
Murine Double Minute Clone 2 (MDM2) plays a key role in the regulation of cell cycle and apoptosis. The MDM2 gene was originally discovered and cloned from a spontaneously transformed BALB/c 3T3 mouse cell line [1], and it was later shown to act as an oncogene in nude mice [2]. The MDM2 protein acts as a p53 antagonist and is linked to p53 in a feedback-loop where p53 enhances MDM2 transcription in response to genotoxic stress, whereas MDM2 binds to p53 and directs it for proteosomal degradation through ubiquitinylation [3, 4]. The vital importance of this MDM2 – p53 interaction is underlined by the fact that although MDM2 null mice uniformly die at an early embryonic stage, MDM2 / p53 null double knock-outs are live born with no obvious developmental defects [5, 6].
MDM2, in addition, inactivates the retinoblastoma protein (pRB) by protein binding [7], preventing pRB from binding and inactivating E2F1. Further, it directly interacts with and stimulates E2F1 [8], a transcription factor with complex functions, both able to promote cell cycle progression through induction of genes coding for e.g DNA polymerase and cyclin E but also apoptosis through induction of genes coding for e.g. APAF1 and caspases [9]. The fact that MDM2 acts as a key regulator of both the p53 and the pRb functional pathways points to MDM2 as a “master” gene potentially governing the balance between cellular processes like continued growth, growth arrest, senescence and apoptosis.
Somatic alterations of MDM2 in malignant tumors
So far, very few somatic mutations in the MDM2 gene has been identified. The gene however, has been found amplified in many tumour forms [reviewed in 10]. In addition, several tumors reveal elevated staining for the MDM2 protein despite harbouring a normal gene copy number. Thus, several mechanisms apart from MDM2 gene amplification has been suggested to influence MDM2 levels and activity. These include enhanced translation through increased activity of BCR/ABL [11-13] and expression of a variety of alternatively or aberrantly spliced MDM2 mRNA variants [14]. While splice variants have been reported to inhibit the function of wild-type MDM2 through dimerization [15, 16], the full function of these alternatively spliced mRNAs remains to be elucidated. Recently, another alternative mechanism of MDM2 upregulation, disturbing proper function of the p53 pathway, was reported: Inuzuka and co-workers demonstrated that MDM2 is phosphorylated by Casein Kinase I (CKI), marking the MDM2 molecule for degradation via the SCFβ-TRCP Ubiquitin ligase [17]. This has been suggested as a potential mechanism of MDM2 upregulation in cases where CKI and / or β-TRCP are inactivated [18], a hypothesis that is supported by the findings of β-TRCP-deletions in several tumor forms [18-20].
Germ line alterations of MDM2 function.
Despite its role as a proto-oncogene, so far, neither MDM2 germ line mutations nor increased gene copy number at the germ line level has been identified in cancer prone families. Thus, one may speculate whether loss of MDM2 function through deleterious mutations may be incompatible with life, resembling the effect in MDM2 null mice.
The MDM2 gene contains two main promoters; the P1 located upstream of exon 1, and the intronic P2 promoter, located between exon 1 and exon 2. P1 is considered responsible for regulating MDM2 expression levels in the “non-stressed” setting. This promoter contains a binding site for PTEN, which has been shown to suppress MDM2 expression. The P2 promoter is considered to be more inducible and regulates MDM2 level in response to cellular stress and stimulation by different ligands including p53, Sp1 and the estrogen receptor (Figure 1) [21, 22].
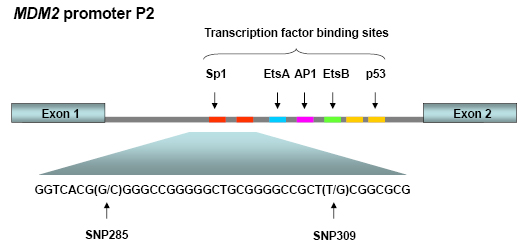
Figure 1: MDM2 promoter P2. This promoter is located between the exons 1 and 2 of the MDM2 gene and is induced upon cellular stress. Promoter P2 harbors binding sites for among others p53 and Sp1. SNP285 and SNP309 are both located within the latter binding sites.
In 2004, the group lead by Arnold Levine identified a polymorphism SNP309 (T>G; rs2279744) in the P2 promoter [21]. The G-allele is found across all ethnic groups albeit at a variable frequency (~10% of MDM2 alleles among African Americans, 40% in Caucasians and 50% in Asians) [23]. It was found to increase the expression levels of MDM2 through enhancing the binding of the Sp1 transcription factor. The G-allele was linked to early age at cancer diagnosis among individuals with Li-Fraumeni syndrome (carrying germline TP53 mutations) and an early age at diagnosis of soft tissue sarcomas, large B-cell lymphomas and colorectal cancers in women not diagnosed with any cancer predisposing germline mutations [21, 24]. In addition, an early diagnosis of estrogen receptor rich (defined as >50% of cells revealing positive ER staining) but not receptor-poor breast cancer was recorded [21]. Although these data are strongly supported by the finding that MDM2SNP309G/G mice are more tumor prone than MDM2SNP309T/T mice [25], case control studies across many tumor forms and ethnic groups have provided conflicting results regarding the role of the SNP309G-allele as a cancer risk factor in humans [23, 26]. Notably, so far most of the studies linking the SNP309G variant allele to enhanced cancer risk or a young age at diagnosis have been performed on Asian or Ashkenazi Jewish populations [23, 26]. In contrast, the majority of studies performed in Caucasians are negative [23, 26]. In addition to SNP309, several other SNPs have also been reported both in the promoter P2 area and the rest of the MDM2 gene. Notably, in the three most thoroughly studied ethnic groups (Caucasians, African Americans and Ashkenazi Jews) the variability in other polymorphic sites is much higher for the SNP309T-allele than for the G-allele [27]. Taken together with the relatively high frequency of SNP309G, this may indicate a positive selection pressure for the G-variant. Notably, the reported polymorphisms in the MDM2 gene are restricted to either SNP309T or G, indicating that they are of younger origin than the 309 variation. To this day, potential biological function of these polymorphisms remains to be elucidated.
SNP285C reduces the risk of breast and ovarian cancer
Studying the potential effects of MDM2 promoter SNP’s on breast cancer therapy, outcome and prognosis, we, concomitantly with a Scottish group [28], discovered a second promoter P2 polymorphism, 285G>C, located 24 bps from SNP309 (Figure 2) [29]. The C-variant of SNP285 is located on the SNP309G allele forming a distinct SNP285C/309G haplotype. Notably, SNP285C was found at a similar frequency (average of 7.8% of individuals) in different Western populations (Dutch, British, Norwegian) but was absent from Asians (Chinese). Interestingly, the frequency of this polymorphism among Finnish individuals living in the Helsinki area (1.6%), was significantly lower compared to the frequency observed among other Western Europeans. Potential interpretations of this finding is discussed below.
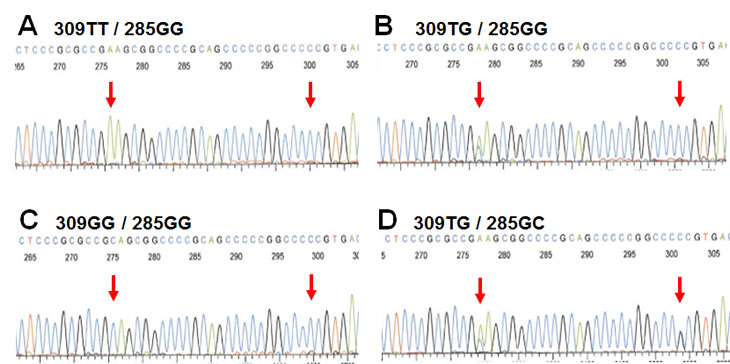
Figure 2: SNP285 and SNP309 genotypes. Electropherograms showing the four different genotypes of the Sp1-binding elements of MDM2 promoter P2 commonly observed among Caucasians. (Electropherograms display sequence as reverse complementary to the open reading frame of MDM2)
Using the highly sensitive surface Plasmon resonance technology (SPR), we confirmed the previous findings of Bond et al [21] that the SNP309G-allele had a stronger binding affinity to the Sp1 transcription factor as compared to the SNP309T-allele (22% increase in binding strength). In contrast, SNP285C significantly reduced Sp1-binding to the MDM2 promoter (51% decreased binding strength). Importantly, the combined SNP285C / SNP309G haplotype (which accounts for about 12% of all 309G alleles in Caucasians) had a reduced affinity towards Sp1 as compared to the “wild-type” SNP285G/309T haplotype (>10% decreased binding strength) [29]. While secondary mutations antagonizing a primary mutation in BRCA2 through excision of the mutated area has been shown to revert platinum drug sensitivity in breast as well as ovarian carcinomas [30, 31], to the best of our knowledge, secondary SNP’s antagonizing the effect of a previous SNP on the same gene, have not previously been reported (Figure 3).
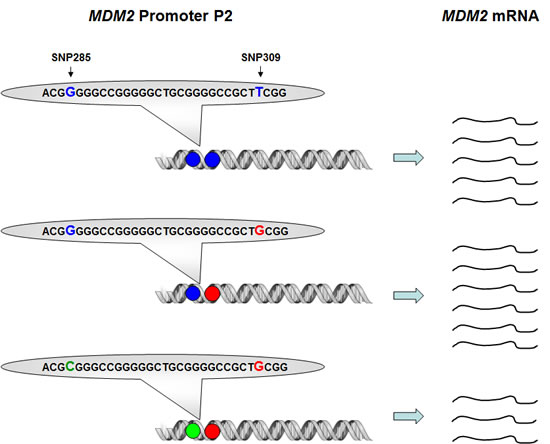
Figure 3: Impact of SNP309 and SNP285 on MDM2 transcription. Presence of the SNP309G-variant (red) enhances the binding strength between MDM2 promoter P2 and the transcription factor Sp1, thus, leading to increased MDM2 transcription as compared to the “wild-type” SNP309T-variant. The effect of the SNP285C-variant (green) is the opposite, reducing the binding strength for Sp1, thus, also reducing the level of MDM2 transcription as compared to the “wild-type” SNP285G-variant. The effect of SNP285C is stronger than SNP309G, thus, presence of the SNP285C/309G haplotype leads to reduced MDM2 transcription as compared to the “wild-type” SNP285G/309T haplotype.
In line with the finding of reduced binding strength to the Sp1 transcription factor and, thereby, reduced transcription, we found the presence of SNP285C to be associated with significantly lowered risk of both breast and ovarian cancer. Among individuals potentially harbouring the SNP285C variant (carriers of SNP309G) we found a 21% reduced risk of breast cancer among individuals carrying SNP285C as compared to those homozygous for SNP285G. Regarding ovarian cancer risk, the effect was even stronger. Here, the presence of a SNP285 allele lead to a reduction in risk of disease of 26%. Among individuals carrying the SNP309TG heterozygous genotype, SNP285C reduced the risk of ovarian cancer by 37%, while no effect was observed in SNP309GG homozygotes. In contrast, the effect of 285C on breast cancer risk was most profound in SNP309GG homozygotes (reduction of 45%), with a small non-significant reduction of 9% in SNP309TG heterozygotes.
The data for ovarian cancer patients fits well with the in vitro data and a hypothesis that one SNP285C may counteract and neutralize the effect of one SNP309G, but not two SNP309Gs. The results from the breast cancer cohorts may seem somewhat harder to understand; notably, it is well known from other genes that heterozygote carriers of a genetic alteration may be at risk of different diseases than the homozygotes [32].
The finding that the SNP285C/309G haplotype accounted for about 12% of all SNP309G alleles among Caucasians may be of importance explaining the potential difference regarding the effect of SNP309 status on cancer risk among Caucasians versus Asians [23, 26]. While studies on Asian populations may represent the “true” effect of SNP309G, studies on Caucasians may need to be corrected for the presence on of the SNP309G-counteracting SNP285C in order to unmask the real effect of SNP309G in these populations.
Importantly, so far the potential effect of SNP285C on the risk of malignancies other than breast and ovarian cancer has not been explored. Considering other risk modulating factors associated with breast and ovarian cancers, notably both diseases are found at high incidence among individuals carrying pathogenic BRCA1 and BRCA2 germline mutations. However, this does not mean that breast and ovarian cancer have a particular and common etiology indicating that the effect of SNP285C may be restricted to these two malignancies. For example estrogen exposition is a major risk factor associated with breast but not ovarian cancer [33]. Thus, it may well be that SNP285C modulate the risk of several other malignancies in addition to breast and ovarian cancer
Evolutionary selection of MDM2 SNPs
Examining MDM2 haplotype status across ethnic groups, Atwal and colleagues recorded multiple polymorphisms with an ethnic diversity between Caucasians (general population), Ashkenazi Jews and American-Africans [27]. They found a single SNP309G haplotype in Africans and Caucasians, but two additional 309G-linked polymorphisms in Ashkenazi’s. In contrast, multiple SNPs were recorded on the T-allele, mostly located outside the promoter area and outside the exons. However, except for the SNP309G and now SNP285C, the potential functional role of the different polymorphisms have not been determined. Thus, we may not at this stage speculate on potential reasons for evolutionary selection (if any) for these additional SNP’s.
The variable distribution of SNP309G and, now, in particular the SNP285C, across ethnic groups may lead to speculations on the phylogenetic selection of these particular MDM2 variants. Considering the key role of MDM2 influencing the p53 as well as retinoblastoma pathway, naturally a fine tuning of its biological activity must be of critical importance, as documented by the influence of SNP285/309 haplotype status on cancer risk. These observations raise further important questions. Considering SNP309G, this variant enhances MDM2 transcription, subsequently antagonising p53 function. The fact that the SNP309G genotype has been found associated with increased cancer risk as well as an early cancer diagnosis [21, 26] may suggest a negative selection for this variant. However, with a few exceptions solid tumours in general are diagnosed after the age of child-birth. Thus, early death related to malignant disease is unlikely to explain evolutionary selection of the SNP309 variants.
Regarding the p53 Pro72Arg polymorphism, the Arginine variant has been shown to reveal stronger pro-apoptotic effects as compared to the Proline allele [34]. In Africa, the p53 proline variant becomes more frequent approaching the equatorial line [35], and Atwal et al postulated that a high frequency of MDM2 SNP309T among Africans may act in concert balancing a weaker p53 codon 72 proline variant [27]. So far however, we know the frequency of the MDM2 SNP309T and G variants among American Africans only; thus, to confirm such a hypothesis, assessing the SNP309 status across different geographical and ethnic groups in Africa is warranted. Further, studies on the distribution of SNP309G across different areas in Africa as well as in Asia may add important information with respect to potential mechanisms selecting for this polymorphism in general. While Fang et al, in two small studies, reported MDM2 SNP309G to be associated with increased risk of missed abortion among Han Chinese [36, 37], this finding needs to be confirmed in larger studies involving other ethnic groups as well.
We do not know the phylogenetic age of either the SNP309G or the SNP285C polymorphism, but some assumptions may be made. The fact that SNP309G exists in all ethnic groups (albeit at different incidence) indicates this variant to be an ancient polymorphism, while the fact that SNP285C locates to the SNP309G allele and is absent in Han Chinese, indicates SNP285C to be a young polymorphism, appearing in Caucasians after the “out of Africa” migration and after separation of the Caucasian and the Asian populations (Figure 4). Further, the fact that SNP285C is found at a low frequency (1.6%) among Finnish habitants, is interesting. The genetic heritage of the Finns is incompletely understood; while their language, similar to that of the Lapps, Estonians and Hungarians belongs to the family of Finno-Uralic languages [38], there is evidence suggesting a more complex genetic background of the population [39], including Y-chromosome migration from Asia [40]. Notably, European migration started out from a few “sanctuaries” at the end of the last ice age about 10.000 years ago. While the first Finnish settlements can be dated around that time [41], there is evidence suggesting the Finnish population may have gone through a “bottleneck” 2-4000 years ago [42, 43], and the Finnish population is characterized by a spectrum of recessive diseases consistent with genetic isolation [41, 44]. Thus, the finding of a low incidence of the SNP285C in the Finnish population may have several explanations: It may have been absent in the original population and come along through subsequent immigration; thus, there is evidence for a second major immigration about 2000 years ago [44]. Further, as the Finnish subjects analysed so far, were collected from the Helsinki area, the polymorphism may also have been brought in through more recent immigrants from other European countries like Sweden. Both hypothesis implicates SNP285C may have arisen after separation of the Finnish ancestors from the main immigrants into Western Europe. In contrast, the possibility exist that SNP285C occurred at an incidence of about 1.6% among Europeans in general 2-4000 years ago. Thereafter, it has expanded through selection pressure in the general European population but, for reasons unexplained, not within the Finnish population.
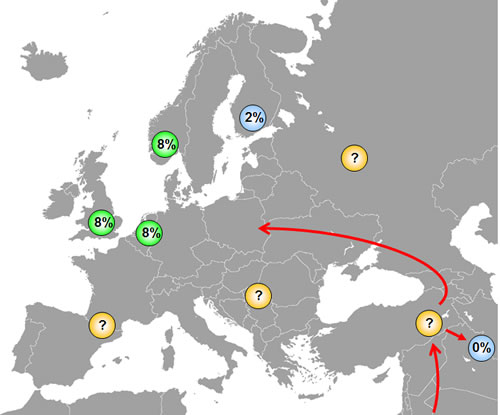
Figure 4: Distribution of SNP285C among different ethnic groups. SNP285 is observed among ~8% of British, Dutch and Norwegians and in ~2% of Finns, while it is absent in Chinese. The presence of SNP285C only among Caucasians indicates that this polymorphism has originated after the separation of Caucasians and Asians approximately 60,000 years ago, but studies on other European populations are warranted to shed light on the origin of SNP285C. Most likely, SNP285C should be absent in all ethnic groups in the Middle-East, but this has yet to be confirmed.
To possibly date the origin of the 285C polymorphism, there is a need to study the distribution of the SNP285C across additional European populations, including Estonians and Hungarians in particular, as well as other European “isolated” populations that have gone through genetic “bottle-necks”, like the Basques and Sardinians [45]. Whatever its exact age may be, its spread in Western European populations strongly suggests a positive selection pressure. As argued with respect to SNP309G, the effect of SNP285C on cancer risk may probably not explain evolutionary selection for this variant in Caucasians.
While there seems to be a positive selection for SNP309G in the Caucasian as well as Asian ethnic groups, it is remarkable with a subsequent selection for the SNP285C, antagonizing SNP309G, among Caucasians. However, the Western European population has passed through distinct evolutionary “bottle-necks” during history. At the end of the ice age, only a limited number of Europeans survived in certain “refugees” like in Northern Spain, Ukraine and Moldovia [46]. As for more recent times, the black plague eradicated between one half and two-thirds of the European population between 1346-50. Further, during the 10-12.000 years passing since the last ice age, there has been severe climate alterations, causing substantial changes in the living conditions in general. It is not unlikely that fine-tuning of MDM2 levels, like the balance between SNP309 and SNP285 status, may have provided survival advantages under some stages of these shifting conditions.
Understanding the mechanisms of evolutionary selection for MDM2 SNP309G as well as SNP285C may add important information to our understanding of the role of this complicated gene in human biology.
References
1. Cahilly-Snyder L, Yang-Feng T, Francke U, George DL. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat Cell Mol Genet 1987; 13:235-244.
2. Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. Embo J 1991; 10:1565-1569.
3. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69:1237-1245.
4. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992; 358:80-83.
5. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206-208.
6. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203-206.
7. Xiao ZX, Chen J, Levine AJ, Modjtahedi N, Xing J, Sellers WR, Livingston DM. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 1995; 375:694-698.
8. Martin K, Trouche D, Hagemeier C, Sorensen TS, La Thangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature 1995; 375:691-694.
9. Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer 2009; 9:738-748.
10. Momand J, Zambetti GP. Mdm-2: “big brother” of p53. J Cell Biochem 1997; 64:343-352.
11. Landers JE, Cassel SL, George DL. Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Research 1997; 57:3562-3568.
12. Sheikh MS, Shao ZM, Hussain A, Fontana JA. The p53-binding protein MDM2 gene is differentially expressed in human breast carcinoma. Cancer Research 1993; 53:3226-3228.
13. Trotta R, Vignudelli T, Candini O, Intine RV, Pecorari L, Guerzoni C, Santilli G, Byrom MW, Goldoni S, Ford LP, Caligiuri MA, Maraia RJ, Perrotti D, Calabretta B. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell 2003; 3:145-160.
14. Bartel F, Harris LC, Wurl P, Taubert H. MDM2 and its splice variant messenger RNAs: expression in tumors and down-regulation using antisense oligonucleotides. Mol Cancer Res 2004; 2:29-35.
15. Chandler DS, Singh RK, Caldwell LC, Bitler JL, Lozano G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res 2006; 66:9502-9508.
16. Evans SC, Viswanathan M, Grier JD, Narayana M, El-Naggar AK, Lozano G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene 2001; 20:4041-4049.
17. Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, Wan L, Ang XL, Mock C, Yin H, Stommel JM, Gygi S, Lahav G, Asara J, Xiao ZX, Kaelin WG, Jr., Harper JW, Wei W. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell 2010; 18:147-159.
18. Inuzuka H, Fukushima H, Shaik S, Wei W. Novel insights into the molecular mechanisms governing mdm2 ubiquitination and destruction. Oncotarget 2010; 1:685-690.
19. Momand J, Wu HH, Dasgupta G. MDM2--master regulator of the p53 tumor suppressor protein. Gene 2000; 242:15-29.
20. Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets 2005; 5:27-41.
21. Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K, Yip L, Hwang SJ, Strong LC, Lozano G, Levine AJ. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004; 119:591-602.
22. Zauberman A, Flusberg D, Haupt Y, Barak Y, Oren M. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res 1995; 23:2584-2592.
23. Hu Z, Jin G, Wang L, Chen F, Wang X, Shen H. MDM2 promoter polymorphism SNP309 contributes to tumor susceptibility: evidence from 21 case-control studies. Cancer Epidemiol Biomarkers Prev 2007; 16:2717-2723.
24. Bond GL, Menin C, Bertorelle R, Alhorpuro P, Aaltonen LA, Levine AJ. MDM2 SNP309 Accelerates colorectal tumour formation in women. J Med Genet 2006.
25. Post SM, Quintas-Cardama A, Pant V, Iwakuma T, Hamir A, Jackson JG, Maccio DR, Bond GL, Johnson DG, Levine AJ, Lozano G. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell 2010; 18:220-230.
26. Economopoulos KP, Sergentanis TN. Differential effects of MDM2 SNP309 polymorphism on breast cancer risk along with race: a meta-analysis. Breast Cancer Res Treat 2010; 120:211-216.
27. Atwal GS, Bond GL, Metsuyanim S, Papa M, Friedman E, Distelman-Menachem T, Ben Asher E, Lancet D, Ross DA, Sninsky J, White TJ, Levine AJ, Yarden R. Haplotype structure and selection of the MDM2 oncogene in humans. Proc Natl Acad Sci U S A 2007; 104:4524-4529.
28. Paulin FE, O’Neill M, McGregor G, Cassidy A, Ashfield A, Ali CW, Munro AJ, Baker L, Purdie CA, Lane DP, Thompson AM. MDM2 SNP309 is associated with high grade node positive breast tumours and is in linkage disequilibrium with a novel MDM2 intron 1 polymorphism. BMC Cancer 2008; 8:281.
29. Knappskog S, Bjornslett M, Myklebust LM, Huijts PE, Vreeswijk MP, Edvardsen H, Guo Y, Zhang X, Yang M, Ylisaukko-Oja SK, Alhopuro P, Arola J, Tollenaar RA, van Asperen CJ, Seynaeve C, Staalesen V, Chrisanthar R, Lokkevik E, Salvesen HB, Evans DG, Newman WG, Lin D, Aaltonen LA, Borresen-Dale AL, Tell GS, Stoltenberg C, Romundstad P, Hveem K, Lillehaug JR, Vatten L, Devilee P, Dorum A, Lonning PE. The MDM2 Promoter SNP285C/309G Haplotype Diminishes Sp1 Transcription Factor Binding and Reduces Risk for Breast and Ovarian Cancer in Caucasians. Cancer Cell 2011; 19:273-282.
30. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008; 451:1111-1115.
31. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008; 451:1116-1120.
32. Smirnov DA, Cheung VG. ATM gene mutations result in both recessive and dominant expression phenotypes of genes and microRNAs. Am J Hum Genet 2008; 83:243-253.
33. Trichopoulos D, MacMahon B, Cole P. Menopause and breast cancer risk. J Natl Cancer Inst 1972; 48:605-613.
34. Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003; 33:357-365.
35. Beckman G, Birgander R, Sjalander A, Saha N, Holmberg PA, Kivela A, Beckman L. Is p53 polymorphism maintained by natural selection? Hum Hered 1994; 44:266-270.
36. Fang Y, Kong B, Yang Q, Ma D, Qu X. MDM2 309 polymorphism is associated with missed abortion. Hum Reprod 2009; 24:1346-1349.
37. Fang Y, Kong B, Yang Q, Ma D, Qu X. The p53-HDM2 gene-gene polymorphism interaction is associated with the development of missed abortion. Hum Reprod 2011.
38. Lahermo P, Sajantila A, Sistonen P, Lukka M, Aula P, Peltonen L, Savontaus ML. The genetic relationship between the Finns and the Finnish Saami (Lapps): analysis of nuclear DNA and mtDNA. Am J Hum Genet 1996; 58:1309-1322.
39. Kittles RA, Perola M, Peltonen L, Bergen AW, Aragon RA, Virkkunen M, Linnoila M, Goldman D, Long JC. Dual origins of Finns revealed by Y chromosome haplotype variation. Am J Hum Genet 1998; 62:1171-1179.
40. Zerjal T, Dashnyam B, Pandya A, Kayser M, Roewer L, Santos FR, Schiefenhovel W, Fretwell N, Jobling MA, Harihara S, Shimizu K, Semjidmaa D, Sajantila A, Salo P, Crawford MH, Ginter EK, Evgrafov OV, Tyler-Smith C. Genetic relationships of Asians and Northern Europeans, revealed by Y-chromosomal DNA analysis. Am J Hum Genet 1997; 60:1174-1183.
41. de la Chapelle A, Wright FA. Linkage disequilibrium mapping in isolated populations: the example of Finland revisited. Proc Natl Acad Sci U S A 1998; 95:12416-12423.
42. de la Chapelle A. Disease gene mapping in isolated human populations: the example of Finland. J Med Genet 1993; 30:857-865.
43. Sajantila A, Salem AH, Savolainen P, Bauer K, Gierig C, Paabo S. Paternal and maternal DNA lineages reveal a bottleneck in the founding of the Finnish population. Proc Natl Acad Sci U S A 1996; 93:12035-12039.
44. Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet 1999; 8:1913-1923.
45. Peltonen L, Palotie A, Lange K. Use of population isolates for mapping complex traits. Nat Rev Genet 2000; 1:182-190.
46. Oppenheimer S. The Origin of the British. London: Constable & Robinson Ltd; 2006.