
Introduction
Immunotoxins (ITs) have emerged as a key weapon in the arsenal against metastatic cancer [1, 2]. Originally described by Ehrlich, ITs are the embodiment of his concept of the magic bullet, combining the specific targeting of antibodies with the exquisite potency of cytotoxins to kill cells [3, 4]. Early studies combining holotoxins and antibodies against proteins with increased expression in cancer failed because of the absence of tumor specificity, producing normal tissue destruction [1, 2]. Improvements in natural toxin payloads have removed their promiscuous binding domains, emulated their linkages to these binding domains, and modified antigenic determinants to evade immune clearance [5]. In addition, novel cytotoxic payloads are emerging, including semiconductor quantum dots for photodynamic therapy and nanoshells which encapsulate traditional drugs but cloak them from normal tissue [6]. However, while cytotoxic payloads and linker technologies have evolved, one limitation to clinical implementation of immunotoxins is the availability of tumor-specific targets with cellular dynamics that effectively deploy cytotoxic payloads.
Guanylyl cyclase C (GUCY2C), the cell surface receptor for diarrheagenic bacterial heat-stable enterotoxins and the endogenous paracrine hormones guanylin and uroguanylin [7], is primarily expressed in apical membranes [8] of intestinal epithelial cells [9-12], segregated from the systemic compartment by the intestinal barrier [13-18]. Similarly, it is expressed by select neurons in hypothalamus, mediating a novel gut brain endocrine axis regulating appetite [19], and separated from the systemic compartment by the blood-brain barrier [20]. Importantly, GUCY2C is over-expressed by primary and metastatic colorectal tumors [9-11, 21, 22]. In that context, GUCY2C has emerged as a novel vaccine target to treat and prevent colorectal cancer metastases without normal tissue damage [23-26]. These characteristics suggest that GCC may qualify as an effective therapeutic target for immunotoxins in metastatic colorectal cancer, the second leading cause of cancer mortality in the United States and the fourth most common cause of cancer worldwide [27]. Here, we define the cellular dynamics of GUCY2C, and leverage those characteristics to develop immunotoxins with therapeutic utility in mouse models of metastatic colorectal cancer.
Results
mAbs Recognize the Extracellular Domain, Without Disrupting Receptor Function, in GUCY2C
We created three unique mouse mAbs (MS7, MS20, MS24) [19] that specifically recognize the extracellular domain of GUCY2C (Supplemental Fig. 1A). MS20 recognized both mouse and human GUCY2C, and was utilized in the present studies. These mAbs did not directly activate GUCY2C and did not block its activation by ST (Supplemental Fig. 1B), demonstrating their inactivity as pharmacophores at the receptor binding site and their utility as inert probes of GUCY2C cellular dynamics.
Lysosomotropic Endocytosis of GUCY2C
CT26 mouse colorectal cancer cells [28], which are devoid of endogenous GUCY2C expression, were engineered to express mouse GUCY2C (CT26.GUCY2C) [23-25]. Unexpectedly, whole cell IF staining with MS20 revealed that GUCY2C primarily resided in the intracellular compartment (Fig. 1A) rather than on the cell surface as suggested by the prevailing paradigm [14, 29-32]. Similarly, GUCY2C in mouse intestine exhibited the same predominant subcellular distribution in wild type (GUCY2C+/+), but not in GUCY2C-deficient (GUCY2C-/-), mice (Fig. 1B). Co-staining with MS20 and Lamp1 demonstrated localization of GUCY2C within lysosomes of colonocytes in wild-type (GUCY2C+/+), but not GUCY2C-deficient (GUCY2C-/-), mice (Fig. 2A, Supplemental Video 1). Also, whole cell IF revealed GUCY2C localization within lysosomes in CT26.GUCY2C cells (Fig. 2B, left). Live cell staining at 4°C, which prevents endocytosis [33], demonstrated that MS20 was limited only to the cell surface of GUCY2C in CT26.GUCY2C cells (Fig. 2B, middle). However, warming to 37°C released MS20-GUCY2C complexes to internalize from the cell surface to lysosomes in the absence of canonical ligands (Fig. 2B, right; Supplemental Video 2). Indeed, MS20-GUCY2C complexes exhibited rapid ligand-independent internalization (Supplemental Fig. 2). Additionally, endogenously expressed GUCY2C internalized to lysosomes in STC1 murine intestinal cancer cells (Supplemental Fig. 3). These observations demonstrate that GUCY2C undergoes rapid ligand-independent internalization from the cell surface into the lysosomal compartment in intestinal epithelial and colorectal cancer cells.
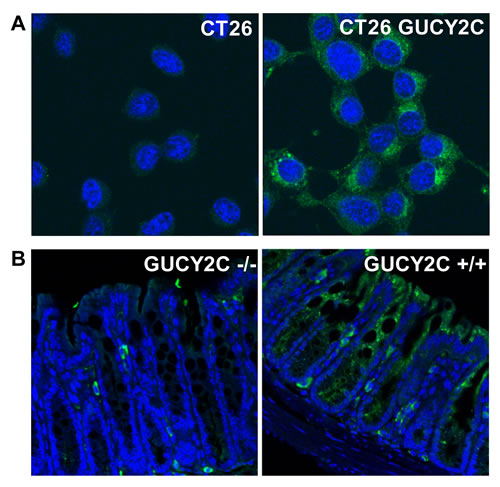
Figure 1: Intracellular localization of GUCY2C in intestinal epithelial and colorectal cancer cells. Immunofluorescent staining with GUCY2CmAb (green), with DAPI counterstain for nuclei (blue), in (A) CT26 and CT26.GUCY2C murine colorectal cancer cells or (B) colon from GUCY2C KO (GUCY2C-/-) or WT (GUCY2C+/+) mice. Results are representative of at least three independent replicates.
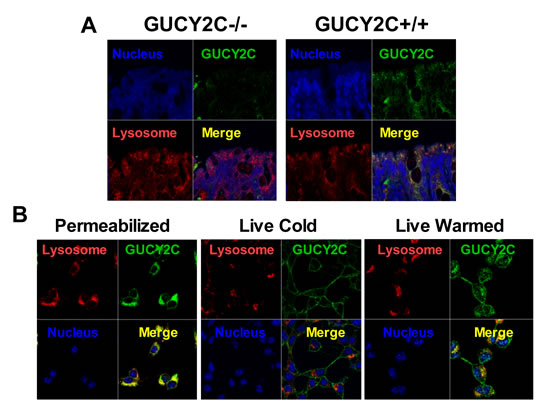
Figure 2: Lysosomotropic endocytosis of GUCY2C. (A) GUCY2C immunofluorescence (green) co-localized (yellow) with lysosomes (red) in colons from GUCY2C+/+, but not GUCY2C-/-, mice (60x magnification). (B) CT26.GUCY2C cells imaged after methanol fixation (left), after 3 h exposure to GUCY2CmAb at 4°C in DMEM, washed with PBS and methanol-fixed (center), or after 3 h exposure to GUCY2CmAb at 4°C in DMEM, washed with PBS, warmed in DMEM at 37°C for 1 h and then methanol-fixed (right). Following methanol fixation and permeabilization, cells were imaged for GUCY2CmAb (green), lysosomes with antibodies to Lamp1 (red), and nuclei with DAPI (blue). Results are representative of at least three independent replicates.
Lysosomotropic Endocytosis Requires Clathrin, But Not Caveolin or the GUCY2C Cytoplasmic Domain
Receptor-dependent endocytosis is mediated by clathrin [34], and reducing expression of this scaffolding protein using three different targeted shRNA constructs proportionately reduced internalization of GUCY2C into lysosomes (Fig. 3A-C). Indeed, these analyses revealed that at least 65% of the complement of GUCY2C resided within the intracellular compartment (Fig. 3C). In contrast, reducing the expression of caveolin, which mediates clathrin-independent endocytosis [35], in CT26.GUCY2C cells did not alter GUCY2C lysosomotropic endocytosis (Supplemental Fig.4). Moreover, a truncation mutant of GUCY2C which lacks all intracellular regions and catalytic activity, but retains extracellular and transmembrane domains and the ability to bind GUCY2C ligands (GUCY2Ctm) [25], also internalized by a clathrin-dependent, caveolin-independent mechanism (Supplemental Fig. 5).
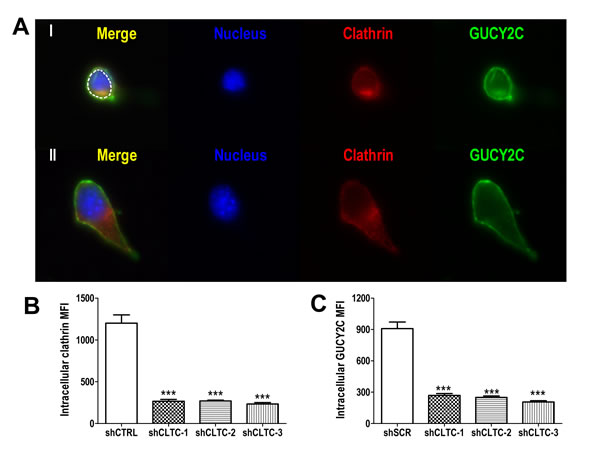
Figure 3: Lysosomotropic endocytosis of GUCY2C is clathrin-dependent. (A) Representative single cell images showing internalized GUCY2C (green) detected as in A-C in cells with stably transfected control (I) or clathrin-specific (II) shRNA. Cells were counterstained with antibodies to clathrin (red) and DAPI for nuclei (blue). The intracellular compartment was selected for quantification (dotted white line). (B) Quantification of clathrin knockdown and (C) GUCY2C internalization in cells treated with control (shCTRL) or three different clathrin-specific shRNAs (shCLTC-1, 2 and 3). Analyses were performed in a blinded fashion and data are mean ± SEM of at least ten cells. *** p<0.0001, one-way ANOVA.
Targeting GUCY2C Lysosomotropic Endocytosis with Immunotoxins
Internalization of GUCY2C-mAb complexes to lysosomes suggests that GUCY2C may be a particularly tractable target for delivery of ITs. We designed an IT in which the sterically-hindered cleavable disulfide linker 4-succinimidyloxycarbonyl-α-methyl-α-[2-pyridyldithio]-toluene (SMPT) [1, 2, 36] joined MS20 to dgRTA by the free sulfhydryl group created after reduction and removal of the ricin B chain (Fig. 4A) [37], resulting in an immunotoxin (ITsmpt) containing 1 dgRTA per IgG which can be liberated in lysosomes. As a negative control, dgRTA was conjugated to MS20 with m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS), producing an immunotoxin with a non-cleavable thioether bond (ITmbs) which cannot be liberated in lysosomes. Immunoblot analysis, probing for dgRTA under non-reducing conditions, revealed a 180 kDa conjugate consisting of the 30 kDa dgRTA and 150 kDa MS20 IgG (Fig. 4B). Slight reduction of immunoglobulin heavy and light chains during electrophoresis produced an 80 kDa species representing dgRTA conjugated to the heavy chain of IgG (Fig. 4B). Reduction released dgRTA from ITsmpt and isotype control IgG immunotoxin (negative control for the cleavable IT; ISOsmpt), but not ITmbs, producing 30 kDa (free dgRTA) and 80 kDa (dgRTA + IgG heavy chain) bands, respectively (Fig. 4B).
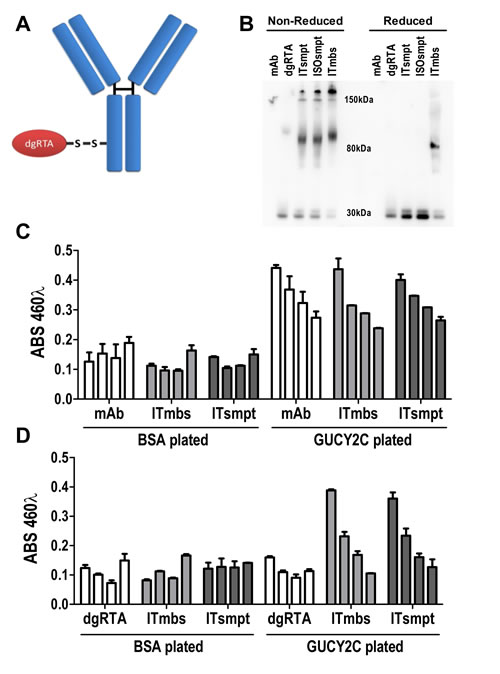
Figure 4: GUCY2C-targeted immunotoxin delivers dgRTA specifically to GUCY2C. (A) The design of cleavable ITsmpt conjugates of dgRTA and GUCY2C-specific mAb. (B) Immunoblot detecting ricin A chain under non-reducing (left) or reducing (right) conditions. Unconjugated GUCY2CmAb (mAb) and dgRTA served as controls for ITsmpt (reducible), ISOsmpt, and ITmbs (non-reducible) conjugates. (C, D) ELISA titration of GUCY2CmAb, ITmbs or ITsmpt on plates coated with BSA or GUCY2C1-430, detected with (C) anti-mouse HRP or (D) anti-ricin/anti-rabbit HRP. Bars indicate means + SD of triplicate wells titrated from 0.01-10 µg/mL.
MS20 conjugated by cleavable or non-cleavable linkers recognized GUCY2C (Fig. 4C) and delivered antigen-targeted dgRTA (Fig. 4D). In that context, ITsmpt (Fig. 5A), but not ISOsmpt (Fig. 5B), specifically killed CT26.GUCY2C, but not CT26, cells (Fig. 5C) in a dose-dependent fashion. The non-cleavable ITmbs was minimally effective compared to ITsmpt (extrapolated Kd ITmbs, 1.158 µg/mL vs Kd ITsmpt, 0.019 µg/mL; p<0.0001; Fig. 5D), supporting liberation of dgRTA from the IT in lysosomes. Further, shRNA targeting clathrin eliminated the cytotoxicity of ITsmpt (Fig. 5E-F, Supplemental Fig. 6). Moreover, ITsmpt, but not ISOsmpt, specifically killed GUCY2Ctm in a clathrin-dependent fashion (Supplemental Fig. 6). As in CT26.GUCY2C cells, the non-cleavable ITmbs was only weakly active, compared to the cleavable ITsmpt, in CT26.GUCY2Ctm cells (extrapolated Kd ITmbs, 0.483 µg/ml vs Kd ITsmpt, 0.016 µg/mL; p<0.0001; Supplemental Fig. 6). Thus, MS20 IT targets antibody-drug conjugate to cell surface GUCY2C which mediates endocytosis of the complex in a ligand- and GUCY2C cytoplasmic domain-independent, but clathrin-dependent, fashion, delivering ITs to lysosomes where they are cleaved, liberating cytotoxic dgRTA.
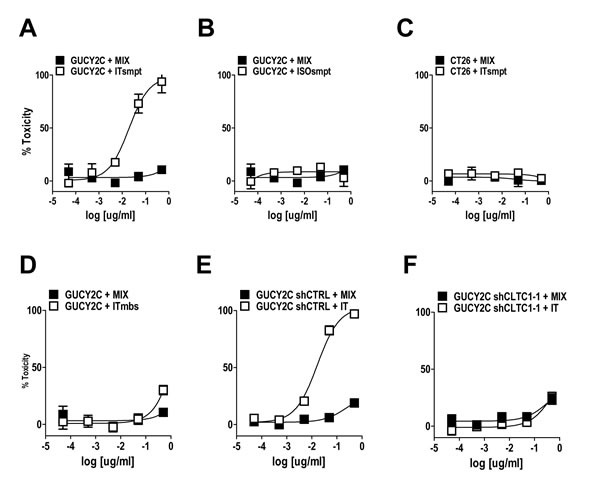
Figure 5: GUCY2C-targeted ITsmpt cytotoxicity depends on GUCY2C, lysosomal cleavage, and clathrin. (A-D) CT26.GUCY2C cells were treated with ITsmpt (A) or the isotype control toxin conjugate ISOsmpt (B) for 48 h prior to MTT assay to quantify cytotoxicity. (C) Parental CT26 cells served as a negative control. (D) CT26.GUCY2C cells also were treated with the non-cleavable ITmbs. (E, F) The clathrin-dependence of ITsmpt cytotoxicity was quantified in CT26.GUCY2C cells treated with (E) control or (F) clathrin-specific shRNAs. All treatments (A-F) were compared to mixed, but not conjugated, molar equivalents of antibody + dgRTA (MIX). Results are the mean ± SEM of at least 3 independent determinations.
GUCY2C-Targeted IT Opposes Colorectal Cancer Metastases Without Injuring Normal Tissues
ITsmpt administered by tail vein to mice bearing CT26.GUCY2C lung metastases in a regimen of 40 mg/kg every other day for 6 doses reduced tumor burden >80% (p<0.001) quantified after 12 days of therapy (Fig. 6A and B). In contrast, ITsmpt treatment of mice inoculated with parental CT26 cells was without effect on tumor metastases in lung (Sup. 7). Moreover, this regimen of ITsmpt increased survival 25% (p<0.001) in mice bearing CT26.GUCY2C lung metastases (Fig. 6C). Importantly, this regimen of ITsmpt was without specific adverse effects in normal tissues, including sites in which GUCY2C is normally expressed (intestine, brain; Fig. 6D). Indeed, thorough histopathologic examination of brain, colon, epididymus, heart, kidney, liver, lung, salivary gland, small intestine, and spleen did not reveal any evidence of significant clinical toxicity specifically produced by the GUCY2C-targeted IT (Fig. 6D).
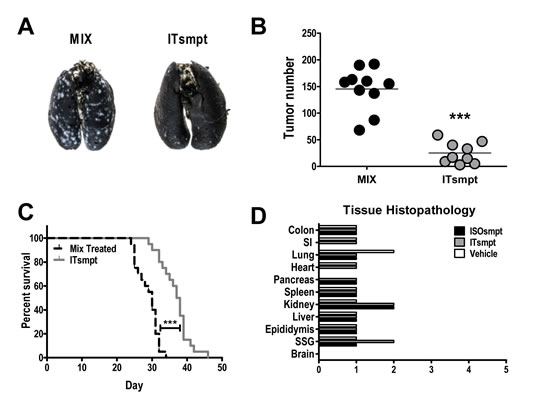
Figure 6: GUCY2C-targeted ITsmpt reduces colorectal cancer metastases to lung and increases survival in mice. Mice were challenged IV with 5 x 105 CT26.GUCY2C cells and treated with 0.4 mg/kg ITsmpt or MIX every other day beginning two days after tumor challenge. (A, B) For some mice, lungs were collected on day 14, (A) stained, and (B) tumors enumerated. (C) Remaining mice (at least 9 per cohort) were followed for survival. **** p<0.0001, one-way ANOVA. (D) Tissue toxicities in mice treated with 0.4 mg/kg ISOsmpt, 0.4 mg/kg ITsmpt, or vehicle control (3 mice per group). Histopathologic injury was scored as described in Materials and Methods.
Discussion
GUCY2C is uniquely suited as a therapeutic target for antibody-drug conjugates for metastatic disease. It is selectively expressed primarily by intestinal epithelia cells, from the duodenum to the rectum, where it is a key regulator of homeostatic processes organizing the crypt-surface axis [38-41]. Also, GUCY2C is expressed by select hypothalamic neurons, where it mediates a novel gut-neural axis regulating appetite and satiety [19]. These anatomical compartments serve as barriers, with tight segregation from the systemic circulation, structurally and functionally insulating them from circulating GUCY2C-targeted cells and macromolecules [9-12, 20]. Indeed, GUCY2C-targeted imaging agents in the circulation do not distribute to the gastrointestinal tract or central nervous system [10]. Moreover, GUCY2C-targeted vaccines generate systemic T cell and antibody responses without affecting the gastrointestinal tract or central nervous system [23-25]. Beyond this compartmentalization, GUCY2C is over-expressed on the surface of primary and metastatic colorectal tumors [9, 22, 42]. These characteristics, in which GUCY2C in normal tissues is compartmentalized and insulated outside the systemic compartment, but over-expressed on metastatic tumors residing within the systemic compartment, make it a highly specific functional tumor target to direct immunotoxins.
While anatomic compartmentalization coupled with general association with metastatic disease suggest unique utility as a therapeutic target, the cellular dynamics of GUCY2C, beyond its expression at the cell surface, has remained incompletely defined. The prevailing paradigm suggests that GUCY2C is primarily deployed in apical brush border membranes of intestinal epithelial cells, available to interact with its cognate paracrine ligands produced in the lumen of intestine [14, 29-32]. GUCY2C internalization has been quantified in vitro with radiolabeled ligand, although the intracellular fate of endocytosed complexes was unknown [43]. Similarly, imaging studies of tumors in mice using receptor-targeted ligands revealed accumulation of label inside tumor cells specifically mediated by GUCY2C although, again, the fate of internalized complexes remained undefined [10].
Here, development of monoclonal antibodies directed to the extracellular ligand-binding domain, but without effect on receptor activities (binding, catalytic activation), provided a unique opportunity to probe the fate of GUCY2C internalized from the cell surface. In striking contrast to the prevailing paradigm [14, 29-32], these analyses revealed that the majority of the GUCY2C associated with intestinal epithelial and colorectal cancer cells resided in the intracellular compartment, primarily in lysosomes. Live cell imaging directly visualized rapid internalization of GUCY2C from the cell surface into lysosomes. Like other guanylyl cyclases [44], endocytosis required clathrin, and disrupting its expression eliminated internalization of GUCY2C from the cell surface into lysosomes and the cytotoxic effects of GUCY2C-targeted ITs. Importantly, GUCY2C internalization was independent of ligands, and trafficking to lysosomes required the extracellular ligand binding, but not the cytoplasmic catalytic, domain, like other guanylyl cyclases [45, 46].
These previously unknown mechanistic elements of GUCY2C endocytosis inform a strategy to employ this receptor as a therapeutic target in metastatic colorectal cancer. Rapid internalization of GUCY2C independent of ligand binding, receptor activation or the cytoplasmic catalytic domain supports therapeutic targeting with inert structural probes of the extracellular domain, like antibodies. Similarly, endocytosis from the cell surface into lysosomes suggests that targeted agents can leverage the unique characteristics of these organelles and incorporate cytotoxins and linkers that optimize liberation to maximize cytotoxicity. In that context, dgRTA cannot access the cytoplasmic compartment of cells in the absence of an internalization partner, like an antibody, enhancing the specificity for targeted killing [1, 2, 37]. Further, dgRTA accesses ribosomal cytotoxic targets by activation in, and translocation from, lysosomes [1, 2, 37]. Moreover, the SMPT linker produces a sterically-hindered disulfide bond which resists disruption in the circulation, but maximizes release of dgRTA in the acidic pH and reductive environment of lysosomes [1, 2, 36].
Employing this mechanism-directed strategy, ITs targeted by MS20 and delivering the cytotoxic dgRTA, linked together with SMPT, killed mouse colorectal cancer cells in vitro. ITsmpt cytotoxicity was highly specific for GUCY2C, and isogenic colorectal cancer cells devoid of GUCY2C resisted cytotoxicity by this IT, while conjugates of non-specific immunoglobulin and dgRTA were inactive. Similarly, ITsmpt cytoxicity required internalization by antibody-GUCY2C complexes, and disrupting clathrin expression specifically eliminated IT efficacy, while mixtures of free MS20 and dgRTA were without effect. Moreover, IT cytotoxicity required activation in, and release from, lysosomes and MS20-dgRTA conjugates linked by the non-cleavable thioether MBS were 10- to 100-fold less potent compared to ITsmpt. Beyond cells in vitro, this mechanism-based approach translated into effective therapy in mice with established colorectal cancer metastases in lung. Indeed, six doses of ITsmpt administered every other day reduced metastatic disease >80%, and improved survival 25%, compared to mice receiving a mixture of free MS20 and dgRTA. Further, the effects of the targeted IT were highly specific for GCC-expressing tumors, and there were no obvious toxicities specifically associated with ITsmpt. In that context, tissues in which GUCY2C normally resides, including intestine and hypothalamus, were not affected by ITsmpt.
These studies reveal a novel biological mechanism mediating endocytosis of cell surface GUCY2C into lysosomes that is independent of receptor activity in normal intestinal epithelial and colorectal cancer cells. This mechanism can be leveraged to develop monoclonal antibody-directed ITs that specifically target GUCY2C-expressing colorectal cancer metastases, maximizing the efficacy of tumor cell cytoxicity while minimizing off target adverse effects in normal tissues, including extra-systemic compartments normally expressing GUCY2C. The emerging evolution in IT platforms, including humanizing monoclonal antibodies [47], novel cytotoxins like mayntansinoids [48], and next-generation scaffolds like pH-dependent boronate-linked linear-hyperbranched polymeric nanovehicles [49], underscores the importance of identifying therapeutic targets like GUCY2C that are tumor-specific, highly associated with disease, and mechanistically tractable to realize the full clinical potential of this therapeutic paradigm. The immediate translation of these approaches can best be appreciated by considering that beyond colorectal cancer, GUCY2C is ectopically expressed by a significant proportion of metastatic gastric, esophageal, and pancreatic tumors and GUCY2C-directed mAb ITs are in clinical development for their treatment [9, 50, 51].
Materials and Methods
GUCY2C mAb Generation
The MS20 mAb was previously described [19]. MS7 and MS24 mAbs were similarly produced. All mAbs were purified with a protein G column (GE HiTrap Protein G HP, #17-0404-01).
Cyclic GMP Accumulation
CT26 mouse colorectal cancer cells engineered to express GUCY2C (CT26.GUCY2C) [23-25], were grown to confluence in DMEM + 10% fetal bovine serum in a 12-well dish washed with three times with PBS, and pretreated with 500uL DMEM containing 1 mM 3-isobutyl-1-methylxanthine (IBMX) and 10 µg/mL antibody at 37°C for 30 min. Media was replaced with 500 µL fresh DMEM/IBMX/antibody mix containing 1 µM ST and incubated for an additional 30 min at 37°C prior to collection. ST is the heat-stable enterotoxin produced by enteroxigenic bacteria that cause Travelers’ diarrhea. It was the first canonical ligand identified that binds to and activates GUCY2C in animals and humans [7]. Media concentrations of cGMP were determined by EIA, and normalized to protein concentration of cell lysates of respective wells determined by BCA assay. Non-transfected CT26 cells were used as a negative control for ST treatment, an irrelevant mouse IgG was used as the control IgG.
Immunotoxin Generation
IT conjugates were generated as previously described [37]. Briefly, cleavable sulfosuccinimidyl 6-[α-methyl-α-(2-pyridyldithio) toluamido] hexanoate (SMPT) or non-cleavable M-maleimid-obenzoyl-N-hydoxysuccinimide ester (MBS) activated the antibody through an amine reaction for 1 h at room temperature. Simultaneously, deglycosylated Ricin Toxin A (dgRTA) domain (Sigma-Aldrich) was reduced with DTT to insure free sulfhydryl groups. Activated antibodies and reduced dgRTA were desalted on C18 columns, and then reacted for 72-96 h at varying stoichiometric ratios. Immunotoxins were then purified by size exclusion chromatography and confirmed by gel electrophoresis.
Immunotoxin ELISA
Binding competence of IT conjugates was confirmed using a GUCY2C extracellular domain (GUCY2C1-430)-based ELISA [25]. ITs were incubated in GUCY2C1-430-coated plates at varying concentrations. The mouse IgG component was detected with HRP-anti-mouse H + L (Jackson ImmunoResearch, #115-035-062). Ricin A chain was detected with rabbit-anti-Ricin antibody (Abcam, ab27169) followed by HRP-anti-rabbit H+L (Jackson ImmunoResearch, #111-035-003). Color was developed with Turbo-TMB substrate (Thermo Scientific, #34022) and quantified at λ480nm.
Cell Imaging
Live imaging of sub-confluent cells plated on glass coverslips in 24 well plates was performed in media containing 25 mM HEPES. Prior to fixation, cells were incubated sequentially with primary antibody and fluorescent secondary antibody, and mounted on slides with DAPI Pro-long anti-fade mounting media and imaged by EVOS FL Auto (Life Technologies) or confocal microscopy (Zeiss 510M and Nikon C1 Plus, Thomas Jefferson University Bioimaging Shared Resource). Whole cell imaging was performed on CT26.GUCY2C cells, with parental CT26 cells as controls [23-25]. Methanol-fixed cells were co-stained with antibodies for lysosomal associated membrane protein 1 (LAMP1, Abcam #24170), clathrin and GUCY2C mAb and reviewed by confocal microscopy. Endocytosis was examined using thermomechanical control, in which internalization was prevented at temperatures <4°C [33]. Cell surface imaging was performed on live cells by sequential exposure to primary antibody and fluorescent secondary antibody at 4°C followed by fixation. GUCY2C internalization was evaluated in live cells by confocal microscopy by sequential exposure to primary and secondary antibodies at 4°C, followed by warming to room temperature while time-lapse imaging. Live cells for time-lapse were counter-stained with live cell-permeable markers Hoechst for nuclei, and lysotracker red for lysosomes.
Targeted Inhibition of Gene Expression
GIPZ lentiviral mouse shRNA control or clathrin heavy chain constructs (Thermo Scientific) were transduced into sub-confluent cells by spinoculation for 1.5 h at 2700 RPM with VSV-G pseudo-typed lentivirus, and clathrin protein expression quantified at 48 h. Mouse siRNA scrambled control or siRNAs to caveolin (Cav-1; Ambion) were transfected into sub-confluent cells by using lipofectamine and Cav-1 protein expression quantified at 48 h.
Immunotoxin Toxicity
Cells, plated in 96 well plates at 80 percent confluence one day prior to treatment, were exposed to various concentrations of ITs for 48 h. Cells were then exposed to MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) for 1 h and then solubilized overnight at 37°C. Absorbance at 590 nm was measured for each well and percent cell kill quantified by comparison to non-treated wells (0% cell kill) and wells incubated with a concentration of cycloheximide which produced 100% cytotoxicity, as follows:
![]()
Metastatic Tumor Model
Mice were inoculated with 5 x 105 CT26.GUCY2C cells by tail vein 24 h prior to administration of 40 mg/kg of IT, or matched concentrations of mixed free antibody and toxin, every other day for 6 doses. Mice were sacrificed on day 13, lungs injected with India ink and fixed for 48 h, and tumors quantified by blinded enumeration.
Tissue Histopathology
Hematoxylin and eosin-stained sections of the following organs were evaluated histologically by a board-certified veterinary pathologist (LDBB): brain, colon, epididymis, heart, kidney, liver, lung, salivary gland, small intestine, and spleen. Sections were specifically evaluated for histopathologic evidence of cell degeneration, necrosis, apoptosis, inflammation, vasculitis, atrophy, and regeneration. The percentage of tissue parenchyma with evidence of histopathologic lesions were scored as follows: None (0% of the parenchyma affected): 0; Rare (<1% of the parenchyma affected): 1; Occasional (1-5% of the parenchyma affected): 2; Minimal (6-10% of the parenchyma affected): 3; Mild (11-30% of the parenchyma affected): 4; Moderate (31-60% of the parenchyma affected): 5; and Marked (61-100% of the parenchyma affected): 6. Tissues from at least 3 mice per group were evaluated to compile scores.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism Software v5.
Funding
These studies were supported by grants from the National Institutes of Health (R01 CA75123, R01 CA95026, RC1 CA146033, R01 CA170533 to SAW; K01 OD010463 to LDBB, and P30 CA56036), the Pennsylvania Department of Health (SAP #4100059197, SAP #4100051723) to SAW, and Targeted Diagnostic and Therapeutics Inc. to SAW. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations or conclusions. A.E.S. was a recipient of the Measey Foundation Fellowship. M.S.M. was a recipient of a Ruth Kirschstein Individual Predoctoral Fellowship Award (F31 CA171672). S.A.W. is the Samuel MV Hamilton Professor of Thomas Jefferson University.
Conflicts of Interest
S.A.W. is the Chair of the Data Safety Monitoring Board for the C-Cure Trial sponsored by Cardio Biosciences, and the Chair (uncompensated) of the Scientific Advisory Board of Targeted Diagnostics & Therapeutics, Inc. which provided research funding that, in part, supported this work and has a license to commercialize inventions related to this work. S.A.W. is an inventor on patents related to this work and could financially benefit from its commercialization.
Abbreviations
Cav-1, caveolin-1; cGMP, cyclic GMP; DTT, dithiothreitol; EIA, enzyme-linked immunoassay; IF, immunofluorescence staining; IT, immunotoxin; GUCY2C, guanylyl cyclase c; HRP, horseradish peroxidase; LAMP1, lysosomal associated membrane protein 1; mAb, monoclonal antibody; RTA, Ricin Toxin A chain; ST, bacterial heat-stable enterotoxin.
References
1. Kreitman RJ. Immunotoxins for targeted cancer therapy. Aaps J. 2006; 8(3):E532-551.
2. Pastan I, Hassan R, Fitzgerald DJ and Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006; 6(7):559-565.
3. Ehrlich P. (1957). The Collected Papers of Paul Ehrlich. (Oxford, UK: Pergamon Press).
4. Sandvig K and van Deurs B. Entry of ricin and Shiga toxin into cells: molecular mechanisms and medical perspectives. The EMBO journal. 2000; 19(22):5943-5950.
5. Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, Tsang KY, Benhar I and Pastan I. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(51):E3597-3603.
6. Samia AC, Chen X and Burda C. Semiconductor quantum dots for photodynamic therapy. Journal of the American Chemical Society. 2003; 125(51):15736-15737.
7. Schulz S, Green CK, Yuen PS and Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990; 63(5):941-948.
8. Hodson CA, Ambrogi IG, Scott RO, Mohler PJ and Milgram SL. Polarized apical sorting of guanylyl cyclase C is specified by a cytosolic signal. Traffic. 2006; 7(4):456-464.
9. Carrithers SL, Barber MT, Biswas S, Parkinson SJ, Park PK, Goldstein SD and Waldman SA. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93(25):14827-14832.
10. Wolfe HR, Mendizabal M, Lleong E, Cuthbertson A, Desai V, Pullan S, Fujii DK, Morrison M, Pither R and Waldman SA. In vivo imaging of human colon cancer xenografts in immunodeficient mice using a guanylyl cyclase C-specific ligand. J Nucl Med. 2002; 43(3):392-399.
11. Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S and Waldman SA. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn. 2005; 5(5):701-713.
12. Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP and Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000; 52(3):375-414.
13. Guerrant RL, Hughes JM, Chang B, Robertson DC and Murad F. Activation of intestinal guanylate cyclase by heat-stable enterotoxin of Escherichia coli: studies of tissue specificity, potential receptors, and intermediates. J Infect Dis. 1980; 142(2):220-228.
14. Guarino A, Cohen MB, Overmann G, Thompson MR and Giannella RA. Binding of E. coli heat-stable enterotoxin to rat intestinal brush borders and to basolateral membranes. Dig Dis Sci. 1987; 32(9):1017-1026.
15. Rao MC, Guandalini S, Smith PL and Field M. Mode of action of heat-stable Escherichia coli enterotoxin. Tissue and subcellular specificities and role of cyclic GMP. Biochimica et biophysica acta. 1980; 632(1):35-46.
16. Almenoff JS, Williams SI, Scheving LA, Judd AK and Schoolnik GK. Ligand-based histochemical localization and capture of cells expressing heat-stable enterotoxin receptors. Mol Microbiol. 1993; 8(5):865-873.
17. Waldman SA, Cagir B, Rakinic J, Fry RD, Goldstein SD, Isenberg G, Barber M, Biswas S, Minimo C, Palazzo J, Park PK and Weinberg D. Use of guanylyl cyclase C for detecting micrometastases in lymph nodes of patients with colon cancer. Dis Colon Rectum. 1998; 41(3):310-315.
18. Cagir B, Gelmann A, Park J, Fava T, Tankelevitch A, Bittner EW, Weaver EJ, Palazzo JP, Weinberg D, Fry RD and Waldman SA. Guanylyl cyclase C messenger RNA is a biomarker for recurrent stage II colorectal cancer. Ann Intern Med. 1999; 131(11):805-812.
19. Valentino MA, Lin JE, Snook AE, Li P, Kim GW, Marszalowicz G, Magee MS, Hyslop T, Schulz S and Waldman SA. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. The Journal of clinical investigation. 2011; 121(9):3578-3588.
20. Ballabh P, Braun A and Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004; 16(1):1-13.
21. Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S and Waldman SA. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol. 2005; 36(2):170-179.
22. Schulz S, Hyslop T, Haaf J, Bonaccorso C, Nielsen K, Witek ME, Birbe R, Palazzo J, Weinberg D and Waldman SA. A validated quantitative assay to detect occult micrometastases by reverse transcriptase-polymerase chain reaction of guanylyl cyclase C in patients with colorectal cancer. Clin Cancer Res. 2006; 12(15):4545-4552.
23. Snook AE, Li P, Stafford BJ, Faul EJ, Huang L, Birbe RC, Bombonati A, Schulz S, Schnell MJ, Eisenlohr LC and Waldman SA. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer research. 2009; 69(8):3537-3544.
24. Snook AE, Magee MS, Schulz S and Waldman SA. Selective antigen-specific CD4 T-cell, but not CD8 T- or B-cell, tolerance corrupts cancer immunotherapy. Eur J Immunol. 2014.
25. Snook AE, Stafford BJ, Li P, Tan G, Huang L, Birbe R, Schulz S, Schnell MJ, Thakur M, Rothstein JL, Eisenlohr LC and Waldman SA. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. Journal of the National Cancer Institute. 2008; 100(13):950-961.
26. Witek M, Blomain ES, Magee MS, Xiang B, Waldman SA and Snook AE. Tumor radiation therapy creates therapeutic vaccine responses to the colorectal cancer antigen GUCY2C. Int J Radiat Oncol Biol Phys. 2014; 88(5):1188-1195.
27. Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ and Thun MJ. Cancer statistics, 2005. CA: a cancer journal for clinicians. 2005; 55(1):10-30.
28. Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, Diken M, Kreiter S, Tureci O and Sahin U. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014; 15(1):190.
29. Albano F, Brasitus T, Mann EA, Guarino A and Giannella RA. Colonocyte basolateral membranes contain Escherichia coli heat-stable enterotoxin receptors. Biochem Biophys Res Commun. 2001; 284(2):331-334.
30. Cohen MB and Giannella RA. Jejunal toxin inactivation regulates susceptibility of the immature rat to STa. Gastroenterology. 1992; 102(6):1988-1996.
31. Cohen MB, Mann EA, Lau C, Henning SJ and Giannella RA. A gradient in expression of the Escherichia coli heat-stable enterotoxin receptor exists along the villus-to-crypt axis of rat small intestine. Biochem Biophys Res Commun. 1992; 186(1):483-490.
32. Cohen MB, Thompson MR, Overmann GJ and Giannella RA. Association and dissociation of Escherichia coli heat-stable enterotoxin from rat brush border membrane receptors. Infect Immun. 1987; 55(2):329-334.
33. Stoneham CA, Hollinshead M and Hajitou A. Clathrin-mediated endocytosis and subsequent endo-lysosomal trafficking of adeno-associated virus/phage. The Journal of biological chemistry. 2012; 287(43):35849-35859.
34. Edeling MA, Smith C and Owen D. Life of a clathrin coat: insights from clathrin and AP structures. Nat Rev Mol Cell Biol. 2006; 7(1):32-44.
35. Williams TM and Lisanti MP. The caveolin proteins. Genome Biol. 2004; 5(3):214.
36. Ueland PM and Refsum H. Plasma homocysteine, a risk factor for vascular disease: plasma levels in health, disease, and drug therapy. The Journal of laboratory and clinical medicine. 1989; 114(5):473-501.
37. Thorpe PE, Wallace PM, Knowles PP, Relf MG, Brown AN, Watson GJ, Blakey DC and Newell DR. Improved antitumor effects of immunotoxins prepared with deglycosylated ricin A-chain and hindered disulfide linkages. Cancer research. 1988; 48(22):6396-6403.
38. Lin JE, Li P, Pitari GM, Schulz S and Waldman SA. Guanylyl cyclase C in colorectal cancer: susceptibility gene and potential therapeutic target. Future Oncol. 2009; 5(4):509-522.
39. Li P, Lin JE, Schulz S, Pitari GM and Waldman SA. Can colorectal cancer be prevented or treated by oral hormone replacement therapy? Curr Mol Pharmacol. 2009; 2(3):285-292.
40. Li P, Lin JE, Snook AE, Gibbons AV, Zuzga DS, Schulz S, Pitari GM and Waldman SA. Colorectal cancer is a paracrine deficiency syndrome amenable to oral hormone replacement therapy. Clin Transl Sci. 2008; 1(2):163-167.
41. Pitari GM, Li P, Lin JE, Zuzga D, Gibbons AV, Snook AE, Schulz S and Waldman SA. The paracrine hormone hypothesis of colorectal cancer. Clin Pharmacol Ther. 2007; 82(4):441-447.
42. Witek ME, Nielsen K, Walters R, Hyslop T, Palazzo J, Schulz S and Waldman SA. The putative tumor suppressor Cdx2 is overexpressed by human colorectal adenocarcinomas. Clin Cancer Res. 2005; 11(24 Pt 1):8549-8556.
43. Urbanski R, Carrithers SL and Waldman SA. Internalization of E. coli ST mediated by guanylyl cyclase C in T84 human colon carcinoma cells. Biochimica et biophysica acta. 1995; 1245(1):29-36.
44. Brackmann M, Schuchmann S, Anand R and Braunewell KH. Neuronal Ca2+ sensor protein VILIP-1 affects cGMP signalling of guanylyl cyclase B by regulating clathrin-dependent receptor recycling in hippocampal neurons. J Cell Sci. 2005; 118(Pt 11):2495-2505.
45. Dickey DM, Flora DR and Potter LR. Antibody tracking demonstrates cell type-specific and ligand-independent internalization of guanylyl cyclase a and natriuretic peptide receptor C. Molecular pharmacology. 2011; 80(1):155-162.
46. Nussenzveig DR, Lewicki JA and Maack T. Cellular mechanisms of the clearance function of type C receptors of atrial natriuretic factor. The Journal of biological chemistry. 1990; 265(34):20952-20958.
47. Nelson AL, Dhimolea E and Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010; 9(10):767-774.
48. Burris HA, 3rd, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, Tan-Chiu E, Krop IE, Michaelson RA, Girish S, Amler L, Zheng M, Chu YW, Klencke B and O’Shaughnessy JA. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011; 29(4):398-405.
49. Jia HZ, Zhu JY, Wang XL, Cheng H, Chen G, Zhao YF, Zeng X, Feng J, Zhang XZ and Zhuo RX. A boronate-linked linear-hyperbranched polymeric nanovehicle for pH-dependent tumor-targeted drug delivery. Biomaterials. 2014; 35(19):5240-5249.
50. Park J, Schulz S, Haaf J, Kairys JC and Waldman SA. Ectopic expression of guanylyl cyclase C in adenocarcinomas of the esophagus and stomach. Cancer Epidemiol Biomarkers Prev. 2002; 11(8):739-744.
51. Zambrano C, Almhanna K, Messersmith W, Ahnert J, Ryan D, Faris J, Jung J, Fasanmade A, Wyant T and Kalebic T. (2014). MLN0264, an investigational antiguanylyl cyclase C (GCC) antibody-drug conjugate (ADC), in patients with advanced gastrointestinal (GI) malignancies: Phase I study. ASCO. (Chicago, IL.