KEY MESSAGE
Pelareorep is an oncolytic virus presently under clinical evaluation in a variety of cancers. A randomized phase II trial of pelareorep with docetaxel in metastic castration resistant prostate cancer was undertaken. Our results show that the combination of docetaxel and pelareorep is not worthy of further evaluation in this setting.
INTRODUCTION
Prostate cancer is the most frequently diagnosed serious cancer, and the second leading cause of cancer death in men [1]. For metastatic castration resistant prostate cancer (mCRPC), docetaxel remains an effective treatment [2], but its benefit is modest. Studies evaluating more effective combinations are warranted.
Pelareorep (Reolysin; Oncolytics Biotech Inc, Calgary, Canada), is a non-attenuated Dearing strain of the double-stranded RNA reovirus serotype 3. Although this is a ubiquitous virus that is not known to cause any serious illness [3], it was found to preferentially infect and exhibit cytotoxic effects on human cancer cells, especially those with activated Ras pathway [4–6]. Recent data suggests that pelareorep infection may also potentiate anti-tumor immune responses [7–9].
Pelareorep was shown to have in vitro activity against many cancers including several prostate cancer cell lines [4] and early studies showed significant in vivo activity in several prostate cancer xenograft models [10]. Intralesional pelareorep injections administered to a cohort of patients with organ-confined prostate cancer demonstrated antitumor activity, increased CD8 T-cell infiltration and increased prostate cancer apoptosis [10]. Sei et al have demonstrated that while pelareorep shows synergism with several agents in chemotherapy sensitive NSCLC cell lines, consistent synergism with taxanes was seen in all cell lines tested [11]. It was observed that the addition of paclitaxel to pelareorep prolonged mitotic arrest and increased the rate of progeny virion production. The proposed underlying mechanism of this synergism is due to viral propensity to exploit microtubules as sites of viral replication [12, 13] and so microtubule stabilizing agents such as taxanes are a natural choice in the evaluation of combination therapies with pelareorep.
A recently published phase I trial evaluating the combination of docetaxel with pelareorep in 24 patients demonstrated excellent tolerability [14]. Of 15 RECIST evaluable patients there was 1 CR, 3 PR and 10 patients with stable disease, 1 evaluable patient with prostate cancer had a 30% decline in PSA from baseline [14].
The combination of pelareorep with taxanes in prostate cancer is therefore an obvious area of interest. The Canadian Cancer Trials Group (CCTG) conducted a randomized phase II clinical trial to evaluate the tolerability and clinical activity of pelareorep and docetaxel in patients with mCRPC.
PATIENTS AND METHODS
Study design
This was a randomized, open label multicenter phase II trial of pelareorep and docetaxel/prednisone or docetaxel/prednisone alone in patients with mCRPC. The primary endpoint was lack of disease progression at 12-weeks (LPD), as per the Prostate Cancer Clinical Trials Working Group guidelines [15]. Secondary endpoints included change in PSA, safety, tolerability, objective response and response duration, and overall survival (OS). Enumeration of circulating tumor cells (CTC) at baseline and after 6 and 12 weeks was also undertaken. This study adhered to the Declaration of Helsinki and Good Clinical Practice. Local ethics committee approval was obtained by all participating institutions and all patients provided written informed consent. This study is registered in www.clinicaltrials.gov as NCT01619813.
Patient selection
Patients had metastatic adenocarcinoma of the prostate with clinically or radiologically documented disease, radiographic or PSA progression and a castrate level of testosterone, a serum PSA of ≥ 5 μg/L, and an Eastern Cooperative Oncology Group (ECOG) performance status of 2 or less. Prior chemotherapy was not permitted, but prior treatment with hormonal agents (including abiraterone acetate or enzalutamide) or radiotherapy was, provided a minimum of 4 weeks had elapsed before enrollment. Patients were not eligible if they had a history of previous invasive cancer (except adequately treated non-melanoma skin cancer, or tumours curatively treated with no evidence of disease for > 5 years), brain metastases, other serious medical illness, or if they were receiving immunosuppressive therapy or had known HIV or active hepatitis B or C infection.
Treatment plan and evaluations
Patients were randomly assigned to receive either docetaxel at 75 mg/m2 IV on day 1 and prednisone 5mg orally twice daily with pelareorep at a dose of 3×1010 TCID50 intravenously daily on days 1 to 5 of a 3-week cycle (arm A), or docetaxel/prednisone alone (arm B). Docetaxel was delayed (up to 2 weeks) or reduced for toxicity, in accordance with the protocol. A single reduction to 1×1010 TCID50 was permitted for pelareoreop toxicity. Patients remained on treatment until protocol completion (10 cycles) or disease progression, intolerable toxicity, start of new cancer therapy, withdrawal of consent, or death. Baseline evaluation included physical exam, laboratory blood tests and radiological imaging with chest/abdominal/pelvic CT scan and bone scan. Laboratory tests for hematology, biochemistry and PSA were repeated on day 1 of every 3-week cycle. Radiological assessments were repeated at the end of every second cycle, and bone scan imaging was performed at 12 weeks, as clinically indicated and at end of study in patients with positive bone scans at baseline. CTC were collected at baseline [16], week 6 and week 12 if patients were still on study treatment. Adverse events were graded according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
Outcome measures
All patients were considered assessable for toxicity from the time of their first treatment. Response and progression were evaluated by RECIST 1.1 criteria [17]. Bone scans were considered to show PD ONLY if > 2 new lesions were noted and confirmed by a second scan a minimum of 6 weeks later; the date of progression was the date of the first PD scan. PSA progression was evaluated following a minimum of 12 weeks on study and was defined as a rise in PSA of 25% (minimum 2 ng/mL) above baseline (or nadir) and confirmed by a second increasing value at least 3 weeks later. CTC counts were scored as favorable (<5 cells/7.5 mL blood) and unfavorable (≥5 cells/7.5 mL blood) at each measurement. Other endpoints included objective response rate in patients with measurable disease at baseline, overall survival and PSA change rate.
Statistical considerations
The primary study endpoint was LPD at 12 weeks, determined by an algorithm based on PSA progression, bone scan progression and RECIST 1.1 progression as described above, as well as survival status. Secondary endpoints were objective response rate (ORR), defined according to the Modified Response Evaluation Criteria in Solid Tumours (RECIST 1.1), overall survival (OS), CTC counts, and potential prognostic or predictive molecular factors by assessment of archival tissue or blood samples.
With 40 patients treated by pelareorep plus docetaxel and prednisone, the study would have 92% power to test the null hypothesis that 12 week LPD rate was < 30% versus the alternative hypothesis that 12 week LPD rate was > 50% at 0.11 significance level. With a total sample size of 80, we would have 58% power to detect a difference in 12-week LPD rate from 30 to 50% with two-sided alpha 0.1 and 90% power to detect difference in 12 week lack of progression rate from 20 to 50% with two-sided alpha 0.1.
All comparisons between treatment arms were carried out at an alpha level of two-sided 10% unless otherwise specified. When appropriate, discrete variables were summarized with the number and proportion of subjects falling into each category, and compared using Fisher’s exact test. Continuous and ordinal categorical variables were summarized using the mean, median, standard error, minimum and maximum values and when appropriate, compared using the Wilcoxon test. All confidence intervals were computed based on normal approximations.
All randomized patients were analyzed on an intention-to-treat basis for efficacy endpoints. Safety analyses included patients who had received at least one dose of protocol therapy.
Time-to-event endpoints (OS) were analyzed using Kaplan-Meier methods. Overall survival was calculated from the day of randomization to death. For alive patients at time of data cutoff, survival was censored as the last recorded date the patient was known alive. Primary comparisons of the treatment group were made using the stratified log-rank; sensitivity analyses adjusted for prespecified baseline factors (performance status [0 vs. 1-2], age [<65 vs. ≥ 65 years]). Hazard ratios (HR) with 80% confidence intervals (CI) were calculated from stratified Cox regression models with treatment group as the single factor. Discrete variables were compared using Fisher’s exact test, and continuous ordinal categorical variables using the Wilcoxon test.
Circulating tumor cells were enumerated as previously described [16] at basline, week 6 and week 12 if patients were still on treatment (CELLSEARCH ®, Janssen Diagnostics, Raritan, NJ, USA). A favorable CTC status was defined as a CTC count < 5 CTC/7.5 mL.
Available archival tumour tissue also underwent mutational analysis as described in Supplemental Appendix 1.
RESULTS
Patient characteristics
Although protocol required accrual of 80 patients, eighty-five patients (41 on arm A and 44 on arm B; see Figure 1) were randomized from June 2012 to September 2015 at 11 centres because two patients on Arm B did not receive protocol treatment and 2 patients on Arm A and 1 on Arm B were found to be ineligible. All of these patients were followed for survival. Only patients who received protocol treatment were included in treatment exposure and safety analyses. Baseline characteristics are listed in Table 1. These were not significantly different between groups except for visceral disease status, LDH and alkaline phosphatase levels which were worse in Arm A. The prognostic model developed by Halabi et al. [18] was calculated, and the mean prognostic score between groups was respectively 145 and 129 on Arm A and Arm B (p=0.005).
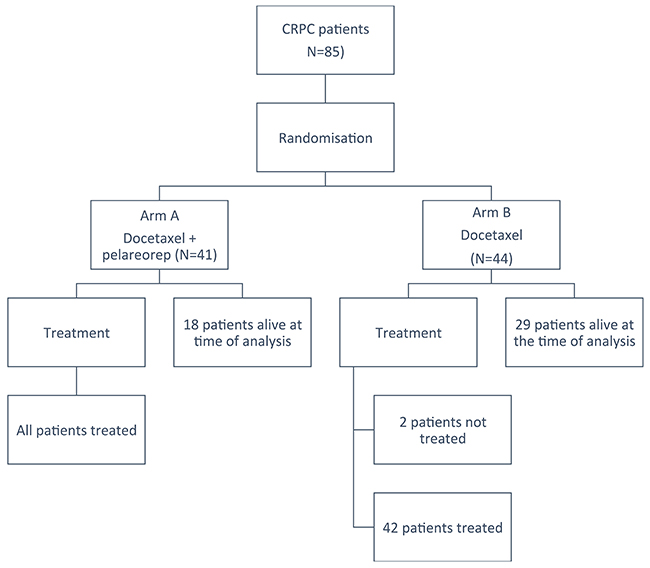
Figure 1: CONSORT diagram outlining subject disposition.
Table 1: Baseline patient demographic and clinical characteristics
Characteristic |
No. patients (%) |
|||
Arm A N=41 |
Arm B N=44 |
P-value |
||
Age, years |
Median |
69.1 |
68.6 |
0.64 |
Range |
50.3 – 83.7 |
49.7 – 86.6 |
||
ECOG Performance Status |
0 |
8 (19.5) |
18 (40.9) |
0.07 |
1 |
31 (75.6) |
25 (56.8) |
||
2 |
2 (4.9) |
1 (2.3) |
||
Gleason Score |
<8 |
13 (31.7) |
12 (26.2) |
0.92 |
8-10 |
26 (63.4) |
21 (68.9) |
||
unknown |
2 (4.9) |
1 (2.3) |
||
Measurable Disease |
No |
26 (63.4) |
21 (47.7) |
0.19 |
Yes |
15 (36.6) |
23 (52.3) |
||
Visceral disease |
No |
23 (56.1) |
32 (72.7) |
0.009 |
Yes |
18 (43.9) |
12 (27.3) |
||
Months from relapse to randomization |
Median |
38.1 |
58.7 |
0.18 |
Range |
3.7 – 148.2 |
7.0 – 126.7 |
||
Type of Progression |
Radiologic |
18 (43.9) |
17 (38.6) |
0.66 |
PSA alone |
23 (56.1) |
27 (61.4) |
||
Prior Therapies |
Androgen Ablation |
41 (100) |
44 (100) |
0.90 |
Abiraterone Acetate |
15 (36.6) |
13 (29.5) |
||
Enzalutamide |
9 (22.0) |
9 (20.5) |
||
Number of Disease Sites |
1 |
27 (65.9) |
18 (40.9) |
0.06 |
2 |
8 (19.5) |
18 (40.9) |
||
3 or more |
6 (14.6) |
8 (18.2) |
||
PSA (ng/mL) |
Mean (SD) |
189.7 (266.8) |
257.1 (473.1) |
0.92 |
LDH |
Mean (SD) |
268.8 (127.2) |
236.6 (97.7) |
0.04 |
≤ ULN |
16 (42.1) |
29 (67.4) |
0.03 |
|
>ULN |
22 (53.6) |
14 (32.6) |
||
Alkaline Phosphatase |
Mean (SD) |
389.4 (590.2) |
214.3 (301.1) |
0.01 |
≤ ULN |
14 (34.1) |
24 (54.5) |
0.08 |
|
>ULN |
27 (65.9) |
20 (45.5) |
||
Hemoglobin |
Mean (SD) |
12.4 (1.6) |
12.7 (1.5) |
0.32 |
≤ LLN |
10 (24.4) |
15 (34.1) |
0.35 |
|
>LLN |
31 (75.6) |
29 (65.9) |
||
Baseline CTC Result |
||||
Favorable (<5) |
17 (41.5) |
20 (45.5) |
0.67 |
|
Unfavorable (≥5) |
22 (53.7) |
20 (45.5) |
||
Missing |
2 (4.9) |
4 (9.1) |
||
Prognostic score |
Mean (SD) |
144 (141) |
129 (127) |
0.005 |
Treatments administered
More than half of patients received 7 cycles of treatment, with the median number of cycles being 7 for arm A (range 1 to 10) and 9 for arm B (range 1 to 13). The median cumulative docetaxel dose was lower in Arm A (457.4 mg/m2 vs 603 mg/m2). Thirty patients (73.2%) on Arm A had all administrations of pelareorep according to protocol, and 68.3% of patients received 90% or more of the planned dose. Patients in Arm A were more likely to discontinue docetaxel therapy prior to completion of all planned cycles (70.7 % vs 61.9%), predominantly because of disease progression. Docetaxel discontinuation due to toxicity was similar in both arms (11 vs 9 patients). More patients on Arm A required docetaxel dose modifications (56% vs 29%), with febrile neutropenia and fatigue being the major contributing factors.
Safety
Adverse events with an incidence of >10% are listed in Table 2. These were generally grade 1-2 and in keeping with known toxicities for these agents. The incidence of acute grade 3 or worse events found in 5% or more in one or both treatment arms included (Table 3): febrile neutropenia, diarrhea, nausea, fatigue, and peripheral neuropathy. The incidence of non-neutropenic fever in arm A was 46% (44% attributed to reovirus) vs. 7% in arm B. Of febrile neutropenic episodes, 10% were related to reovirus while 27% (arm A) and 31% (arm B) were related to docetaxel. No grade 5 adverse events were observed.
Table 2: Adverse events occurring in >10% of patients in one of arms
Adverse event |
Number of patients (%) with an event of any grade |
|
Arm A (n=41) |
Arm B (n=42) |
|
Febrile neutropenia |
11 (27) |
13 (31) |
Watering eyes |
8 (20) |
10 (24) |
Abdominal pain |
8 (20) |
3 (7) |
Constipation |
17 (41) |
19 (45) |
Diarrhea |
30 (73) |
19 (45) |
Dry mouth |
5 (13) |
3 (7) |
Dyspepsia |
8 (20) |
8 (19) |
Mucositis oral |
22 (54) |
21 (50) |
Nausea |
28 (68) |
21 (50) |
Vomiting |
18 (44) |
11 (26) |
Chills |
15 (37) |
3 (7) |
Edema limbs |
18 (44) |
19 (45) |
Fatigue |
40 (98) |
40 (95) |
Fever |
19 (46) |
5 (12) |
Flu-like symptoms |
12 (29) |
1 (2) |
Skin Infection |
4 (10) |
5 (12) |
Upper respiratory infection |
7 (17) |
5 (12) |
Bruising |
6 (15) |
6 (14) |
Anorexia |
27 (66) |
21 (50) |
Dehydration |
7 (17) |
2(5) |
Arthralgia |
15 (37) |
9 (21) |
Back pain |
16 (39) |
20 (48) |
Bone pain |
15 (37) |
12 (29) |
Generalized muscle weakness |
11 (27) |
6 (14) |
Muscle weakness lower limb |
8 (20) |
10 (24) |
Myalgia |
11 (27) |
11 (26) |
Pain in extremity |
18 (44) |
12 (29) |
Dizziness |
12 (29) |
6 (14) |
Dysgeusia |
21 (51) |
24 (57) |
Headache |
16 (39) |
7 (17) |
Peripheral sensory neuropathy |
18 (44) |
28 (67) |
Anxiety |
3 (7) |
5 (12) |
Insomnia |
17 (41) |
14 (33) |
Hematuria |
5 (12) |
3 (7) |
Urinary frequency |
23 (56) |
17 (40) |
Urinary incontinence |
5 (12) |
4 (10) |
Erectile dysfunction |
9 (22) |
10 (24) |
Cough |
10 (24) |
13 (31) |
Dyspnea |
19 (46) |
17 (40) |
Epistaxis |
4 (10) |
5 (12) |
Productive cough |
8 (20) |
3 (7) |
Sore throat |
3 (7) |
6 (14) |
Alopecia |
33 (80) |
30 (71) |
Dry skin |
4 (10) |
5 (12) |
Hyperhidrosis |
5 (12) |
4 (10) |
Nail discoloration |
6 (15) |
5 (12) |
Rash maculo-papular |
7 (17) |
1 (2) |
Hot flashes |
19 (46) |
19 (45) |
Hypertension |
5 (12) |
7 (17) |
Hypotension |
8 (20) |
3 (7) |
Table 3: Adverse events grade ≥3 in >5% of patients
Adverse event |
Number of patients (%) with grade ≥3 event |
|
Arm A N=41 |
Arm B N=42 |
|
Febrile neutropenia |
11 (27) |
13 (31) |
Diarrhea |
4 (10) |
0 (0) |
Nausea |
3 (7) |
0 (0) |
Fatigue |
8 (20) |
6 (14) |
Sepsis |
3 (7) |
0 (0) |
Peripheral sensory neuropathy |
2 (5) |
3 (7) |
Syncope |
4 (10) |
3 (7) |
Erectile dysfunction |
7 (17) |
6 (14) |
Hypertension |
2 (5) |
4 (10) |
Hypotension |
3 (7) |
3 (7) |
Thromboembolic event |
1 (2) |
3(7) |
Anemia |
6 (15) |
2 (5) |
Elevated Alkaline Phosphatase |
7 (17) |
3 (7) |
Efficacy
The 12-week LPD rate was 61% for arm A and 52.4% for arm B and this was not statistically significant (p=0.51). Thirty-five patients were evaluable for objective response; response rates were higher in arm B (26.7% vs 40%; adjusted OR 0.53; 95% CI 0.12 to 2.38, p=0.41). CTC enumeration revealed no significant differences or changes between arms Supplementary Table 1.
A total of 38 deaths (23 Arm A; 15 Arm B) were observed at the time of data cut-off. The median survival was respectively 19.1 months (95% CI 14.8 to 22.6 months) for patients on Arm A and 21.1 months (95% CI 18.8 to undefined) on Arm B (Figure 2). The hazard ratio for survival of Arm A to Arm B was 1.83 (95% CI 0.96 to 3.52; p=0.06). After adjusting for the potential prognostic factors prespecified in the analysis plan the adjusted hazard ratio of Arm A to Arm B was 1.86 (95% CI 0.97 to 3.58; p=0.06). In a sensitivity analysis adjusting for age and a prognostic score calculated based on baseline performance status, Gleason score, LDH level, alkaline phosphatase level, PSA level, hemoglobin level, and presence of visceral disease, the adjusted hazard ratio of Arm A to Arm B was 1.95 (95% CI 0.95 to 4.01; p=0.07). The median prognostic score was associated significantly with survival (adjusted hazard ratio of Arm A to Arm B was 7.55 with a 95% CI 1.09 to 52.2; p=0.04).
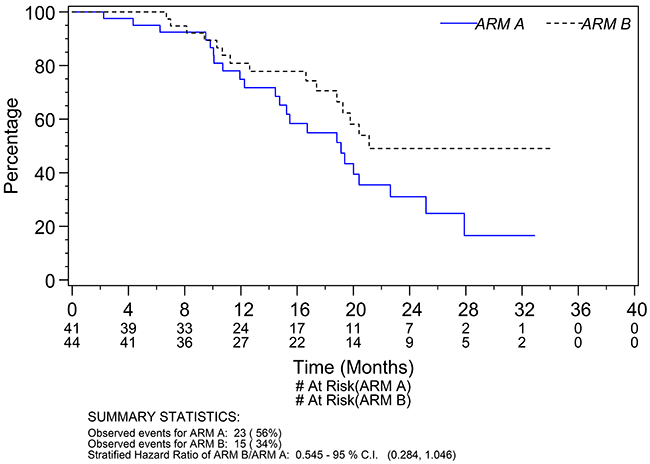
Figure 2: Overall survival.
DISCUSSION
The rationale for combining pelareorep with a taxane in CRPC was based upon the observations that: pelareorep has documented activity against prostate cancer cell lines [4], and in intratumoral injections in humans [10]; and that, in keeping with the proposed mechanism of viral replication occurring at microtubules [12, 13] consistent pelreorep synergism with microtubule stabilizing agents such as taxanes is observed in vitro [11].
Unfortunately, the results show that this combination has no evidence of activity worthy of further study. While OS was not a primary endpoint, note must be made of a trend toward worse survival in the combination arm. Several factors may have contributed to this. First, not unexpected in a small, albeit randomized, study there were some imbalances in baseline prognosic factors favoring arm B. It is unlikely that this minor imbalance would have overcome a promising signal of activity for arm A, and the strong trend to inferior survival remained after adjusted and sensitivity analyses. Second, this is the first study where pelareorep has been combined with daily steroids at potentially immunosuppressive doses. While there is some data that pelareorep may enhance immune-mediated anti-tumor effects [19], it has not been established whether this is the case for prostate cancer nor whether low-dose prednisone may have adversely affected any anti-tumor effect of pelareorep. Third, patients on arm A received fewer cycles and a lower dose intensity of docetaxel compared to arm B. The reason for this is not clear; discontinuation due to toxicity was similar in both arms (11 vs 9 patients), but more patients on Arm A required early docetaxel dose modifications due to toxicities. Certainly there are data supporting a decrease in outcomes for patients who received less than the standard dose intensity of chemotherapy [20]. While this might be ameliorated through dosing schedules avoiding virus exposure at neutropenic points in a cycle, the fact remains that the combination as tested was either not feasible to deliver, or not more efficacious than docetaxel. Finally, our study was a small RCT and was not powered to detect OS differences.
This study was one of four randomized phase II trials run by the CCTG that evaluated pelareorep in the metastatic setting (NCT01622543, NCT01708993, NCT01656538). In the two trials enrolling both genders, men appeared to fare worse than women, suggesting a possible impact of gender on outcomes [21], which may reflect on proposed gender based differences in immunity and response to immune-targeted therapies [22, 23]. Indeed there is an increasing body of evidence that oncolytic viruses are potent stimulators of immune activation, neo-antigen presentation, and PD-1/PD-L1 over-expression [24], Clinical trials evaluating pelareorep with checkpoint inhibitors are now planned or under way (NCT02620423).
In conclusion, while pelareorep given with docetaxel and prednisone is safe and tolerable in men with mCRPC this combination does not show activity worthy of further study in this setting.
CONFLICTS OF INTEREST
LS declares the receipt of institutional support for the conduct of this study from Oncolytics Biotech, Inc. All other authors declare that they have no conflicts of interest.
FUNDING
This work was supported by the Canadian Cancer Society Research Institute [grant numbers 021039, 704970] and Oncolytics Biotech Inc.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108. https://doi.org/10.3322/caac.21262.
2. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004; 351:1502–12. https://doi.org/10.1056/NEJMoa040720.
3. Rosen L, Evans HE, Spickard A. Reovirus infections in human volunteers. Am J Hyg. 1962; 77:29–37.
4. Coffey M, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998; 282:1332–4.
5. Hirasawa K, Nishikawa S, Norman KL, Alain T, Kossakowska A, Lee PW. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002; 62:1696–701.
6. Norman KL, Coffey M, Hirasawa K, Demetrick DJ, Nishikawa S, DiFrancesco LM, Strong JE, Lee PW. Reovirus oncolysis of human breast cancer. Hum Gene Ther. 2002.
7. Gujar SA, Clements D, Dielschneider R, Helson E, Marcato P, Lee PW. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br J Cancer. 2014; 110:83–93. https://doi.org/10.1038/bjc.2013.695.
8. Gujar SA, Marcato P, Pan D, Lee PW. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol Cancer Ther. 2010; 9:2924–33. https://doi.org/10.1158/1535-7163.MCT-10-0590.
9. Steele L, Errington F, Prestwich R, Ilett E, Harrington K, Pandha H, Coffey M, Selby P, Vile R, Melcher A. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer. 2011; 10:20. https://doi.org/10.1186/1476-4598-10-20.
10. Thirukkumaran CM, Nodwell MJ, Hirasawa K, Shi ZQ, Diaz R, Luider J, Johnston RN, Forsyth PA, Magliocco AM, Lee P, Nishikawa S, Donnelly B, Coffey M, et al. Oncolytic viral therapy for prostate cancer: efficacy of reovirus as a biological therapeutic. Cancer Res. 2010; 70:2435–44. https://doi.org/10.1158/0008-5472.can-09-2408.
11. Sei S, Mussio JK, Yang QE, Nagashima K, Parchment RE, Coffey MC, Shoemaker RH, Tomaszewski JE. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol Cancer. 2009; 8:47. https://doi.org/10.1186/1476-4598-8-47.
12. Broering TJ, Parker JS, Joyce PL, Kim J, Nibert ML. Mammalian reovirus nonstructural protein microNS forms large inclusions and colocalizes with reovirus microtubule-associated protein micro2 in transfected cells. J Virol. 2002; 76:8285–97.
13. Parker JS, Broering TJ, Kim J, Higgins DE, Nibert ML. Reovirus core protein mu2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J Virol. 2002; 76:4483–96.
14. Comins C, Spicer J, Protheroe A, Roulstone V, Twigger K, White CM, Vile R, Melcher A, Coffey MC, Mettinger KL, Nuovo G, Cohn DE, Phelps M, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010; 16:5564–72. https://doi.org/10.1158/1078-0432.ccr-10-1233.
15. Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008; 26:1148–59. https://doi.org/10.1200/JCO.2007.12.4487.
16. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008; 14:6302–9. https://doi.org/10.1158/1078-0432.ccr-08-0872.
17. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228–47. https://doi.org/10.1016/j.ejca.2008.10.026.
18. Halabi S, Lin CY, Kelly WK, Fizazi KS, Moul JW, Kaplan EB, Morris MJ, Small EJ. Updated prognostic model for predicting overall survival in first-line chemotherapy for patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2014; 32:671–7. https://doi.org/10.1200/JCO.2013.52.3696.
19. Gong J, Sachdev E, Mita AC, Mita MM. Clinical development of reovirus for cancer therapy: an oncolytic virus with immune-mediated antitumor activity. World J Methodol. 2016; 6:25–42. https://doi.org/10.5662/wjm.v6.i1.25.
20. Meisel A, Vogt D, de Wit R, de Bono J, Sartor O, Eisenberger M, Stenner-Liewen F. (2016). Neutropenia grade >3 during treatment with docetaxel (DOC) is associated with an improved overal survival (OS): a retrospective analysis of the TAX327 phase III trial. ESMO Congress. Copenhagen, Denmark.
21. Tang PA, Jonker DJ, Kennecke H, Welch SA, Cripps C, Asmis T, Chalchal H, Tomiak AT, Ko Y, Chen EX, Alcindor T, Goffin JR, Theis A, et al. IND210: a randomized phase II trial of FOLFOX/bevacizumab with or without pelareorep in metastatic colorectal cancer. Proc ASCO. 2016.
22. Dinesh RK, Hahn BH, Singh RP. PD-1, gender, and autoimmunity. Autoimmun Rev. 2010; 9:583–7. https://doi.org/10.1016/j.autrev.2010.04.003.
23. Dronca RS, Dong H. A gender factor in shaping T-cell immunity to melanoma. Front Oncol. 2015; 5:8. https://doi.org/10.3389/fonc.2015.00008.
24. Rajani K, Parrish C, Kottke T, Thompson J, Zaidi S, Ilett L, Shim KG, Diaz RM, Pandha H, Harrington K, Coffey M, Melcher A, Vile R. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther. 2015. https://doi.org/10.1038/mt.2015.156.