INTRODUCTION
From a clinical and molecular standpoint, melanoma is a complex, heterogeneous and unpredictable disease. The incidence of malignant melanoma (MM) has drastically increased in the past decades [1]. Approximately 70% of new cases of MM are thin melanomas (TM), which are lesions ≤1.00 mm in Breslow thickness, and TM diagnosis are increasing around the world [2]. After local excision, thin tumors have a 10-year survival rate of 85-90% [3]. However, the high percentage of TM without recurrences is in contrast with a group that could develop recurrences and regional or distant metastases [4–7]. The TM histopathological parameters have been extensively studied in the literature and they have been associated with prognosis [8].
Several studies showed numerous parameters indicative of unfavorable prognosis for TM patients including male sex, advanced age, trunk and head/neck anatomic sites, III and IV Clark levels, presence of marked regression, absence of inflammatory infiltrate, presence of ulceration areas and high mitotic rate [9–11]. However, existing prognostic factors cannot reliably differentiate high- and low-risk TM patients. To the best of our knowledge, only a few studies have analyzed the molecular profile of TM and to date there is no molecular predictor of disease progression [12, 13]. In recent years, significant advances in the genetic field have led to the identification of specific driver mutations in melanocytic tumors [14–16]. BRAF, NRAS and TP53 mutations are the most prevalent pathogenic alterations in melanoma [17], but key genetic changes are also identified in CDKN2A, KIT, GNAQ and GNA11 genes [18–21]. Mutation frequencies of these genes change according to the different personal data and pathological features of MM [22], revealing a complex mutational pattern that could be evaluated to predict the risk of developing metastases.
In this study, we aimed at characterizing the clinical and molecular profile of metastatic and not-metastatic TM cases. First, we evaluated a possible association between the presence of metastases and the pathological features. Furthermore, we used a Next Generation Sequencing (NGS) gene panel including 15 genes, relevant in tumorigenesis and targeted therapies, with the aim to investigate multiple somatic mutations in metastatic and not-metastatic TM cases. The identification of patients with high-risk of developing metastatic disease could be potentially used in the clinical practice, in order to gain the best prognostic classification and to potentially define the best therapeutic choices.
RESULTS
Clinical-pathologic features and association with the presence of metastases
Twenty-one TM cases were included in this study. As shown in Table 1, seven of the 21 selected cases were metastatic and 14 were not-metastatic TM. The median age at diagnosis was 49.0 years for metastatic cases (from 37 to 74 years) and 50.5 years for not-metastatic cases (from 29 to 85 years). Four metastatic TM cases were males and three females (57.1%, 42.9%), whereas six not- metastatic TM cases were males and eight females (42.9%, 57.1%). The majority of the metastatic and not-metastatic cases (85.7% and 64.3%, respectively) were localized in trunk (specifically shoulders, abdomen or back) considered site of intermittent sun exposure. All the metastatic cases showed Clark level III, whereas among the not-metastatic cases six showed Clark level II and eight Clark level III (42.9%, 57.1%). All metastatic cases had Breslow thickness ≥0.6 mm, by contrast among not-metastatic cases seven had Breslow thickness ≤0.5 mm and seven ≥0.6 mm. A statistically significant association emerged between the presence of metastases and Breslow thickness ≥0.6 mm (p=0.003). The TIL grade was classified as absent in three of seven metastatic cases (42.9%) and mild to marked in all not-metastatic cases. A mitotic rate ≥1/mm2 was detected in three of seven metastatic cases (42.9%), by contrast all not-metastatic cases showed mitotic rate <1/mm2. The association between the presence of metastases and mitotic rate showed a p-value close to the statistical significance (p=0.051). No ulceration was observed in our TM series.
Table 1: Clinical-pathologic features of the 21 thin melanoma cases analyzed
Clinical-pathologic characteristics |
Total cases |
Metastatic cases N=7 (%) |
Not-Metastatic cases N=14 (%) |
p-value |
|---|---|---|---|---|
Median age at diagnosis (range) |
49.0 (29-85) |
49.0 (37-74) |
50.5 (29-85) |
1.000 |
Sex |
||||
Male |
10 (47.6) |
4 (57.1) |
6 (42.9) |
|
Female |
11 (52.4) |
3 (42.9) |
8 (57.1) |
0.659 |
Sun exposurea |
||||
Chronic (head and neck) |
1 (4.8) |
0 (0.0) |
1 (7.1) |
|
Continuous (lower/upper extremities) |
5 (23.8) |
1 (14.3) |
4 (28.6) |
|
Intermittent (shoulders/abdomen/back) |
15 (71.4) |
6 (85.7) |
9 (64.3) |
0.741 |
Clark level |
||||
II |
6 (28.6) |
0 (0.0) |
6 (42.9) |
|
III |
15 (71.4) |
7 (100.0) |
8 (57.1) |
0.061 |
Breslow thicknessb |
||||
≤0.5 mm |
7 (33.3) |
0 (0.0) |
7 (50.0) |
|
0.6-1.00 mm |
14 (66.7) |
7 (100.0) |
7 (50.0) |
0.003 |
TIL grade c |
||||
0 |
3 (17.6) |
3 (42.9) |
0 (0.0) |
|
1 |
5 (29.4) |
1 (14.3) |
4 (40.0) |
|
2 |
7 (41.2) |
2 (28.6) |
5 (50.0) |
|
3 |
2 (11.8) |
1 (14.3) |
1 (10.0) |
0.195 |
Mitotic rate/mm2c |
||||
<1 |
14 (82.4) |
4 (57.1) |
10 (100.0) |
|
≥1 |
3 (17.6) |
3 (42.9) |
0 (0.0) |
0.051 |
Ulcerationc |
||||
0 |
17 (100.0) |
7 (100.0) |
10 (100.0) |
|
1 |
0 (0.0) |
0 (0.0) |
0 (0.0) |
- |
aAccording to Whiteman et al 2006; baccording to Richetta et al 2014; csome data for each pathologic feature are not available; in bold p-value <0.05 considered statistically significant. Abbreviations: TIL, Tumor-infiltrating lymphocyte; N= number of cases.
Somatic mutation profile and association with the presence of metastases
The somatic mutation profile of the 15 most frequently mutated genes in solid tumors was screened in all 21 TM samples by TruSight Tumor 15 panel, using NGS technology. Figure 1 reports an overview of the percentages of mutations identified in BRAF, NRAS, TP53, KIT, and ERBB2 genes, the distribution of point mutations in metastatic and non-metastatic TM cases analyzed, labeled by Breslow thickness (≤ 0.5 mm and ≥0.6 mm) and the presence or absence of metastases. We detected a total of 22 mutations in 19 of the 21 (90.5%) TM cases analyzed. All seven metastatic and 12 of 14 not-metastatic TM cases had at least one mutation in the genes analyzed (Table 2). We did not find a statistically significant difference between metastatic and not-metastatic cases and the presence of all mutations; the same results were obtained when the most frequently mutated genes (BRAF, NRAS and TP53) were evaluated separately (Table 2).
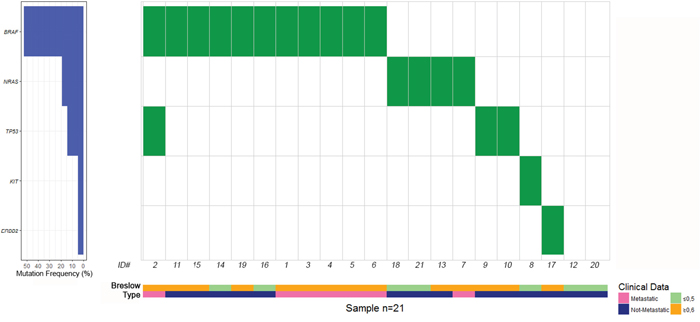
Figure 1: Distribution of gene mutations in metastatic and non-metastatic thin melanoma cases, labeled by pink and blue bars, respectively. Thin melanoma cases with a Breslow thickness of ≤ 0.5 mm and 0.6-1 mm are showed by light green and yellow bars, respectively. To the left of the figure are the percentages of mutations identified in BRAF, NRAS, TP53, KIT and ERBB2 genes.
Table 2: Distribution of 21 metastatic and not-metastatic thin melanoma cases according to all gene mutations and specific mutated genes
Total cases |
Metastatic cases |
Not-metastatic cases |
p-value |
||||
|---|---|---|---|---|---|---|---|
Mutated |
Wild-type |
Mutated |
Wild-type |
Mutated |
Wild-type |
||
All mutations |
19 (90.5) |
2 (9.5) |
7 (100.0) |
0 (0.0) |
12 (85.7) |
2 (14.3) |
0.533 |
BRAF |
11 (52.4) |
10 (47.6) |
6 (85.7) |
1 (14.3) |
5 (35.7) |
9 (64.3) |
0.063 |
NRAS |
4 (19.0) |
17 (81.0) |
1 (14.3) |
6 (85.7) |
3 (21.4) |
11(78.6) |
1.000 |
TP53 |
3 (14.3) |
18 (85.7) |
1 (14.3) |
6 (85.7) |
2 (14.3) |
12 (85.7) |
1.000 |
KIT |
1 (4.8) |
20 (95.2) |
0 (0.0) |
7 (100.0) |
1 (7.1) |
13 (92.9) |
- |
ERBB2 |
1 (4.8) |
20 (95.2) |
0 (0.0) |
7 (100.0) |
1 (7.1) |
13 (92.9) |
- |
Abbreviation: N= number of cases.
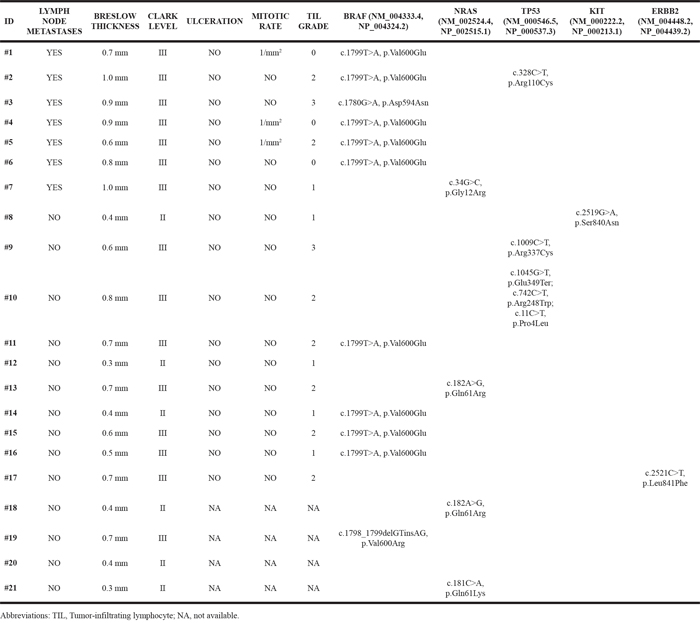
As shown in Table 2, BRAF mutations were detected in 11 cases (52.4%), NRAS in four (19.0%), TP53 in three (14.3%), KIT and ERBB2 in one case (4.8%) each. BRAF and NRAS were the most frequently mutated genes and their mutations were mutually exclusive. Overall, BRAF mutations were identified in six of seven metastatic cases (85.7%) and in five of 14 not-metastatic cases (35.7%). No mutations were identified in the other genes examined. The pathological features and the mutations identified in the series of TM cases analyzed are reported in Table 3. BRAF c.1799T>A (p.Val600Glu) was the most frequent mutation identified. Overall, nine of 21 (42.9%) of TM cases harbored this mutation, in particular it was detected in five of seven (71.4 %) metastatic and in four of 14 (28.6 %) not-metastatic cases. Two additional BRAF mutations, c.1780G>A (p.Asp594Asn) and c.1798_1799GT>CG (p.Val600Arg), were identified in two TM cases. Three NRAS mutations, c.182A>G (p.Gln61Arg), c.181C>A (p.Gln61Lys) and c.34G>C (p.Gly12Arg), were identified in four TM cases. Five TP53 mutations, c.328C>T (p.Arg110Cys), c.1009C>T (p.Arg337Cys), c.1045G>T (p.Glu349Ter), c.742C>T (p.Arg248Trp) and c.11C>T (p.Pro4Leu), were detected in three TM cases. No mutations were observed in two TM cases whereas mutations in more than one gene were detected in one TM case (Table 3). Specifically, no mutations were detected in two not-metastatic cases with the lowest values of Breslow thickness (0.3 mm and 0.4 mm, respectively) whereas BRAF c.1799T>A (p.Val600Glu) and TP53 c.328C>T (p.Arg110Cys) mutations were identified in one metastatic case with the highest value of Breslow thickness (1.00 mm). Associations between gene mutations and Breslow thickness (≤0.5 mm and 0.6-1 mm) distribution (Supplementary Table 1) and TIL grade distribution (Supplementary Table 2) were also tested and no statistically significant associations emerged.
Table 3: Pathological features and mutations identified in metastatic and not-metastatic thin melanoma cases
DISCUSSION
In this study we analyzed a series of TM, in order to investigate clinical-pathologic and molecular differences in metastatic and not-metastatic cases. The strategy of early detection and the greater incidence of melanomas in the last years has contributed to increase the diagnosis of TM [25]. Unfortunately, despite the excellent prognosis of TM, a small group of patients could develop metastases, fatal in 5% of the cases [26]. It is therefore of great interest to identify predictors of poor prognosis in TM patients [8]. Several authors highlighted an increased risk of progression for TM with Breslow thickness ≥0.75 or ≥0.5 mm [6, 25, 27]. Most studies analyzing TM risk progression identified ulceration and mitotic rate as main predictors [6, 27], while others indicated other risk factors [28–30]. Our results showed that the presence of lymph-node metastases was associated with Breslow thickness ≥0.6 mm (p=0.003). In particular, none of TM with lymph-node involvement had Breslow thickness <0.6 mm. These observations confirm Breslow thickness as the most powerful predictor of developing metastatic disease. Most studies showed that the presence of lymph-node metastases is very rare (<5%) in melanomas with Breslow thickness <0.8 mm and occur in approximately 5% to 12% of patients with primary melanomas with Breslow thickness from 0.8 to 1.0 mm [6, 31–33].
In the present study, we have also attempted to identify a different mutational pattern between metastatic and not-metastatic TM cases. We characterized multiple mutations in 15 most frequently mutated genes in solid tumors by NGS technology. The most frequent mutations identified were BRAF mutations (52.4%), followed by mutations in NRAS (19.0%) and TP53 (14.3%). BRAF mutations were mainly detected in metastatic TM. This result is in line with the literature [34], confirming a more aggressive behavior for melanomas associated to BRAF mutations, including thin lesions.
Mutations were also found in KIT and ERBB2 (4.8%). Overall, our data are in agreement with literature data showing that BRAF is mutated in about 50% of melanomas [14, 35, 36] and that NRAS is the second most frequently mutated gene [37]. In agreement with literature data [38], we also showed that the most frequent BRAF mutation was the c.1799T>A (p.Val600Glu). In addition, we identified two different BRAF mutations, c.1780 G>A (p.Asp594Asn) which results in an inactivation of BRAF gene [39] and c.1798_1799GT>CG (p.Val600Arg), which rarely occurs in melanoma [40]. In our series, one metastatic case showed the concurrent presence of BRAF and TP53 mutations. Recent Whole Exome Sequencing (WES) study attributed a TP53 mutation frequency of 19.0% in melanoma [14]. We identified one not-metastatic TM case with multiple TP53 variants (c.1045G>T, p.Glu349Ter; c.742C>T, p.Arg248Trp; c.11C>T, p.Pro4Leu). To date, there are conflicting data about the role of TP53 alterations in melanoma [41, 42] and further studies could help to clarify the role of this gene in melanoma progression. We identified the ERBB2 c.2521C>T (p.Leu841Phe) mutation in one case. This mutation is located in the kinase domain and, to our knowledge, it has not been previously reported. In recent study no ERBB2 mutations were found in melanoma, however the importance of ERBB2 mutations in the kinase domain is well documented in a wide variety of human cancers [43]. Further studies are needed to clarify the role of ERBB2 mutations in melanoma, as it may help for considering targeted therapy. We also identified the KIT c.2519G>A (p.Ser840Asn) mutation in one case. This mutation was found for the first time in a 2-year-old boy affected by cutaneous mastocytosis [44]. KIT mutation is most frequently observed in acral, mucosal and chronically sun-exposed melanomas [45, 46]. In our study, KIT mutation was identified in a TM located in acral site. KIT is another important checkpoint for the targeted therapy in melanomas, since molecules as imatinib, nilotinib and apatinib can affect melanoma cells with KIT mutations [44–46]. Notably, in our series only two cases were negative for the presence of mutations in the genes analyzed. Both these cases were not-metastatic cases and had low Breslow thickness (<0.4 mm). On the other hand, we detected mutations in more than one gene (BRAF and TP53) in one metastatic case with a high Breslow thickness (1.0 mm).
In conclusion, our findings support and confirm Breslow thickness as a valid prognostic factor, able to distinguish TM with a high- or low-risk of developing metastases and suggest that Breslow thickness ≥0.6 mm may be considered as a threshold value where Sentinel Lymph Node Biopsy should be discussed and considered. Future studies on a larger cohort of TM patients examined by NGS analyses are needed to provide a genetic profile that could be useful as a prognostic and predictive factor.
MATERIALS AND METHODS
Sample information and DNA extraction
We performed a retrospective observational study on a selected series of 21 TM cases including seven metastatic and 14 not-metastatic cases. Cases were selected to include: melanoma patients with lesions ≤1.00 mm in Breslow thickness, comparable median age at diagnosis and a comparable number of female and male individuals. The series included in the study was collected between January 2009 and December 2015 at the Department of Dermatology and Venerology, Sapienza University of Rome and at the Laboratory of Dermatopathology of the “San Gallicano” Dermatological Institute and at the Dermato-oncology and preventive Unit of San Gallicano Institute of Rome. All patients involved in the current study signed an informed consent form with a detailed description of the study protocol. The study was approved by The Local Ethical Committee (Sapienza University of Rome, Protocol 873/13). The study was performed according to the Helsinki’s declaration. All cases were characterized by the personal data (sex and age at diagnosis) and by the main clinical-pathologic features including presence of metastases, anatomic site based on sun exposure (chronic, continuous or intermittent), Clark level, Breslow thickness, mitotic rate (number of mitoses/mm2), ulceration status and tumor-infiltrating lymphocyte (TIL) grade based on assessment of absent, mild, moderate, or marked density (TIL grade 0-3). Genomic DNA of both tumor and normal tissue samples was extracted from 10 μm-thick microdissected formalin fixed paraffin-embedded (FFPE) sections, using QIAamp DNA FFPE tissue kit (Qiagen), according to the manufacturer’s instructions. DNA quantification was performed with Qubit dsDNA HS Assay Kit (Invitrogen), according to the instructions provided by the manufacturer.
Target sequencing and variant classification
Genomic screening for 21 TM cases was performed with TruSight Tumor 15 panel (Illumina), including relevant regions in 15 frequently mutated genes in solid tumors (AKT1, BRAF, EGFR, ERBB2, FOXL2, GNA11, GNAQ, KIT, KRAS, MET, NRAS, PDGFRA, PIK3CA, TP53, RET). Briefly, genomic regions were prepared in paired-end libraries, pooled and loaded into the MiniSeq system (Illumina) for automated cluster generation, sequencing and data analysis, including variant calling. The results were annotated and filtered using Illumina Variant Studio software. Somatic mutations were identified by directly comparing the mutation profile of tumor samples with their matched normal samples. Furthermore, we filtered out somatic single-nucleotide variants (SNVs) with allele frequency <5% and those with <500 reads. In silico bioinformatics analyses, SIFT and Polyphen [23, 24], were used to determine the potential functional effects of identified somatic SNVs. The presence of these variants was also examined in the Catalogue of Somatic Mutations in Cancer (COSMIC). In order to direct inspect mutations, sequenced reads were visualized with the Integrative Genomics Viewer (IGV) tool, using hg19 as reference genome.
Statistical analysis
Mann-Whitney and Fisher’s exact test were used (where appropriate) in order to evaluate the potential associations between the presence of metastases and pathological features and the potential association between the presence of metastases and the mutation profile. A p-value ≤0.05 was considered statistically significant. All statistical analyses were performed with the R software (www.r-project.org).
Abbreviations
MM: Malignant melanoma; TM: Thin Melanoma; NGS: Next Generation Sequencing; TIL: tumor-infiltrating lymphocyte; FFPE: formalin fixed paraffin-embedded; SNVs: single-nucleotide variants; COSMIC: Catalogue of Somatic Mutations in Cancer; IGV: Integrative Genomics Viewer.
Author contributions
Antonio G Richetta: collection of clinical-pathologic features, drafting and reviewing the manuscript, agreement to submit the manuscript for publication. Virginia Valentini and Federica Marraffa: performing experiments, drafting and reviewing the manuscript, agreement to submit the manuscript for publication. Giovanni Paolino: collection of clinical-pathologic features, reviewing the manuscript, agreement to submit the manuscript for publication. Piera Rizzolo, Valentina Silvestri and Veronica Zelli: data acquisition, statistical analyses, reviewing the manuscript, agreement to submit the manuscript for publication. Anna Carbone, Cinzia Di Mattia, Stefano Calvieri, Pasquale Frascione and Pietro Donati: collection of clinical-pathologic features, reviewing the manuscript, agreement to submit the manuscript for publication. Laura Ottini: study concept, design and coordination, data acquisition, drafting and reviewing the manuscript, agreement to submit the manuscript for publication.
ACKNOwLEDGMENTS
Virginia Valentini contributed to this paper as recipient of the PhD program of Biotechnologies and Clinical Medicine Sapienza University of Rome.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
Italian Association for Cancer Research (AIRC-IG16933) to Laura Ottini.
REFERENCES
1. Rigel DS, Carucci JA. Malignant melanoma: prevention, early detection, and treatment in the 21st century. CA Cancer J Clin. 2000; 50:215–36. https://doi.org/10.3322/canjclin.50.4.215.
2. Gimotty PA, Guerry D, Ming ME, Elenitsas R, Xu X, Czerniecki B, Spitz F, Schuchter L, Elder D. Thin primary cutaneous malignant melanoma: a prognostic tree for 10-year metastasis is more accurate than American Joint Committee on Cancer staging. J Clin Oncol. 2004; 22:3668–76. https://doi.org/10.1200/JCO.2004.12.015.
3. Roncati L, Pusiol T, Piscioli F. Thin Melanoma: A Generic Term Including Four Histological Subtypes of Cutaneous Melanoma. Acta Dermatovenerol Croat. 2016; 24:169–74.
4. Ranieri JM, Wagner JD, Wenck S, Johnson CS, Coleman JJ 3rd. The prognostic importance of sentinel lymph node biopsy in thin melanoma. Ann Surg Oncol. 2006; 13:927–32. https://doi.org/10.1245/ASO.2006.04.023.
5. Balch CM, Balch GC, Sharma RR. Identifying early melanomas at higher risk for metastases. J Clin Oncol. 2012; 30:1406–07. https://doi.org/10.1200/JCO.2011.40.6983.
6. Han D, Zager JS, Shyr Y, Chen H, Berry LD, Iyengar S, Djulbegovic M, Weber JL, Marzban SS, Sondak VK, Messina JL, Vetto JT, White RL, et al. Clinicopathologic predictors of sentinel lymph node metastasis in thin melanoma. J Clin Oncol. 2013; 31:4387–93. https://doi.org/10.1200/JCO.2013.50.1114.
7. Durham AB, Schwartz JL, Lowe L, Zhao L, Johnson AG, Harms KL, Bichakjian CK, Orsini AP, McLean SA, Bradford CR, Cohen MS, Johnson TM, Sabel MS, Wong SL. The natural history of thin melanoma and the utility of sentinel lymph node biopsy. J Surg Oncol. 2017; 116:1185–92. https://doi.org/10.1002/jso.24765.
8. Richetta AG, Bottoni U, Paolino G, Clerico R, Cantisani C, Ambrifi M, Corsetti P, Calvieri S. Thin melanoma and late recurrences: it is never too thin and never too late. Med Oncol. 2014; 31:909. https://doi.org/10.1007/s12032-014-0909-4.
9. Månsson-Brahme E, Carstensen J, Erhardt K, Lagerlöf B, Ringborg U, Rutqvist LE. Prognostic factors in thin cutaneous malignant melanoma. Cancer. 1994; 73:2324–32. https://doi.org/10.1002/1097-0142(19940501)73:9<2324::AID-CNCR2820730914>3.0.CO;2-5.
10. Kalady MF, White RR, Johnson JL, Tyler DS, Seigler HF. Thin melanomas: predictive lethal characteristics from a 30-year clinical experience. Ann Surg. 2003; 238:528–35. https://doi.org/10.1097/01.sla.0000090446.63327.40.
11. Sinnamon AJ, Neuwirth MG, Yalamanchi P, Gimotty P, Elder DE, Xu X, Kelz RR, Roses RE, Chu EY, Ming ME, Fraker DL, Karakousis GC. Association Between Patient Age and Lymph Node Positivity in Thin Melanoma. JAMA Dermatol. 2017; 153:866–73. https://doi.org/10.1001/jamadermatol.2017.2497.
12. Montagnani V, Benelli M, Apollo A, Pescucci C, Licastro D, Urso C, Gerlini G, Borgognoni L, Luzzatto L, Stecca B. Thin and thick primary cutaneous melanomas reveal distinct patterns of somatic copy number alterations. Oncotarget. 2016; 7:30365–78. https://doi.org/10.18632/oncotarget.8758.
13. Hothem Z, Bayci A, Thibodeau BJ, Ketelsen BE, Fortier LE, Uzieblo AF, Cosner D, Totoraitis K, Keidan RD, Wilson GD. Using global gene expression to discriminate thin melanomas with poor outcomes. Mol Cell Oncol. 2016; 4:e1253527. https://doi.org/10.1080/23723556.2016.1253527.
14. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150:251–63. https://doi.org/10.1016/j.cell.2012.06.024.
15. Richetta AG, Silvestri V, Giancristoforo S, Rizzolo P, D’Epiro S, Graziano V, Mattozzi C, Navazio AS, Falchetti M, Calvieri S, Ottini L. Mutational profiling in melanocytic tumors: multiple somatic mutations and clinical implications. Oncology. 2014; 86:104–08. https://doi.org/10.1159/000356878.
16. Carlson JA, Caldeira Xavier JC Jr, Tarasen A, Sheehan CE, Otto G, Miller VA, Stephens PJ, Elvin JA, Vergilio JA, Suh J, Gay LM, Ross JS. Next-Generation Sequencing Reveals Pathway Activations and New Routes to Targeted Therapies in Cutaneous Metastatic Melanoma. Am J Dermatopathol. 2017; 39:1–13. https://doi.org/10.1097/DAD.0000000000000729.
17. Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, Malke J, Haydu L, Prieto VG, Tetzlaff M, Ivan D, Wang WL, Torres-Cabala C, Curry J, et al. Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol. 2015; 135:508–15. https://doi.org/10.1038/jid.2014.366.
18. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006; 24:4340–46. https://doi.org/10.1200/JCO.2006.06.2984.
19. Takata M, Saida T. Genetic alterations in melanocytic tumors. J Dermatol Sci. 2006; 43:1–10. https://doi.org/10.1016/j.jdermsci.2006.05.002.
20. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009; 457:599–602. https://doi.org/10.1038/nature07586.
21. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010; 363:2191–99. https://doi.org/10.1056/NEJMoa1000584.
22. Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012; 132:854–63. https://doi.org/10.1038/jid.2011.421.
23. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001; 11:863–74. https://doi.org/10.1101/gr.176601.
24. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–49. https://doi.org/10.1038/nmeth0410-248.
25. Quarto G, Sivero L, Benassai G, Bucci L, Desiato V, Perrotta S, Benassai G, Massa S. Is 1 mm thickness sec. Breslow the correct cut-off for sentinel node biopsy in melanoma? Report of six cases of metastasis by thin melanoma. Ann Ital Chir. 2013; 84:575–78.
26. Mihic-Probst D, Shea C, Duncan L, de la Fouchardiere A, Landman G, Landsberg J, ven den Oord J, Lowe L, Cook MG, Yun SJ, Clarke L, Messina J, Elder DE, et al. Update on Thin Melanoma: Outcome of an International Workshop. Adv Anat Pathol. 2016; 23:24-9. https://doi.org/10.1097/PAP.0000000000000100.
27. Han D, Yu D, Zhao X, Marzban SS, Messina JL, Gonzalez RJ, Cruse CW, Sarnaik AA, Puleo C, Sondak VK, Zager JS. Sentinel node biopsy is indicated for thin melanomas ≥0.76 mm. Ann Surg Oncol. 2012; 19:3335–42. https://doi.org/10.1245/s10434-012-2469-1.
28. Venna SS, Thummala S, Nosrati M, Leong SP, Miller JR 3rd, Sagebiel RW, Kashani-Sabet M. Analysis of sentinel lymph node positivity in patients with thin primary melanoma. J Am Acad Dermatol. 2013; 68:560–67. https://doi.org/10.1016/j.jaad.2012.08.045.
29. Green AC, Baade P, Coory M, Aitken JF, Smithers M. Population-based 20-year survival among people diagnosed with thin melanomas in Queensland, Australia. J Clin Oncol. 2012; 30:1462–67. https://doi.org/10.1200/JCO.2011.38.8561.
30. Faries MB, Wanek LA, Elashoff D, Wright BE, Morton DL. Predictors of occult nodal metastasis in patients with thin melanoma. Arch Surg. 2010; 145:137–42. https://doi.org/10.1001/archsurg.2009.271.
31. Andtbacka RH, Gershenwald JE. Role of sentinel lymph node biopsy in patients with thin melanoma. J Natl Compr Canc Netw. 2009; 7:308–17. https://doi.org/10.6004/jnccn.2009.0023.
32. Cordeiro E, Gervais MK, Shah PS, Look Hong NJ, Wright FC. Sentinel Lymph Node Biopsy in Thin Cutaneous Melanoma: A Systematic Review and Meta-Analysis. Ann Surg Oncol. 2016; 23:4178–88. https://doi.org/10.1245/s10434-016-5137-z.
33. Murali R, Goumas C, Kricker A, From L, Busam KJ, Begg CB, Dwyer T, Gruber SB, Kanetsky PA, Orlow I, Rosso S, Thomas NE, Berwick M, et al, and GEM Study Group. Clinicopathologic features of incident and subsequent tumors in patients with multiple primary cutaneous melanomas. Ann Surg Oncol. 2012; 19:1024–33. https://doi.org/10.1245/s10434-011-2058-8.
34. Hugdahl E, Kalvenes MB, Puntervoll HE, Ladstein RG, Akslen LA. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br J Cancer. 2016; 114:801–08. https://doi.org/10.1038/bjc.2016.44.
35. Siroy AE, Aung PP, Torres-Cabala CA, Tetzlaff MT, Nagarajan P, Milton DR, Curry JL, Ivan D, Prieto VG. Clinical significance of BRAF V600E mutational status in capsular nevi of sentinel lymph nodes in patients with primary cutaneous melanoma. Hum Pathol. 2017; 59:48–54. https://doi.org/10.1016/j.humpath.2016.09.010.
36. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012; 44:1006–14. https://doi.org/10.1038/ng.2359.
37. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353:2135–47. https://doi.org/10.1056/NEJMoa050092.
38. Lovly CM, Dahlman KB, Fohn LE, Su Z, Dias-Santagata D, Hicks DJ, Hucks D, Berry E, Terry C, Duke M, Su Y, Sobolik-Delmaire T, Richmond A, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012; 7:e35309. https://doi.org/10.1371/journal.pone.0035309.
39. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010; 140:209–21. https://doi.org/10.1016/j.cell.2009.12.040.
40. Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, Yao H, Lazar AJ, Aldape KD, Medeiros LJ, Luthra R. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013; 15:220–26. https://doi.org/10.1016/j.jmoldx.2012.10.002.
41. Viros A, Sanchez-Laorden B, Pedersen M, Furney SJ, Rae J, Hogan K, Ejiama S, Girotti MR, Cook M, Dhomen N, Marais R. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature. 2014; 511:478–82. https://doi.org/10.1038/nature13298.
42. Kim DW, Haydu LE, Joon AY, Bassett RL Jr, Siroy AE, Tetzlaff MT, Routbort MJ, Amaria RN, Wargo JA, McQuade JL, Kemnade J, Hwu P, Woodman SE, et al. Clinicopathological features and clinical outcomes associated with TP53 and BRAFNon-V600 mutations in cutaneous melanoma patients. Cancer. 2017; 123:1372–81. https://doi.org/10.1002/cncr.30463.
43. Wen W, Chen WS, Xiao N, Bender R, Ghazalpour A, Tan Z, Swensen J, Millis SZ, Basu G, Gatalica Z, Press MF. Mutations in the Kinase Domain of the HER2/ERBB2 Gene Identified in a Wide Variety of Human Cancers. J Mol Diagn. 2015; 17:487–95. https://doi.org/10.1016/j.jmoldx.2015.04.003.
44. Hoffmann KM, Moser A, Lohse P, Winkler A, Binder B, Sovinz P, Lackner H, Schwinger W, Benesch M, Urban C. Successful treatment of progressive cutaneous mastocytosis with imatinib in a 2-year-old boy carrying a somatic KIT mutation. Blood. 2008; 112:1655–57. https://doi.org/10.1182/blood-2008-03-147785.
45. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F 3rd, Azar S, Rubin BP, Muller S, West R, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008; 14:6821-8. https://doi.org/10.1158/1078-0432.CCR-08-0575.
46. Luo C, Shen J, Ying J, Fang X, Wang X, Fu Z, Liu P. Case report of a KIT-mutated melanoma patient with an excellent response to apatinib and temozolomide combination therapy. Onco Targets Ther. 2017; 10:4553-4557. https://doi.org/10.2147/OTT.S146409.