INTRODUCTION
Lung cancer is a leading cause of cancer-related deaths worldwide, and it ranks first in China [1, 2]. Despite novel therapeutic strategies and anticancer agents for lung cancer treatment, the clinical efficacy of these strategies is not optimal and high mortality and poor patient quality of life are problems that remain [3–5]. Thus, we need novel lung cancer drugs.
Mitophagy, an autophagic process that selectively removes damaged or excessive mitochondria via ubiquitination degradation in autophagolysosomes, is critical for maintaining the mitochondrial population and cellular homeostasis [6–8]. Defective mitophagic regulation results in abnormal cellular function due to the accumulation of damaged mitochondria, leading to many pathophysiological states [9–11]. Data show that the deregulation of mitophagy may contribute to cancer progression and cell survival in various cancers [12, 13]. Also, the inhibition of mitophagy enhances chemosensitivity in cancer cells [14].
Toxicarioside H (ToxH) is extracted from the seeds of the tropical medicinal plant Antiaris toxicaria (Pers.) Lesch (Moraceae) [15]. Structural analysis by MS and NMR spectral data and chemical evidence show that ToxH is a cardiac glycoside with a special cardenolide structure (Figure 1A) [15]. Recent research results indicate that cardenolides extracted from natural sources can have anticancer activity and can block tumor cell proliferation and induce tumor apoptosis via the regulation of several cell signaling pathways and sodium pump inhibition [16–19], and even through the induction of immunogenic cell death [20–22]. At this time, the anticancer activity of cardenolides has not been linked to mitophagy.
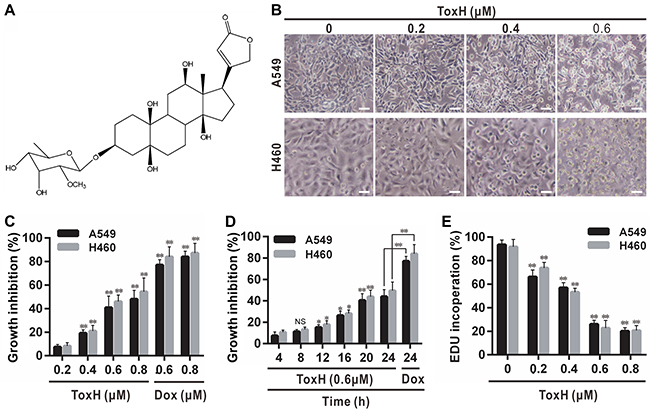
Figure 1: ToxH inhibits growth and proliferation of lung cancer cells. (A) Structure of ToxH. (B) A549 and H460 cells treated as indicated for 24 h observed microscopically. (C and D) Cells treated as indicated with ToxH or doxorubicin (Dox, as the positive control) for 24 h (C) or with ToxH (0.6 μM), and growth inhibition (% compared to the controls) was measured. (E) Cells were treated as indicated with (B) and an EDU proliferation assay was used. Data are means ± SD of three independent experiments. **p < 0.001 vs the left non-marked group. Bar = 10 μm. ToxH (0 μM), vehicle (DMSO) control.
Because ToxH is cytotoxic, it may cause stress or mitochondrial damage as do chemotherapeutics [7, 23]. Therefore, it is reasonable to consider that mitophagic machinery may be responsible for anticancer activity induced by ToxH. Here, we report that ToxH induced apoptosis and mitophagy and this was associated with the high expression of sirtuin-3 (Sirt3) and inhibition of Sirt3 expression by siRNA increased apoptosis in ToxH-treated lung cancer cells, indicating an apoptosis-resistant role of mitophagy induced by ToxH. Our findings indicate that ToxH and a mitophagy inhibitor may offer a novel approach to treat lung cancer.
RESULTS
Toxicarioside H (ToxH) inhibits growth and proliferation of lung cancer cells
After treatment of A549 and H460 cells with ToxH for 24 h, the cells stopped growing and some dead cells floated to the top of the medium. Apoptotic cells increased in a dose-dependent manner (Figure 1B). Also, cell viability decreased in a dose- and time-dependent manner (Figure 1C and 1D). Cytotoxicity was confirmed with EDU assays (Figure 1E). Thus, ToxH can inhibit growth and proliferation of lung cancer cells, and 0.6 μM was the optimal dose (Figure 1B-1E) so it was chosen for subsequent experiments.
ToxH induces mitochondrion-mediated apoptosis in lung cancer cells
Figure 2A and 2B show that the apoptotic cells increased with increasing dose of ToxH but ~38% cells were apoptotic after ToxH treatment for 24 h at 0.6 μM (Figure 2B). JC-1 stain was used to measured changes in mitochondrial transmembrane potential. Figure 2C shows that compared to the controls, ToxH-treated cells had green fluorescence (Figure 2C) and the ratio of green fluorescence intensity significantly increased or red fluorescence decreased in a dose-dependent manner (Figure 2D and 2E), suggesting that ToxH induced a significant decrease in mitochondrial membrane potential in lung cancer cells. Moreover, marker proteins of mitochondrion-mediated apoptosis, such as Cyt c, PARP, and caspase-3 and -9, were measured with Western blot. Data show that Cyt c increased in the cytoplasm and decreased in the mitochondria after ToxH treatment for 12 h (Figure 2F), suggesting that ToxH caused the release of Cyt c into the cytoplasm from the mitochondria. In addition, increased activation in PARP and caspase-3 and -9 was observed at 12 h after ToxH (0.6 μM) treatment (Figure 2F). However, caspase-8 (a death receptor pathway marker), was not influenced by ToxH treatment (Figure 2F). Thus, ToxH induced apoptosis in lung cancer cells via the mitochondrion-mediated pathway.
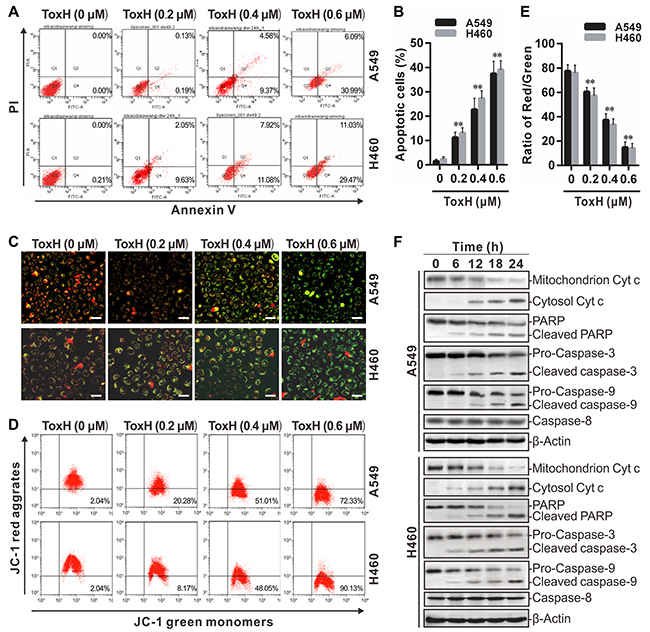
Figure 2: ToxH induces mitochondrion-dependent apoptosis in lung cancer cells. (A and B) A549 and H460 cells were treated as indicated and Annexin V and PI staining was used after flow cytometry. (C) Cells treated as in (A) were stained with JC-1 and observed under a fluorescent microscope. (D and E) JC-1-stained cells were quantified using flow cytometry. (F) Cells were treated with ToxH (0.6 μM) at the indicated times and mitochondrion-dependent apoptotic proteins were assayed using Western blotting. Data are means ± SD of three independent experiments, **p < 0.001 vs non-ToxH-treated (0 μM, DMSO control) group. Bar = 10 μm.
ToxH induces mitophagy in lung cancer cells
A549 and H460 lung cancer cells were treated with (0.6 μM) or without (UT, 0 μM, DMSO control) ToxH for 24 h and mitochondrial proteins (Tom20 for the outer membrane; TIM23 for the inner membrane and Hsp60 for mitochondrial matrix) were assessed with Western blotting. Data show that Tom20, TIM23, and Hsp60 were less abundant in the ToxH-treated lung cancer cells compared to the controls (Figure 3A-3D) Mitophagy was measured using co-localization of autophagosomes with mitochondria and the ratio of punctate green fluorescent protein (GFP-LC3) localized with mitochondria was significantly higher in the lung cancer cells treated with ToxH compared to the controls (Figure 3E-3G). We assessed the recruitment of p62 (an autophagic/mitophagic receptor) to damaged mitochondria after ToxH treatment by assaying the co-localization of p62 with damaged mitochondria (Mito-Red). Figure 3H and 3I show that p62 displayed diffuse cytoplasmic distribution in non-ToxH-treated (UT) cells, but p62-containing aggregates were either co-localized with or adjacent to the mitochondria in the ToxH-treated cells. Cell fractionation data confirmed these results. In UT cells, little p62 and LC3 II co-sedimented with the mitochondria, but more p62 and LC3 II were present in the mitochondrial fraction in the ToxH-treated cells (Figure 3J). Thus, ToxH treatment caused mitophagy in lung cancer cells.
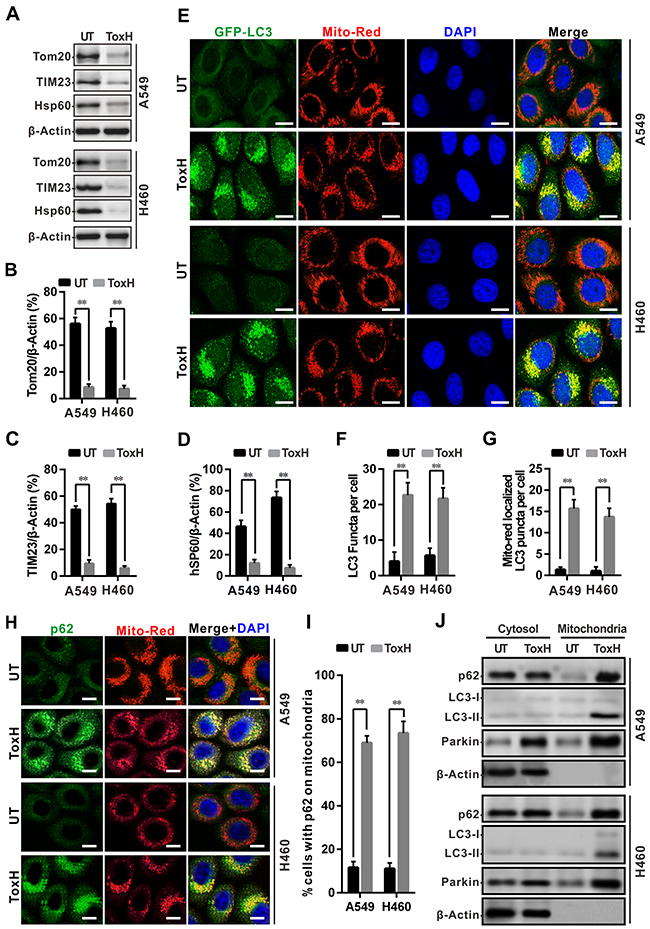
Figure 3: ToxH induces mitophagy in lung cancer cells. (A) A549 and H460 cells were treated with (0.6 μM) or without (UT, DMSO control) ToxH, and the indicated mitochondrial proteins were measured with Western blot. (B-D) Band density from (A) was analyzed with ImageJ software and normalized to β-actin. (E) Cells were transfected with GFP-LC3 plasmids and treated with (0.6 μM) or without (UT, DMSO control) ToxH for 24 h, followed by staining with Mito-Red and DAPI. Co-localization of GFP-LC3 with mitochondria (Mito-Red) observed under fluorescent microscopy. (F and G) Quantification of GFP-LC3 puncta (F) and co-localization with mitochondria/cell (G) in C. (H) Cells were treated with (0.6 μM) or without (UT, DMSO control) ToxH for 24 h, and then stained with antibody against p62 (FITC-conjugated), Mito-Red, and DAPI. Co-localization of p62 (green) with mitochondria (red) was observed under fluorescent microscopy. (I) Quantification of co-localization of p62 with mitochondria/cells in H. (J) Cells were treated with/without (UT, DMSO control) ToxH for 24 h, and cytosol and mitochondria proteins were measured with Western blotting, respectively. Data are means ± SD of three independent experiments. **p < 0.001. Bar = 5 μm.
ToxH induces mitophagy by up-regulation of sirt-3 expression in lung cancer cells
Sirt3 was assayed with respect to ToxH-induced mitophagy by Western blotting. Data show that Sirt3 was expressed in ToxH-treated lung cancer cells, and that the decreased expression of Tom20 occurred (Figure 4A-4B). To confirm that the high expression of Sirt3 was involved in ToxH effects, we transfected lung cancer cells with siRNA against Sirt3 (siSirt3) and treated with/without ToxH for 24 h and measured Sirt3 and Tom20 with Western blotting. Figure 4C-4E shows that interference with siSirt3 significantly decreased the expression of Sirt3, and ToxH treatment no longer induced the expression of Sirt3 in the A549 and H460 cells (Figure 4C and 4D). Tom20 was not influenced by ToxH treatment when the lung cancer cells were treated with siSirt3 (Figure 4C and 4E). In addition, we studied the co-localization of GFP-LC3 puncta with mitochondria (Mito-Red) in lung cancer cells after siSirt3 treatment. Figure 4F-4G shows that control siRNA (siCtrl) did not influence mitophagy induced by ToxH treatment, but siSirt3 decreased mitophagy in ToxH-treated lung cancer cells. Moreover, cell fractionation after interference with siParkin and inhibition with 3-MA was used to measure the mitophagy/autophagy markers p62 and LC3 II. More p62 and LC3 II were present in the mitochondrial fraction in cells cotreated with siCtrl and ToxH, respectively, but little p62 and LC3 II co-sedimented with the mitochondria in the cells transfected with siParkin or cells co-treated with ToxH and 3-MA (Figure 4I and 5A). Thus, ToxH induces mitophagy by the up-regulation of Sirt3 expression in lung cancer cells.
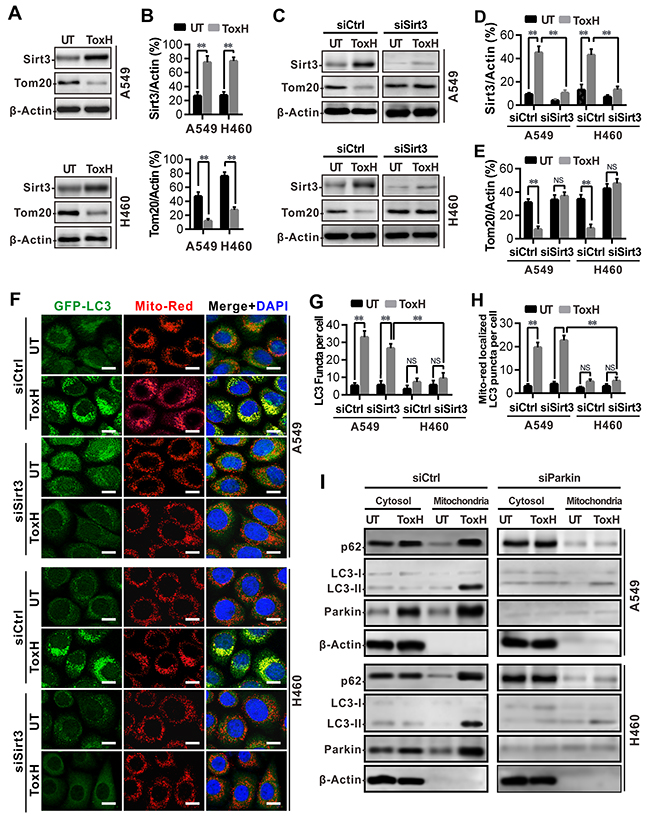
Figure 4: ToxH induces mitophagy by up-regulation of Sirt3 expression in lung cancer cells. (A) A549 and H460 cells were treated with (0.6 μM) or without (UT) ToxH, Sirt3, and Tom20 were measured with Western blotting. (B) Band density (Sirt3 and Tom20) from (A) was analyzed with ImageJ software and normalized to β-actin. (C) A549 and H460 cells treated with (0.6 μM) or without (UT) ToxH were interfered with (siSirt3) or without (siCtrl) siRNA, Sirt3, and Tom20 were then quantified with Western blotting. (D and E) Band density (Sirt3 and Tom20) from (C) was analyzed with ImageJ software and normalized to β-actin. (F) Cells were treated as indicated (transfected with GFP-LC3 plasmids, interfered with (siSirt3) or without (siCtrl) siRNA, and treated with (0.6 μM) or without (UT) ToxH) for 24 h, followed by staining with Mito-Red and DAPI. Co-localization of GFP-LC3 with mitochondria (Mito-Red) observed under fluorescent microscopy. (G and H) Quantification of GFP-LC3 puncta (G) and their co-localized with mitochondria (H) /cells in (F). (I) Cells were interfered with siCtrl or siSirt3 and co-treated with/without (UT, DMSO control) ToxH, ctytosol, and mitochondria proteins were measured with Western blotting. Data are means ± SD in three independent experiments. **p < 0.001. Bar = 5 μm.
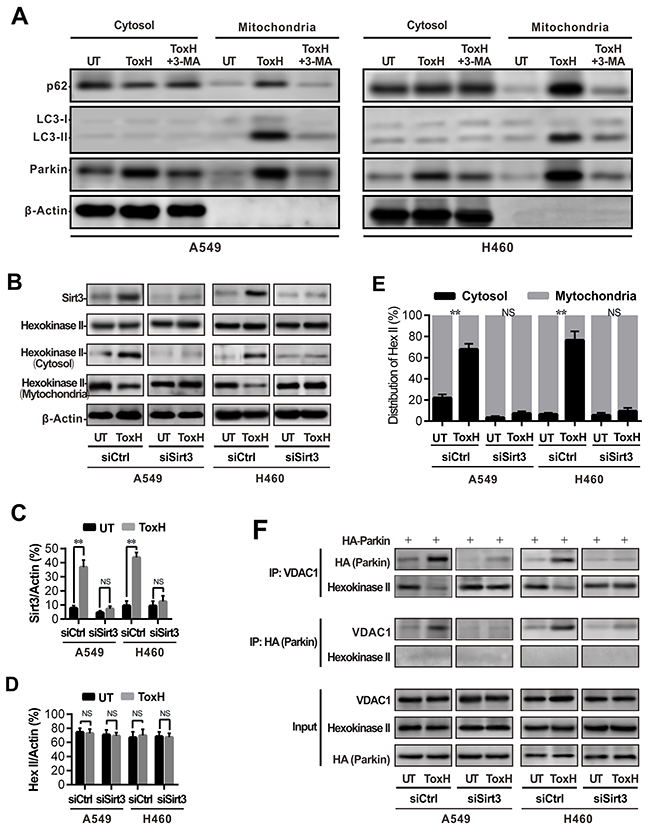
Figure 5: Sirt3 induces mitophagy by promoting association of parkin with VDAC1. (A) Cells were treated with/without (UT, DMSO control) ToxH or cotreated with ToxH and 3-MA, ctytosol and mitochondria proteins were measured with Western blotting. (B) A549 and H460 cells were transfected with a siRNA against Sirt3 (siSirt3) or control siRNA (siCtrl), and treated with/without (UT) ToxH for 24 h. Sirt3 and total hexokinase II, including mitochondrial and cystol hexokinase II, were measured with Western blotting. (C and D) Sirt3 and hexokinase II (Hex) band density in (A) were analyzed with ImageJ software and normalized to β-actin. (E) Cytosol (black) and mitochondrial hexokinase II (Hex II) band density in (A) were analyzed with ImageJ software and normalized to β-actin, and calculated as 100%. (F) A549 and H460 cells were transfected with HA-parkin plasmid and treated as in (B) for 24 h, and cell lysates were immunoprecipitated with antibodies against VDAC1 or HA. Immunoprecipitates were assayed with Western blotting with antibodies against HA, hexokinase II, and VDAC1, respectively. Data were expressed as mean ± SD in three independent experiments. **p<0.001.
Sirt3 activates mitophagy by enhancing the association of parkin with VDAC1
We investigated the relationship among Sirt3, hexokinase II, parkin, and VDAC1. Western blot analysis showed that ToxH treatment increased Sirt3 expression, but not the expression of hexokinase II protein (Figure 5B-5D); RNA interference of Sirt3 with siSirt3 blocked the expression of Sirt3, but it had no effect on the expression of the hexokinase II protein (Figure 5B-5D). However, ToxH treatment led to the redistribution of hexokinase II from the mitochondria to the cytosol, but redistribution of hexokinase II from the mitochondria to the cytosol disappeared with siSirt3 (Figure 5B and 5E). Co-immunoprecipitation assays confirmed that the binding of VDAC1 with hexokinase II occurred in the lung cancer cells that were not treated with ToxH, but ToxH treatment led to the disassociation of VDAC1 with hexokinase II and resulted in the binding of VDAC1 with parkin (Figure 5F). Figure 5F shows that the binding of VDAC1 with parkin was related to the ToxH-induced high expression of Sirt3, as evidenced by low-level binding of VDAC1 with parkin after treatment with siSirt3 in ToxH-treated lung cancer cells (Figure 5F). Thus, Sirt3 activates mitophagy by decreasing the interactions of hexokinase II with VDAC1 but increasing the interactions of parkin with VDAC1.
Interruption of Sirt3-induced mitophagy promotes apoptosis in ToxH-treated lung cancer cells
Figure 4C-4H shows that the interference of Sirt3 with siSirt3 significantly suppressed Sirt3 expression and blocked the formation of ToxH-induced mitophagy in A549 and H460 lung cancer cells. Next, Figure 6A shows that the inhibition of ToxH-induced mitophagy by siSirt3 interference more significantly suppressed proliferation in lung cancer cells. Similar results were found with an EDU incorporation assay; that inhibition of autophagy by siSirt3 could sensitize lung cancer cells to ToxH treatment, leading to at least a one-fold decrease in the inhibition of cell proliferation in lung cancer cells (Figure 6B). We then measured apoptotic cells using Annexin-V/PI double staining followed by flow cytometry, and data show that apoptosis increased in ToxH-treated lung cancer cells when ToxH-induced mitophagy was blocked with siSirt3 interference (Figure 6C and 6D). Moreover, proteins of mitochondrion-mediated apoptosis, PARP, and caspase-3, were measured with Western blotting and data show increased activation (cleaved) of PARP and caspase-3 observed in the ToxH-treated lung cancer cells treated with siSirt3 (Figure 6E). In addition, ToxH-induced mitophagy was blocked with 3-MA and data were similar to siSirt3. Consistently, the inhibition of ToxH-induced mitophagy by 3-MA increased the inhibition of cell growth and proliferation according to Figure 6F and EDU assays in Figure 6G, respectively. Flow cytometry showed that apoptosis was increased in lung cancer cells co-treated with ToxH and 3-MA (Figure 6H and 6I). Moreover, marker proteins of mitochondrion-mediated apoptosis had increased activation (cleaved) in cells co-treated with ToxH and 3-MA (Figure 6J). Thus, ToxH-induced mitophagy plays a drug-resistant role against ToxH-induced apoptosis in lung cancer cells.
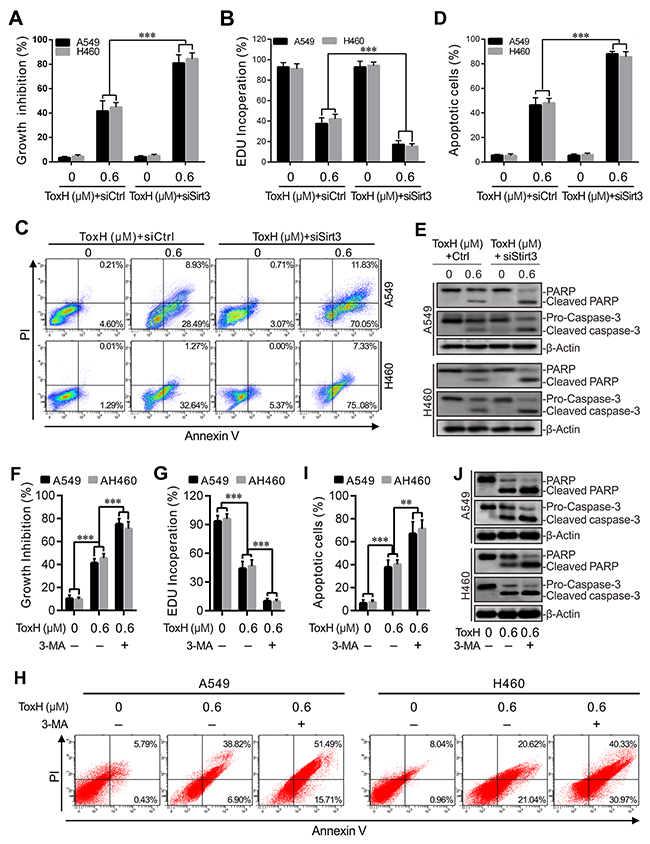
Figure 6: Interruption of Sirt3-induced mitophagy promotes apoptosis in ToxH-treated lung cancer cells. (A) A549 and H460 cells were treated with/without ToxH (0.6 μM) for 24 h, and with/without siRNA against Sirt3. MTT assay was used to measure growth inhibition. (B) Cells were treated as in (A), and cell proliferation was measured using EDU assay. (C and D) Cells were treated as in (A), and apoptosis was measured using PI and Annexin V double staining and flow cytometry. (E) Cells were treated as in (A), and mitochondrion-dependent apoptotic proteins were quantified using Western blotting. (F) Cells were treated with/without ToxH (0.6 μM) or cotreated with 3-MA for 24 h, and MTT assay data are depicted. (G) Cells were treated as in (F), and cell proliferation was measured (EDU). (H and I) Cells were treated as in (F), and apoptosis was measured as before with double staining and flow cytometry. (J) Cells were treated as in (F), and mitochondrion-dependent apoptotic proteins were quantified using Western blotting. Data are means ± SD in three independent experiments. **p < 0.001 vs non-ToxH-treated (UT) group.
DISCUSSION
Natural products derived from plants, such as cardenolides, flavonoids, and terpenes, have diverse pharmacological properties, and they may potentially be used to treat cancer [24–26]. ToxH was extracted from the seeds of Antiaris toxicaria (Pers.) Lesch (Moraceae) [15] and structural analysis with MS and NMR confirmed that ToxH is a cardiac glycoside with a special cardenolide structure (Figure 1A). Recent studies suggest that cardenolides derive anticancer activities via the induction of cancer apoptosis [16–18]. Since ToxH is a new cardenolide structure, its cytotoxicity was confirmed using an MTT assay [15]. We also found that ToxH inhibited cell growth and proliferation, and induced mitophagy and apoptosis in A549 and H460 lung cancer cells. ToxH treatment of A549 and H460 lung cancer cells increased the expression of Sirt3, and inhibited Sirt3 expression with siSirt3 blocked mitophagy and caused more apoptosis. Thus, ToxH activates drug-resistant mitophagy in lung cancer cells. ToxH-mediated drug-resistant mitophagy was caused by the expression of Sirt3 by ToxH treatment, and the high expression of Sirt3 increased the binding of parkin with VDAC1.
Mitophagy is necessary for the maintenance of a functioning mitochondrial network and the prevention of the accumulation of damaging mitochondrial DNA mutations and reactive oxygen species [27]. The parkin pathway has been thought to be a major initiator of mitophagy [28–30]. Parkin, an E3 ubiquitin ligase, can ubiquitinate numerous mitochondrial substrates (including VDAC1) and enroll them to commit mitochondria to mitophagic turnover [28–30]. Here, we measured whether ToxH inhibited cancer cell growth (proliferation) and the induction of apoptosis and mitophagy, and evaluated their relationship. ToxH mediated cell-growth inhibition, apoptosis, and mitophagy in A549 and H460 lung cancer cells but only ~38% of the ToxH-treated cells become apoptotic, which is consistent with the inhibition of proliferation induced by ToxH treatment (Figure 1C-1E). Thus, ToxH may not be a suitable chemotherapeutic unless its antitumor activity can be improved. Previous studies suggest that the induction of apoptosis is a chief form of cell death from which cardenolides derive anticancer activity [31–33]. Thus, we blocked ToxH-mediated mitophagy with siSirt3 and 3-MA, respectively, and the data show that when ToxH-mediated mitophagy is interrupted, enhanced inhibition of cell growth and significantly increased apoptosis occurred (Figure 6). Thus ToxH-induced mitophagy may be cytoprotective to allow lung cells to combat ToxH-induced apoptosis and inhibit cell growth and proliferation. Since silencing Sirt3 or 3-MA treatment significantly halted ToxH-induced mitophagy and enhanced sensitivity to ToxH in lung cancer cells, we concluded that this mitophagy has a drug-resistant role in A549 and H460 lung cancer cells. Therefore, ToxH plus a mitophagic inhibitor may be a feasible strategy for treating lung cancer but the complicated intrinsic relationship between mitophagy and apoptosis induced by ToxH is not clear and warrants further studies.
Recent studies have linked parkin, Sirt3, VDAC1, and hexokinase II with drug-resistant mitophagy [34]. Sirt3 is a sirtuin and nicotinamide adenine dinucleotide protein deacetylase that mainly localizes to the mitochondria and controls acetylation of mitochondrial proteins [34, 35]. Data show that Sirt3 is implicated in the regulation of numerous cellular processes, including the stress response, aging, and energy metabolism, mainly through its effect on protein acetylation [36–38]. Sirt3 can protect cells against various types of stress-induced death by maintaining mitochondrial integrity or by mitophagy [34, 39]. VDAC1 is an outer mitochondrial membrane protein, and it acts as a mitochondrial binding site for hexokinase II [40]. VDAC1 can promote hexokinase II binding to mitochondria, which contributes to cancer cell metabolism, and inhibits damage to the mitochondria [40]. In addition, recent studies report that VDAC1 induces mitophagy via recruiting parkin to defective mitochondria to promote their degradation [34, 41, 42]. Sirt3 expression is increased by ToxH treatment, and the inhibition of Sirt3 with siSirt3 significantly suppressed the induction of mitophagy triggered by ToxH treatment. Moreover, ToxH treatment redistributes hexokinase II from the mitochondria to the cytosol in A549 and H460 lung cancer cells, and it decreases the interactions of hexokinase II with VDAC1, which is accompanied by increased interactions of VDAC1 with parkin in lung cancer cells. Also, the disruption of Sirt3 by siSirt3 decreases the association of VDAC1 and parkin. These data suggest that ToxH induces mitophagy by enhancing the expression of Sirt3, leading to the dissociation of hexokinase II from VDAC1, which increased the binding of VDAC1 with parkin.
Thus, ToxH is a potential treatment for lung cancer as it induces drug-resistant mitophagy by increasing Sirt3 expression, leading to the release of hexokinase II from the mitochondria into the cytosol. Increased hexokinase II in the cytosol results in VDAC1 (a parkin substrate) binding to parkin. Data suggest that a combination therapy of ToxH and an mitophagic inhibitor may be a potential alternative approach for treating lung cancer.
MATERIALS AND METHODS
Reagents and antibodies
ToxH was isolated and purified by our group [15]. Purity was > 95% according to chromatographic analysis. Other reagents used in this study included: MTT (Sigma, M2128) and DMSO (Sigma, D2650). The following antibodies were purchased: β-actin (Santa Cruz, sc-1616), caspase 3, caspase 8, and caspase 9 (Millipore, MAB4703), PARP (Millipore, 04-575), and HA-tag (Santa Cruz, sc-53516). In addition, antibodies to Tom20, Tim23, Hsp60, Sirt3, hexokinase II, and VDAC1, were purchased from Sigma. All of the cell culture media were purchased from Invitrogen (Gibco). Western blot reagents were obtained from Sigma.
Cell lines and cell culture
The human lung cancer cell lines A549 (CRM-CCL-185) and H460 (HTB-177) were obtained from ATCC. All of the cells were cultured in RPMI 1640 or DMEM (Gibco), supplemented with 10% fetal bovine serum (Gibco, 100 U/ml penicillin (Sigma), and 100 μg/ml streptomycin (Sigma, P0781) at 37°C in an atmosphere containing 5% CO2.
Cell viability (MTT) assay
The cells were plated in 96-well plates and incubated overnight, and then the cells were exposed to the test compound for 24 h. Subsequently, the cells were evaluated using an MTT assay. Briefly, 10 μl of MTT was added to each well and incubated for 4 h. The medium was removed, and 150 μl of DMSO was added to each well to dissolve crystal formazan dye. Absorbance was measured at 570 nm using an ELISA reader (ELX808IU, Bio-Tek). In this study, ToxH (0 μM) and UT (untreated) cells were treated with DMSO unless otherwise indicated.
EDU proliferation assay
Cells were seeded in 96-well plates and exposed to the test compound under standard conditions in complete media for 24 h. Cell proliferation was assayed using EDU and a Cell Proliferation Assay Kit (Ribobio, Guangzhou, China). Briefly, the cells were incubated with 50 mM EDU for 6 h before fixation, permeabilization, and EDU staining, which were performed according to the kit’s instructions. Cell nuclei were stained with DAPI (Sigma) at 1 mg/ml for 20 min. The proportion of the cells that incorporated EDU was measured with fluorescent microscopy (Nikon, 80i, Japan).
Preparation of cytosolic and mitochondrial extracts
Cytoplasmic and mitochondrial proteins were collected using a cell mitochondrial kit (Beyotime, China) according to the kit’s instructions. In brief, harvested pellets were incubated in cell lysis buffer (250 mM of sucrose; 1 mM of DTT; 10 mM of KCl; 1 mM of EDTA; 1 mM of EGTA; 1.5 mM of MgCl2; phenylmethyl-sulfonyl fluoride; and 20 mM of HEPES, pH 7.4) at 4°C and homogenized with a glass homogenizer. Cell lysate was centrifuged at 800 x g for 10 min to remove any unbroken cells, and the supernatant was centrifuged at 15,000 x g for 10 min at 4°C. The resulting supernatant contained the cytoplasmic fraction and the pellet contained the mitochondrial fraction. The mitochondrial pellet was then resuspended in a mitochondrial lysis buffer at 4°C, and both the resuspended mitochondrial pellet solution and the supernatant were centrifuged at 24,000 x g at 4°C to remove the nuclei. Protein in the cytoplasm and mitochondria was quantified using the Bradford method with BSA as a standard.
Mitochondrial membrane potentials assay
JC-1 probe was used to measure mitochondrial depolarization in lung cancer cells. Six-well plates were used to culture cells treated with ToxH or DMSO. Cells were incubated with JC-1 staining solution (5 μg/mL) at 37°C for 20 min and washed twice with PBS. Cells were observed by a Nikon (80i, Tokyo, Japan) fluorescent microscope at an argon ion wavelength of 488 nm laser excitation.
Flow cytometry
Treated cells were incubated with JC-1 working solution for 20 min in 5% CO2 humidity at 37°C, washed twice with PBS, and resuspended in 500 μL PBS. Then, flow cytometry (FACS Canto II, BD Biosciences) was used to measure red/green fluorescence and FlowJo Software (BD Biosciences) was used to analyze the data. Cells were cultured and treated for 24 h and harvested by trypsinization and washed with PBS. Cells were resuspended and incubated in PI/Annexin-V solution (KeyGEN Biotech, KGA106) for apoptosis analysis. At least 10,000 live cells were analyzed with BD flow cytometry and software.
Western blotting
Cells were lysed with RIPA buffer (50 mM Tris, 1.0 mM EDTA, 150 mM NaCl, 0.1% Triton X-100, 1% sodium deoxycholate, and 1 mM PMSF). Protein was measured using a BCA protein assay kit (Pierce, 23225). Proteins were resolved on 12% SDS-PAGE, and then transferred to PVDF (BioRad) membranes. After blocking, the membranes were incubated with primary antibodies at 4°C overnight, and incubated with secondary antibodies at room temperature for 1 h. Target proteins were examined using ECL reagents (Millipore, WBKLS0100). Semi-quantitative analysis was performed by measuring band density using ImageJ software (V6, NIH).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 30 min, washed with PBS, and then incubated with 0.1% Triton X-100 for permeabilization. Cells were stained with anti-LC3B polyclonal antibody at 4°C overnight, and subsequently with Alexa Fluor 488-conjugated goat anti-rabbit LgG (Abcam, ab150077) at 37°C for 1 h. Nuclei were stained with DAPI for 5 min. Images were captured using confocal laser scanning microscopy (Olympus, FV3000).
Small RNA interference and plasmid transfection
siRNA targeting Sirt3 (siSirt3) and its Scrambled non-targeting siRNA (siCtrl, as a control) were purchased from Santa Cruz Biotechnology. GFP-LC3 and HA-parkin plasmids were presents from Professor Canhau Huang (Sichuan University, China). siRNA and plasmids were introduced into cells using Lipofectamine 2000 according to the manufacturer’s instructions.
Co-immunoprecipitation
Cells were lysed with RIPA buffer supplemented with protease inhibitor cocktail. Whole cell lysates obtained by centrifugation were incubated with 1–4 μg of the indicated antibodies overnight at 4°C, followed by incubation for 2 h at 4°C with protein G-Sepharose beads (40 μL, GE Healthcare). The immune complexes were washed 3 times with RIPA buffer and then analyzed by immunoblotting with the indicated antibodies.
Statistical analysis
Statistical analysis was performed using Prism 6. Statistical differences were determined by a nonparametric 2-tailed Student’s t test, and they are expressed as means ± SD. Data were deemed to be statistically significant if p < 0.05. Error bars indicate SD unless otherwise indicated.
CONFLICTS OF INTEREST
There is no any competing interest.
FUNDING
This work was funded by the National Natural Science Foundation of China (81460557, 81560599, 81460020 and 81360482) and Jiangsu Province Science Foundation for Youths, China (BK2012101).
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–E386.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66:115–132.
3. Iacono D, Chiari R, Metro G, Bennati C, Bellezza G, Cenci M, Ricciuti B, Sidoni A, Baglivo S, Minotti V, Crino L. Future options for ALK-positive non-small cell lung cancer. Lung Cancer. 2015; 87:211–219.
4. Qiu YF, Liu ZG, Yang WJ, Zhao Y, Tang J, Tang WZ, Jin Y, Li F, Zhong R, Wang H. Research progress in the treatment of small cell lung cancer. J Cancer. 2017; 8:29–38.
5. Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. 2015; 12:511–526.
6. Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010; 6:871–878.
7. Yan C, Luo L, Guo CY, Goto S, Urata Y, Shao JH, Li TS. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017; 388:34–42.
8. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011; 12:9–14.
9. Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012; 393:547–564.
10. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010; 12:119–131.
11. Jin S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy. 2006; 2:80–84.
12. Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy. 2013; 9:1720–1736.
13. Maes H, Agostinis P. Autophagy and mitophagy interplay in melanoma progression. Mitochondrion. 2014; 19:58–68.
14. Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, Huang C, Li P, Gao N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy. 2015; 11:1259–1279.
15. Dai HF, Gan YJ, Que DM, Wu J, Wen ZC, Mei WL. A new cytotoxic 19-nor-cardenolide from the latex of Antiaris toxicaria. Molecules. 2009; 14:3694–3699.
16. Roberts DM, Gallapatthy G, Dunuwille A, Chan BS. Pharmacological treatment of cardiac glycoside poisoning. Br J Clin Pharmacol. 2016; 81:488–495.
17. Kaushik V, Azad N, Yakisich JS, Iyer AK. Antitumor effects of naturally occurring cardiac glycosides convallatoxin and peruvoside on human ER+ and triple-negative breast cancers. Cell Death Discov. 2017; 3:17009.
18. Karasneh RA, Murray LJ, Cardwell CR. Cardiac glycosides and breast cancer risk: a systematic review and meta-analysis of observational studies. Int J Cancer. 2017; 140:1035–1041.
19. Dietze R, Konrad L, Shihan M, Kirch U, Scheiner-Bobis G. Cardiac glycoside ouabain induces activation of ATF-1 and StAR expression by interacting with the alpha4 isoform of the sodium pump in Sertoli cells. Biochim Biophys Acta. 2013; 1833:511–519.
20. Diederich M, Muller F, Cerella C. Cardiac glycosides: from molecular targets to immunogenic cell death. Biochem Pharmacol. 2017; 125:1–11.
21. Menger L, Vacchelli E, Kepp O, Eggermont A, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: cardiac glycosides and cancer therapy. Oncoimmunology. 2013; 2:e23082.
22. Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, Schlemmer F, Sulpice E, Locher C, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012; 4:143ra199.
23. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014; 74:647–651.
24. Chew AL, Jessica JJ, Sasidharan S. Antioxidant and antibacterial activity of different parts of Leucas aspera. Asian Pac J Trop Biomed. 2012; 2:176–180.
25. Mann CD, Neal CP, Garcea G, Manson MM, Dennison AR, Berry DP. Phytochemicals as potential chemopreventive and chemotherapeutic agents in hepatocarcinogenesis. Eur J Cancer Prev. 2009; 18:13–25.
26. Seeka C, Sutthivaiyakit S. Cytotoxic cardenolides from the leaves of calotropis gigantea. Chem Pharm Bull (Tokyo). 2010; 58:725–728.
27. Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene. 2017; 36:1315–1327.
28. Grenier K, Kontogiannea M, Fon EA. Short mitochondrial ARF triggers Parkin/PINK1-dependent mitophagy. J Biol Chem. 2014; 289:29519–29530.
29. Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015; 29:989–999.
30. Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell. 2014; 55:451–466.
31. Cerella C, Muller F, Gaigneaux A, Radogna F, Viry E, Chateauvieux S, Dicato M, Diederich M. Early downregulation of Mcl-1 regulates apoptosis triggered by cardiac glycoside UNBS1450. Cell Death Dis. 2015; 6:e1782.
32. Frese S, Frese-Schaper M, Andres AC, Miescher D, Zumkehr B, Schmid RA. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res. 2006; 66:5867–5874.
33. McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000; 60:3807–3812.
34. Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li L, Chen X, Li F, Chen AF, Zhou J, Yang JM, Cheng Y. Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget. 2016; 7:43390–43400. https://doi.org/10.18632/oncotarget.9717.
35. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol CellBiol. 2007; 27:8807–8814.
36. Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011; 19:416–428.
37. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010; 143:802–812.
38. Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010; 17:41–52.
39. Pellegrini L, Pucci B, Villanova L, Marino ML, Marfe G, Sansone L, Vernucci E, Bellizzi D, Reali V, Fini M, Russo MA, Tafani M. SIRT3 protects from hypoxia and staurosporine-mediated cell death by maintaining mitochondrial membrane potential and intracellular pH. Cell Death Differ. 2012; 19:1815–1825.
40. Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008; 15:521–529.
41. Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012; 287:40652–40660.
42. Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sc U S A. 2010; 107:5018–5023.