of MSC markers and decrease in neural markers observed in NDs treated with JQ1.
Furthermore, the induction of MSCs into NDs resulted in the increased expression of neuronal markers, TUJ1, Nestin, and PAX6, as well as BRD4 and c-MYC. BRD4 has been shown to activate transcription in neurons and is typically expressed in cells positive for NeuN but not GFAP [19]. Additionally, c-MYC has been shown to promote neuronal differentiation [42]. JQ1 treatment led to downregulation or loss of these markers. Again, this was presumably due to the selective toxicity of JQ1 to NDs.
We further investigated JQ1 induced cell death in NDs. The results indicated increased expression of Caspase 9 in the JQ1 treated but not the untreated NDs. In addition, the western blot analysis showed activation of Caspase 9 but not Caspase 8 (data not shown), indicating that death of neuronal cells was caused by Caspase 9 mediated apoptosis. This is in line with previous reports that showed that JQ1 activated Caspase 9 in established cancer cell lines, including hepatocellular carcinoma and glioblastoma cells [43, 44]. In addition, the results indicated upregulation of Cytochrome C, which is released by the mitochondria to cause cleavage of Caspase 9 in JQ1 treated NDs.
Further investigation of the mechanism of action of JQ1 in NDs showed downregulation of p53 and p21. This corresponded with the near significant downregulation of NOXA, and PUMA, which are transcriptional targets of p53 [45–47]. We also investigated the expression of BAX, a pro-apoptotic protein that triggers intrinsic apoptosis through the release of Cytochrome C [48, 49], and found it to be significantly upregulated in NDs treated with JQ1. These results indicated that NDs are sensitive to JQ1, and undergo Caspase 9 mediated apoptosis. JQ1 sensitivity may be due to high levels of c-MYC expression in NDs following induction of neuronal differentiation. In fact, JQ1 is well known to cause downregulation of c-MYC [6, 28, 50] and has been shown to significantly decrease cell proliferation and preferentially induce apoptosis in medulloblastoma derived cell lines expressing high levels of MYC [16].
In sensitive cells, JQ1 treatment inhibits the BRD4 mediated transcription of RTKs [29, 51], resulting in the inactivation of the phosphoinositide 3-kinase (PI3K)/Akt pathway, which leads to the downstream dephosphorylation of BAX, thus allowing it to enter the mitochondria resulting in the release of Cytochrome C [52]. This is consistent with the Caspase 9 mediated cell death observed in NDs. On the other hand, transcriptional analysis showed upregulation of p53 and p21 in MSCs treated with JQ1, which is consistent with our previous study indicating that MSCs are undergoing cell cycle arrest [22]. We propose that JQ1 treated MSCs are undergoing p21 mediated cell cycle arrest, which is in agreement with other studies showing that JQ1 induced G1 cell cycle arrest caused by upregulation of p21 [15, 53–55].
Overall, these results led us to propose the mechanism of action of JQ1 in MSCs and NDs as depicted in Figure 4B. Briefly, JQ1 caused cell cycle arrest and differentiation in MSCs, but neural toxicity in NDs. These results are likely to prompt further investigation into the molecular mechanism of action of JQ1. Furthermore, they suggest a more careful evaluation of the use of BET inhibitors as therapeutic agents, since they may cause unwanted damage to non-target cells and tissues.
MATERIALS AND METHODS
Cell culture
Human umbilical cord MSCs were isolated and characterized as previously described [56]. MSCs (Passage 7) were grown in culture medium (CM) containing high glucose Dulbecco’s modified Eagle’s medium (DMEM) with 4500 mg/L glucose and 2 mM L-glutamine (Fisher Scientific, Pittsburgh, PA, USA). CM was supplemented with 10% fetal bovine serum (FBS) (VWR International, Radnor, PA, USA) and 5.6% of antibiotic solu tion (0.1% gentamicin, 0.2% streptomycin, and 0.12% penicillin) (Sigma Aldrich, St. Louis, MO, USA). MSCs were differentiated into ADs using high glucose DMEM containing 0.5 μM IBMX (Sigma), 1 μM dexamethasone (Sigma), 10 μM insulin (PeproTech, Rocky Hill, NJ, USA), and 200 μM indomethacin (Sigma). MSCs were differentiated into CDs using high glucose DMEM supplemented with 20 ng TGFβ1 (PeproTech), 10 ng insulin (PeproTech), 100 nM dexamethasone (Sigma), and 100 μM ascorbic acid (Sigma). Differentiation into ODs was achieved using high glucose DMEM supplemented with 0.1 μM dexamethasone (Sigma), 10 μM β-glycerophosphate (Sigma), and 50 μM ascorbate-phosphate (Sigma). To differentiate MSCs into NDs, MSCs were cultured in neural induction medium (NM) containing neurobasal medium (Fisher Scientific), supplemented with 2 mM L-glutamine, 2% B27 (Fisher Scientific), 10−5 M retinoic acid (Sigma), 100 μM ascorbic acid (Sigma), and 5 μM IBMX (Sigma), 20 ng/mL NGF, 30 ng/mL EGF, and 10 ng/mL bFGF (PeproTech). Cells were grown in a 5% CO2 incubator.
Treatment of cells with JQ1
MSCs were cultured in CM as controls. For differentiation, MSCs were grown in culture plates (2.5 × 104 cells/25 flasks) in cell specific media for 2 (NDs) or 5 (ADs, CDs, ODs, and NDs) days. They were then treated in the absence or presence of 500 nM JQ1 (Bradner’s Laboratory, Harvard Medical School, Boston, MA, USA) dissolved in dimethyl sulfoxide (Fisher Scientific) and further incubated for 48 hours before analysis for morphological and biological changes.
Determination of cell viability and proliferation
After MSCs and their derivatives were cultured in their respective media for 2 (MSCs and NDs) or 5 (ADs, CDs, and ODs) days, they were then treated in the absence or presence of JQ1 for 48 hours followed by staining with trypan blue solution (Sigma) to determine cell proliferation and viability. Relative growth was normalized to untreated controls. The stained cells were considered non-viable and counted using a hemocytometer.
MSC characterization using flow cytometry
MSCs were cultured in CM or NM for 2 days, treated in the absence or presence of JQ1 for 48 hours, dissociated with TrypLE (Life Technologies, Carlsbad, CA, USA), centrifuged, and re-suspended in PBS. Cells were incubated with mouse antibodies against specific markers including CD90 and CD44 (conjugated with FITC), and CD73 and CD105 (conjugated with APC) (Fisher Scientific) and analyzed by flow cytometry and Diva Software (BD Canto II, BD Biosciences, San Jose, CA, USA).
Apoptosis assay
MSCs were cultured in CM and NM for 2 days, treated in the absence or presence of JQ1 for 48 hours, and then subjected to apoptosis assay. In brief, cells suspended in 100 μL of Annexin-V binding buffer were stained with 5 μL of Annexin-V/FITC (Biolegend, San Diego, CA, USA) and 10 μL of propidium iodide (PI, Sigma, St. Louis, MO, USA). The stained samples were analyzed by flow cytometry. Cells that were positive for Annexin-V staining only were considered apoptotic while cells that were positive for PI were considered necrotic, and cells positive for both Annexin-V and PI were defined as late apoptotic. Viable cells were negative for both Annexin-V and PI staining.
Immunocytochemistry analysis
MSCs cultured in CM and NM for 5 days were then treated in the absence or presence of JQ1 for 48 hours before analysis. For immunocytochemistry, cells were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.5% TritonX100 (Sigma) for 10 minutes, and blocked with 2% bovine serum albumin (Sigma) dissolved in PBS for 1 hour to block non-specific binding. Cells were then incubated with primary antibodies including TUJ1 (Santa Cruz Biotechnology, Dallas, TX, USA), NeuN (Invitrogen, Carlsbad, CA, USA), Nestin (Santa Cruz Biotechnology), or Caspase 9 (Santa Cruz) overnight at 4° C. Primary antibodies were then removed, cells were washed with PBS three times and stained with the respective secondary antibodies (Cy3-labelled goat anti-mouse IgG (KPL, Gaithersburg, MD, USA), and Alexa Fluor 488 goat anti-rabbit IgG (Fisher Scientific) for 1 hour at room temperature. Secondary antibodies were then removed and cells were washed with PBS and stained with 4′,6-diamidino-2-phenylindole (DAPI, Fisher Scientific) to stain the nuclei. Stained cells were analyzed using fluorescence microscopy (NIKON Instruments Inc., Melville, NY, USA). Protein expression was quantified with ImageJ software for the corrected total cell fluorescence (CTCF) using the following equation: CTCF = Integrated Density – (Area of selected cell x Mean fluorescence of background readings). Fluorescence intensities were normalized to the expression of NDs without JQ1.
qRT-PCR
MSCs cultured in CM and NM for 5 days were treated in the absence or presence of JQ1 for 48 hours. Total mRNA was isolated from these cells using the GeneJET RNA purification kit (Fisher Scientific) by following the instructions given by manufacturer. RNA was purified by incubating the isolated RNA at 37° C for 30 minutes with DNase (Promega, Madison, WI, USA) in a thermocycler (MJ Research PTC-100 Thermal Cycler; GMI, Ramsey, MN, USA). The purified RNA was used to synthesize cDNA by using BioRad iScript kit (BioRad, Hercules, CA, USA). Sso-Advanced Universal SYBR Green Supermix Kit (BioRad) was used for qRT-PCR using the CFX96 Real-Time System (BioRad) with 5 μL of SYBR green, 3 μL of distilled water, 1 μL of cDNA that is diluted 1:10 in distilled water, and 0.5 μL of forward primer and reverse primer. GAPDH and β-ACTIN were used as reference genes to normalize the amplification of the target genes. Primer sequences are listed in Table 1.
Table 1: List of human primer sequences used in qRT-PCR
Gene |
Primer Sequence |
||
|---|---|---|---|
Forward (5′-3′) |
Reverse (5′-3′) |
Product Length |
|
TUJ1 |
GGCCAAGTTCTGGGAAGTCA |
CGAGTCGCCCACGTAGTTG |
70 |
Nestin |
GAAACAGCCATAGAGGGCAAA |
TGGTTTTTCCAGAGTCTTCAGTGA |
168 |
PAX6 |
CTTTGCTTGGGAAATCCGAG |
AGCCAGGTTGCGAAGAACTC |
103 |
BRD4 |
AGGCAAAAGGAAGAGGACG |
CGATGCTTGAGTTGTGTTTGG |
86 |
c-MYC |
CGGAACTCTTGTGCGTAAGG |
CTCAGCCAAGGTTGTGAGGT |
123 |
p53 |
GGGAGCACTAAGCGAGCA |
ACGCCCACGGATCTGAAG |
102 |
p21 |
GCAGACCAGCATGACAGATTT |
GGATTAGGGCTTCCTCTTGGA |
70 |
PUMA |
GACGACCTCAACGCACAGTA |
CTAATTGGGCTCCATCTCG |
147 |
NOXA |
GAGATGCCTGGGAAGAAGG |
TTCTGCCGGAAGTTCAGTTT |
125 |
BAX |
GGGTGGTTGGGTGAGACTC |
AGACACGTAAGGAAAACGCATTA |
191 |
GAPDH |
GAAGGTGAAGGTCGGAGTC |
GAAGATGGTGATGGGATTTC |
226 |
β-ACTIN |
AATCTGCGACCACACCTTCTAC |
ATAGCACAGCCTGGATAGCAAC |
170 |
Western blot analysis
MSCs cultured in CM or NM for 5 days and then treated with or without JQ1 were lysed for protein using RIPA buffer (Sigma) and quantified using the Pierce 660 nm protein assay on the NanoDrop 1000 spectrophotometer (Fisher Scientific). The lysate (30 μg of total proteins) was resolved using SDS-PAGE with 12% resolving gel and 6% stacking gel and transferred to a nitrocellulose membrane (BioRad) at a continuous current of 100V for 90 minutes. For antibody staining, the membrane was blocked with 5% nonfat dry milk dissolved in TBS 1X containing 0.1% Tween-20 (TBST) for 30 minutes and incubated with primary antibodies (Caspase 9, Cytochrome C, β-Actin; Santa Cruz Biotechnology) at a 1:500 dilution in the blocking solution overnight at 4° C. The membrane was then washed with TBST and incubated with the secondary antibody conjugated with HRP (Santa Cruz Biotechnology) at a 1:10,000 dilution in the blocking solution for 2 hours at room temperature. After washing with TBST, the blot was stained with BioRad chemiluminescence for 5 minutes and bands were visualized using a chemidoc (BioRad). Band intensities were quantified using ImageJ software (NIH, Bethesda, MD, USA) and normalized to β-Actin.
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM). All experiments were performed in triplicate and three independent experiments were carried out. One-way ANOVA analysis was performed and results with a p-value ≤ 0.05 were considered statistically significant (*p < 0.05 and **p < 0.01) as compared to untreated control cells. All analyses were performed using SPSS version 11.5 (SPSS Inc.).
Abbreviations
BET: Bromodomain and extra-terminal domain; JQ1: (S)-tert-butyl2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2 f][1,2,4]triazolo[4,3a][1,4] diazepin-6-yl)acetate; MSCs: Mesenchymal stem cells; CM: Culture medium; ADs: Adipogenic derivatives; CDs: Chondrogenic derivatives; ODs: Osteogenic derivatives; NM: Neural induction medium; NDs: Neuronal derivatives; qRT-PCR: Quantitative reverse transcriptase polymerase chain reaction; PI: Propidium iodide; RTK: Receptor tyrosine kinase; PI3K: Phosphoinositide 3-kinase; FBS: Fetal bovine serum; SEM: Standard error of the mean.
Author contributions
S.B. and C.M. contributed to the experimentation and data analysis as well as assisted in manuscript preparation. K.W. and C.B. assisted in experiments. R.C. designed and supervised the study. He was also involved in preparing the manuscript. All authors have reviewed and approved the final manuscript.
ACKNOWLEDGMENTS
This study was supported by Oakland University, the Oakland University William Beaumont Institute for Stem Cell and Regenerative Medicine (OU-WB ISCRM), and the Michigan Head and Spine Institute (MHSI). S. Bakshi received the Provost Undergraduate Research Award from Oakland University for this project. The authors would also like to thank Dr. James Bradner (Harvard Medical School) for providing JQ1.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
FUNDING
None.
REFERENCES
1. Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007; 26:5521–27. https://doi.org/10.1038/sj.onc.1210618.
2. Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002; 22:3794–802. https://doi.org/10.1128/MCB.22.11.3794-3802.2002.
3. Taniguchi Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int J Mol Sci. 2016; 17:17. https://doi.org/10.3390/ijms17111849.
4. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067–73. https://doi.org/10.1038/nature09504.
5. Shang E, Salazar G, Crowley TE, Wang X, Lopez RA, Wang X, Wolgemuth DJ. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr Patterns. 2004; 4:513–19. https://doi.org/10.1016/j.modgep.2004.03.002.
6. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146:904–17. https://doi.org/10.1016/j.cell.2011.08.017.
7. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011; 108:16669–74. https://doi.org/10.1073/pnas.1108190108.
8. Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012; 12:465–77. https://doi.org/10.1038/nrc3256.
9. Chung C, Tough DF. Bromodomains: a new target class for small molecule drug discovery. Drug Discov Today Ther Strateg. 2012; 9:e111–20. https://doi.org/10.1016/j.ddstr.2011.12.002.
10. Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med. 2011; 13:e29. https://doi.org/10.1017/S1462399411001992.
11. Pérez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics. 2017; 12:323–39. https://doi.org/10.1080/15592294.2016.1265710.
12. Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, Heslin MJ, Arnoletti JP, Richardson JH, Chen D, Hanna CA, Cramer SL, Yang ES, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016; 35:833–45. https://doi.org/10.1038/onc.2015.126.
13. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016; 529:413–17. https://doi.org/10.1038/nature16508.
14. Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-molecule inhibition of BRDT for male contraception. Cell. 2012; 150:673–84. https://doi.org/10.1016/j.cell.2012.06.045.
15. Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res. 2013; 19:1748–59. https://doi.org/10.1158/1078-0432.CCR-12-3066.
16. Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, Speleman F, Althoff K, Schäfers S, Schramm A, Sure U, Fleischhack G, Eggert A, Schulte JH. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget. 2013; 4:2080–95. https://doi.org/10.18632/oncotarget.1534.
17. Li J, Zhao L, Urabe G, Fu Y, Guo LW. Epigenetic intervention with a BET inhibitor ameliorates acute retinal ganglion cell death in mice. Mol Vis. 2017; 23:149–59.
18. Magistri M, Velmeshev D, Makhmutova M, Patel P, Sartor GC, Volmar CH, Wahlestedt C, Faghihi MA. The BET-Bromodomain Inhibitor JQ1 Reduces Inflammation and Tau Phosphorylation at Ser396 in the Brain of the 3xTg Model of Alzheimer’s Disease. Curr Alzheimer Res. 2016; 13:985–95. https://doi.org/10.2174/1567205013666160427101832.
19. Korb E, Herre M, Zucker-Scharff I, Darnell RB, Allis CD. BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat Neurosci. 2015; 18:1464–73. https://doi.org/10.1038/nn.4095.
20. Lee DU, Katavolos P, Palanisamy G, Katewa A, Sioson C, Corpuz J, Pang J, DeMent K, Choo E, Ghilardi N, Diaz D, Danilenko DM. Nonselective inhibition of the epigenetic transcriptional regulator BET induces marked lymphoid and hematopoietic toxicity in mice. Toxicol Appl Pharmacol. 2016; 300:47–54. https://doi.org/10.1016/j.taap.2016.03.013.
21. Horne GA, Stewart HJ, Dickson J, Knapp S, Ramsahoye B, Chevassut T. Nanog requires BRD4 to maintain murine embryonic stem cell pluripotency and is suppressed by bromodomain inhibitor JQ1 together with Lefty1. Stem Cells Dev. 2015; 24:879–91. https://doi.org/10.1089/scd.2014.0302.
22. Alghamdi S, Khan I, Beeravolu N, McKee C, Thibodeau B, Wilson G, Chaudhry GR. BET protein inhibitor JQ1 inhibits growth and modulates WNT signaling in mesenchymal stem cells. Stem Cell Res Ther. 2016; 7:22. https://doi.org/10.1186/s13287-016-0278-3.
23. Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai K, Herait P, Kahatt C, Lokiec F, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016; 3:e196–204. https://doi.org/10.1016/S2352-3026(16)00021-1.
24. Takeda YS, Xu Q. Neuronal Differentiation of Human Mesenchymal Stem Cells Using Exosomes Derived from Differentiating Neuronal Cells. PLoS One. 2015; 10:e0135111. https://doi.org/10.1371/journal.pone.0135111.
25. Yu P, McKinney EC, Kandasamy MM, Albert AL, Meagher RB. Characterization of brain cell nuclei with decondensed chromatin. Dev Neurobiol. 2015; 75:738–56. https://doi.org/10.1002/dneu.22245.
26. Braun T, Gardin C. Investigational BET bromodomain protein inhibitors in early stage clinical trials for acute myelogenous leukemia (AML). Expert Opin Investig Drugs. 2017; 26:803–11. https://doi.org/10.1080/13543784.2017.1335711.
27. Sun B, Shah B, Fiskus W, Qi J, Rajapakshe K, Coarfa C, Li L, Devaraj SG, Sharma S, Zhang L, Wang ML, Saenz DT, Krieger S, et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015; 126:1565–74. https://doi.org/10.1182/blood-2015-04-639542.
28. Zhou X, Fan LX, Peters DJ, Trudel M, Bradner JE, Li X. Therapeutic targeting of BET bromodomain protein, Brd4, delays cyst growth in ADPKD. Hum Mol Genet. 2015; 24:3982–93. https://doi.org/10.1093/hmg/ddv136.
29. Kurimchak AM, Shelton C, Duncan KE, Johnson KJ, Brown J, O’Brien S, Gabbasov R, Fink LS, Li Y, Lounsbury N, Abou-Gharbia M, Childers WE, Connolly DC, et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Reports. 2016; 16:1273–86. https://doi.org/10.1016/j.celrep.2016.06.091.
30. Brown JD, Feldman ZB, Doherty SP, Reyes JM, Rahl PB, Lin CY, Sheng Q, Duan Q, Federation AJ, Kung AL, Haldar SM, Young RA, Plutzky J, et al. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc Natl Acad Sci USA. 2018; 115:2144–49. https://doi.org/10.1073/pnas.1711155115.
31. Lee JE, Park YK, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W, Ge K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun. 2017; 8:2217. https://doi.org/10.1038/s41467-017-02403-5.
32. Najafova Z, Tirado-Magallanes R, Subramaniam M, Hossan T, Schmidt G, Nagarajan S, Baumgart SJ, Mishra VK, Bedi U, Hesse E, Knapp S, Hawse JR, Johnsen SA. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017; 45:127–41. https://doi.org/10.1093/nar/gkw826.
33. Niu N, Shao R, Yan G, Zou W. Bromodomain and Extra-terminal (BET) Protein Inhibitors Suppress Chondrocyte Differentiation and Restrain Bone Growth. J Biol Chem. 2016; 291:26647–57. https://doi.org/10.1074/jbc.M116.749697.
34. Roberts TC, Etxaniz U, Dall’Agnese A, Wu SY, Chiang CM, Brennan PE, Wood MJ, Puri PL. BRD3 and BRD4 BET Bromodomain Proteins Differentially Regulate Skeletal Myogenesis. Sci Rep. 2017; 7:6153. https://doi.org/10.1038/s41598-017-06483-7.
35. Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997; 90:169–80. https://doi.org/10.1016/S0092-8674(00)80323-2.
36. Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990; 60:585–95. https://doi.org/10.1016/0092-8674(90)90662-X.
37. Fanarraga ML, Avila J, Zabala JC. Expression of unphosphorylated class III beta-tubulin isotype in neuroepithelial cells demonstrates neuroblast commitment and differentiation. Eur J Neurosci. 1999; 11:517–27. https://doi.org/10.1046/j.1460-9568.1999.00459.x.
38. Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992; 116:201–11.
39. Li J, Ma J, Meng G, Lin H, Wu S, Wang J, Luo J, Xu X, Tough D, Lindon M, Rioja I, Zhao J, Mei H, et al. BET bromodomain inhibition promotes neurogenesis while inhibiting gliogenesis in neural progenitor cells. Stem Cell Res (Amst). 2016; 17:212–21. https://doi.org/10.1016/j.scr.2016.07.006.
40. Kumar K, DeCant BT, Grippo PJ, Hwang RF, Bentrem DJ, Ebine K, Munshi HG. BET inhibitors block pancreatic stellate cell collagen I production and attenuate fibrosis in vivo. JCI Insight. 2017; 2:e88032. https://doi.org/10.1172/jci.insight.88032.
41. Shao Z, Yao C, Khodadadi-Jamayran A, Xu W, Townes TM, Crowley MR, Hu K. Reprogramming by De-bookmarking the Somatic Transcriptional Program through Targeting of BET Bromodomains. Cell Reports. 2016; 16:3138–45. https://doi.org/10.1016/j.celrep.2016.08.060.
42. Zinin N, Adameyko I, Wilhelm M, Fritz N, Uhlén P, Ernfors P, Henriksson MA. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. EMBO Rep. 2014; 15:383–91. https://doi.org/10.1002/embr.201337424.
43. Ishida CT, Bianchetti E, Shu C, Halatsch ME, Westhoff MA, Karpel-Massler G, Siegelin MD. BH3-mimetics and BET-inhibitors elicit enhanced lethality in malignant glioma. Oncotarget. 2017; 8:29558–73. https://doi.org/10.18632/oncotarget.16365.
44. Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP, Ma XX, Zhang G, Li J, Yan B, Tang HW, Li SS, Wang LD, Zhang SJ. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget. 2016; 7:2462–74. https://doi.org/10.18632/oncotarget.6275.
45. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001; 7:683–94. https://doi.org/10.1016/S1097-2765(01)00214-3.
46. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000; 288:1053–58. https://doi.org/10.1126/science.288.5468.1053.
47. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001; 7:673–82. https://doi.org/10.1016/S1097-2765(01)00213-1.
48. Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou JC. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol. 1998; 143:217–24. https://doi.org/10.1083/jcb.143.1.217.
49. Pastorino JG, Chen ST, Tafani M, Snyder JW, Farber JL. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem. 1998; 273:7770–75. https://doi.org/10.1074/jbc.273.13.7770.
50. Wang H, Cheng F, Cheng J, Villagra A, Tao J, Bradner JE, Sotomayor EM. JQ1, a selective bromodomain inhibitor, augment the immunogenicity of mantle cell lymphoma by influencing the expression of PD-L1. Blood. 2015; 126:822.
51. Wang J, Liu Z, Wang Z, Wang S, Chen Z, Li Z, Zhang M, Zou J, Dong B, Gao J, Shen L. Targeting c-Myc: JQ1 as a promising option for c-Myc-amplified esophageal squamous cell carcinoma. Cancer Lett. 2018; 419:64–74. https://doi.org/10.1016/j.canlet.2018.01.051.
52. Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002; 277:14040–47. https://doi.org/10.1074/jbc.M108975200.
53. Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, Tremblay È, Nadeau V, Lambert C, et al. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ Res. 2015; 117:525–35. https://doi.org/10.1161/CIRCRESAHA.115.307004.
54. Shao Q, Kannan A, Lin Z, Stack B, Suen JY, Gao L. BET protein inhibitor JQ1 attenuates MYC-amplified MCC tumor growth in vivo. Cancer Res. 2014; 74:7090-102. https://doi.org/10.1158/0008-5472.CAN-14-0305.
55. Zhang HT, Gui T, Sang Y, Yang J, Li YH, Liang GH, Li T, He QY, Zha ZG. The BET Bromodomain Inhibitor JQ1 Suppresses Chondrosarcoma Cell Growth via Regulation of YAP/p21/c-Myc Signaling. J Cell Biochem. 2017; 118:2182–92. https://doi.org/10.1002/jcb.25863.
56. Beeravolu N, Khan I, McKee C, Dinda S, Thibodeau B, Wilson G, Perez-Cruet M, Bahado-Singh R, Chaudhry GR. Isolation and comparative analysis of potential stem/progenitor cells from different regions of human umbilical cord. Stem Cell Res (Amst). 2016; 16:696–711. https://doi.org/10.1016/j.scr.2016.04.010.