
INTRODUCTION
Loss of DNA methyltransferase 3A (DNMT3A) activity has been shown to lead to hematopoietic stem cell differentiation defects and the development of myeloid malignancies. DNMT3A is commonly mutated in myeloid diseases, with mutations found in over 20% of all acute myeloid leukemia (AML) patients [1], in 8% of myelodysplastic syndrome (MDS) patients [2], and in smaller frequencies in other leukemias. For AML patients, 60% of those with a DNMT3A mutation are heterozygous at Arginine 882 (R882), a dominant negative mutation that results in less than 80% protein activity [3]. The remaining patients usually demonstrate a compound heterozygous or homozygous mutation.
In contrast to AML, individuals with clonal hematopoiesis commonly bear a loss-of-function mutation in only one copy of DNMT3A [4]. In this pre-leukemic state, almost all DNMT3A mutations are nonsynonymous, truncating, or splicing, and mutations in R882 are rare [5]. These otherwise healthy individuals are at an increased risk of developing MDS/AML due to acquisition of a second driver mutation [6] and tend to have a poor overall prognosis [1]. Thus, although DNMT3A protein function must be almost completely lost in order to cause malignancy [7], heterozygous loss-of-function mutations are often found combined with a driver mutation in the development of frank leukemia. Furthermore, Dnmt3a haploinsufficiency alone with no other lesions is sufficient for mice to develop myeloid malignancies when aged to 18–24 months [8].
Here, we studied the effects of Dnmt3a haploinsufficiency combined with Ptpn11D61Y, a gain-of-function mutation in the protein tyrosine phosphatase SHP2, which is the most commonly mutated gene in juvenile myelomonocytic leukemia (JMML) and is used as a mouse model of myeloproliferative disease [9]. Concurrent mutations in both DNMT3A and PTPN11, albeit rare, have been reported in AML [10, 11] and in JMML patients [12]. JMML is an aggressive myeloproliferative neoplasm (MPN) of early childhood with no effective chemotherapeutic treatment and a poor prognosis.
RESULTS
We crossed Ptpn11D61Y/+;Mx1-Cre+ mice [9] with Dnmt3a+/− mice [13] to produce Mx1-Cre- (WT), Dnmt3a+/−;Ptpn11+/+;Mx1-Cre+ (Dnmt3a+/−), Dnmt3a+/+;Ptpn11D61Y/+;Mx1-Cre+ (D61Y), and Dnmt3a+/−;Ptpn11D61Y/+;Mx1-Cre+ (Dnmt3a+/−;D61Y) mice. Six cohorts of mice, with 1 to 4 mice per genotype, were treated with polyI:polyC to knockout one copy of Dnmt3a and to knock-in the mutant Ptpn11D61Y allele. All mice were followed until the Dnmt3a+/−;D61Y mice became moribund and the entire cohort was euthanized for analysis, which occurred at an average of 24 weeks after polyI:polyC treatment. We found that at the time when the Dnmt3a+/−;D61Y mice appeared moribund (thin, hunched, increased respiratory rate and effort, abdominal distension, ruffled fur, pale extremities), the Mx1-Cre− or single mutant mice of the same cohort remained healthy. Upon euthanasia, the Dnmt3a+/−;D61Y mice showed obvious splenomegaly and the spleen to body weight percentage was significantly increased compared to the other three genotypes (Figure 1A). In addition to splenomegaly, the double mutant mice also showed significantly higher peripheral blood WBC counts compared to WT or Dnmt3a+/− mice (Figure 1B).
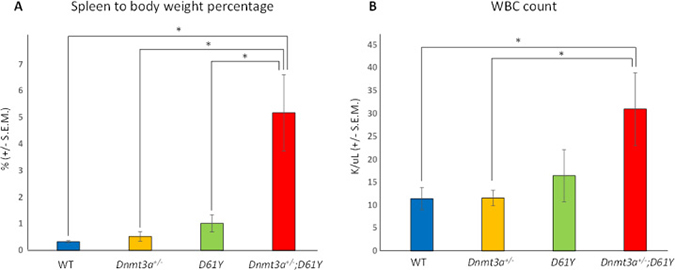
Figure 1: Dnmt3a+/−;D61Y mice show splenomegaly and leukocytosis at the time of death. (A) Average spleen to body weight ratio of mice at the time of euthanasia; n = 13 for WT, n = 9 for Dnmt3a+/−, n = 10 for D61Y, n = 6 for Dnmt3a+/−;D61Y, *p < 0.0001 comparing Dnmt3a+/−;D61Y to WT, *p = 0.003 comparing Dnmt3a+/−;D61Y to Dnmt3a+/−, *p = 0.003 comparing Dnmt3a+/−;D61Y to D61Y; statistical analyses performed by unpaired, two-tailed, Student’s t-test. (B) Average WBC count in peripheral blood of mice immediately prior to euthanasia; n = 8 for WT, n = 7 for Dnmt3a+/−, n = 5 for D61Y, n = 5 for Dnmt3a+/−;D61Y; *p = 0.0157 comparing Dnmt3a+/−;D61Y to WT, *p = 0.018 comparing Dnmt3a+/−;D61Y to Dnmt3a+/−; statistical analyses performed by unpaired, two-tailed, Student’s t-test.
Flow cytometric analysis of the spleen and peripheral blood to assess the frequency of Gr1+Mac1+ myeloid cells revealed a significantly higher percentage of these mature myeloid cells in the Dnmt3a+/−;D61Y mice compared to WT and Dnmt3a+/− mice in the spleen, and compared to WT mice in the peripheral blood (Figure 2A–2D). In contrast, the myeloid cell frequencies in the single mutant mice were not statistically different from WT mice except for D61Y mice in the peripheral blood (Figure 2C and 2D). The absolute number of Gr1+Mac1+ cells were also significantly greater in double mutant mice relative to the WT and Dnmt3a+/− mice (Figure 2E). In contrast to the increase in myeloid cells, the T and B cells were decreased in the spleen and peripheral blood of Dnmt3a+/−;D61Y mice relative to other groups (Figure 3A and 3B).
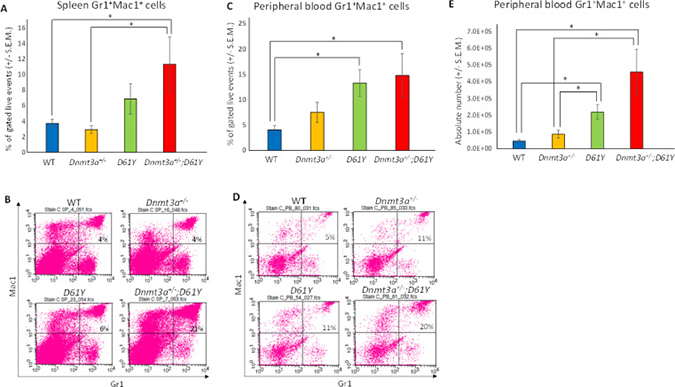
Figure 2: Dnmt3a+/−;D61Y mice have increased myeloid cells in the periphery at the time of death. (A) Spleen average percentage of myeloid cells (Gr1+Mac1+) gated on live events and (B) representative flow diagrams; n = 10 for WT, n = 7 for Dnmt3a+/−, n = 9 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.0095 comparing Dnmt3a+/−;D61Y to WT, *p = 0.0177 comparing Dnmt3a+/−;D61Y to Dnmt3a+/−; statistical analyses performed by unpaired, two-tailed, Student’s t-test. (C) Peripheral blood average percentage of myeloid cells (Gr1+Mac1+) gated on live events and (D) representative flow diagrams; n = 12 for WT, n = 9 for Dnmt3a+/−, n = 9 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.0025 comparing Dnmt3a+/−;D61Y to WT, *p = 0.0009 comparing D61Y to WT; statistical analyses performed by unpaired, two-tailed, Student’s t-test. (E) Peripheral blood average absolute number of myeloid cells (Gr1+Mac1+) calculated by multiplying percentage and WBC count; n = 12 for WT, n = 9 for Dnmt3a+/−, n = 9 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.0002 comparing Dnmt3a+/−;D61Y to WT, *p = 0.0034 comparing Dnmt3a+/−;D61Y to Dnmt3a+/−, *p = 0.0003 comparing D61Y to WT, *p = 0.0159 comparing D61Y to Dnmt3a+/−; statistical analyses performed by unpaired, two-tailed, Student’s t-test.
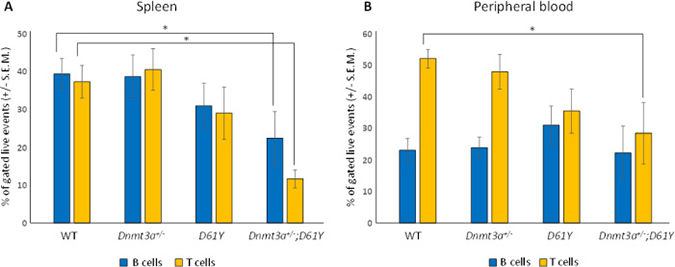
Figure 3: Dnmt3a+/−;D61Y mice have decreased B and T cells in the periphery at the time of death. (A) Spleen average percentage of B cells (CD19+ or B220+) and T cells (CD4+ or CD8+) gated on live events; n = 13 for WT, n = 9 for Dnmt3a+/−, n = 9 for D61Y, n = 6 for Dnmt3a+/−;D61Y, *p = 0.0386 comparing Dnmt3a+/−;D61Y to WT B cells, *p = 0.0013 comparing Dnmt3a+/−;D61Y to WT T cells; statistical analyses performed by unpaired, two-tailed, Student’s t-test. (B) Peripheral blood average percentage of B cells (CD19+ or B220+) and T cells (CD4+ or CD8+) gated on live events; n = 12 for WT, n = 9 for Dnmt3a+/−, n = 9 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.0071 comparing Dnmt3a+/−;D61Y to WT T cells; statistical analyses performed by unpaired, two-tailed, Student’s t-test.
In an effort to explain the increase in mature myeloid cells in the double mutant mice, we performed flow cytometric analysis on bone marrow cells from all four genotypes to assess the number of granulocyte macrophage progenitors (GMPs). As seen in Figure 4, Dnmt3a+/−;D61Y mice showed a significant increase in the absolute number of GMPs compared to WT and Dnmt3a+/− mice. The double mutant mice also had significantly more absolute numbers of megakaryocyte erythrocyte progenitors (MEPs) compared to WT mice, but no significant differences in common myeloid progenitors (CMPs) were observed among any of the four groups (Figure 4). The double mutant mice also showed signs of anemia, with significant decreases in peripheral red blood cell counts, hemoglobin levels, as well as hematocrits relative to WT mice (Figure 5A–5C). Consistent with these observations, within the erythroid lineage, the CD36+CD71+ erythroid progenitors (EPs) were significantly increased in the spleens of the Dnmt3a+/−;D61Y mice, suggesting compensatory erythropoiesis (Figure 5D and 5E). Although EP frequency was not increased in the Dnmt3a+/−;D61Y bone marrow compartment (data not shown), the increased splenic erythropoiesis may explain the increased bone marrow MEP numbers.
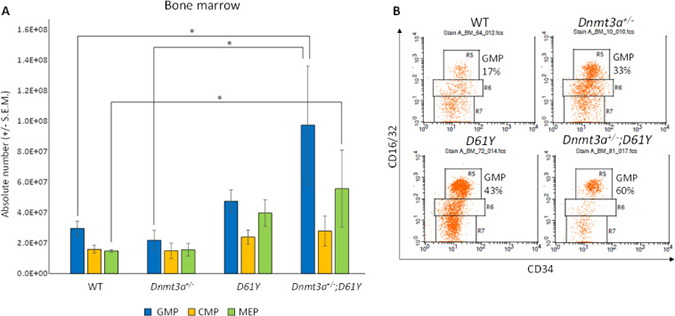
Figure 4: Dnmt3a+/−;D61Y mice have increased GMPs and MEPs in the bone marrow at the time of death. (A) Absolute numbers of granulocyte monocyte progenitors (GMPs), common myeloid progenitors (CMPs), and megakaryocyte erythrocyte progenitors (MEPs) in bone marrow, gated on lineage−cKit+Sca1− events and (B) representative flow diagrams; n = 10 for WT, n = 6 for Dnmt3a+/−, n = 6 for D61Y, n = 4 for Dnmt3a+/−;D61Y, *p = 0.0159 comparing Dnmt3a+/−;D61Y GMPs to WT GMPs, *p = 0.0445 comparing Dnmt3a+/−;D61Y GMPs to Dnmt3a+/− GMPs, *p = 0.0186 comparing Dnmt3a+/−;D61Y MEPs to WT MEPs; statistical analyses performed by unpaired, two-tailed, Student’s t-test.
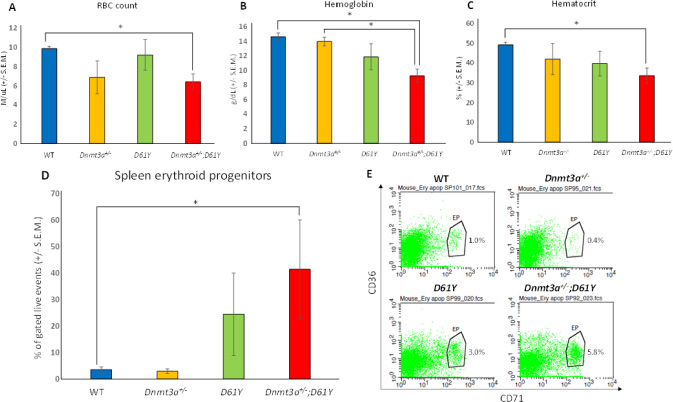
Figure 5: Dnmt3a+/−;D61Y mice have anemia with compensatory erythropoiesis in spleen. (A) Average peripheral blood red blood cell (RBC) count in mice immediately prior to euthanasia; n = 8 for WT, n = 7 for Dnmt3a+/−, n = 5 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.0005 comparing Dnmt3a+/−;D61Y to WT. (B) Average peripheral blood hemoglobin values in mice immediately prior to euthanasia; n = 8 for WT, n = 6 for Dnmt3a+/−, n = 5 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.0002 comparing Dnmt3a+/−;D61Y to WT, *p = 0.0016 comparing Dnmt3a+/−;D61Y to Dnmt3a+/−. (C) Average peripheral blood hematocrit values in mice immediately prior to euthanasia; n = 8 for WT, n = 7 for Dnmt3a+/−, n = 5 for D61Y, n = 5 for Dnmt3a+/−;D61Y, *p = 0.001 comparing Dnmt3a+/−;D61Y to WT, statistical analyses performed by unpaired, two-tailed, Student’s t-test. (D) Average percentage of erythroid progenitors (CD36+CD71+) in spleen gated on live events, with (E) representative flow diagrams; n = 6 for WT, n = 4 for Dnmt3a+/−, n = 3 for D61Y, n = 3 for Dnmt3a+/−;D61Y; *p = 0.0177 comparing Dnmt3a+/−;D61Y to WT, statistical analyses performed by unpaired, two-tailed, Student’s t-test.
DISCUSSION
AML patients often have complete loss of DNMT3A enzyme activity, but its role when only partially inhibited and in combination with a second driver mutation of leukemia is not known. Here we show that mice with heterozygous loss of Dnmt3a combined with a gain-of-function Shp2 mutation, D61Y, develop more rapid disease progression and earlier mortality. Mice expressing Shp2D61Y do not usually succumb to leukemia until 45 weeks after induction of expression [9] and mice with Dnmt3a haploinsufficiency do not develop disease until approximately 80 weeks [8]. We found that mice with the two mutations together become moribund much earlier at 24 weeks, indicating that they cooperate to promote myeloid leukemia progression and to shorten survival.
The most marked phenotypic changes we observed in the Dnmt3a+/−;D61Y mice were splenomegaly, mature myeloid cell expansion in the periphery as well as increased GMPs in the bone marrow. These mice also exhibited signs of anemia, perhaps due to defects in erythrocyte cell maturation in the bone marrow. In support of this notion, MEPs in the bone marrow of compound mutant mice were increased and there were more erythroid progenitors present in the spleen, which most likely contributed to the splenomegaly in these mice.
The disease course of JMML also exhibits pronounced splenomegaly, sometimes even in the absence of highly elevated WBC count. It has been previously reported that mice with Dnmt3a−/− combined with KrasG12D/+ mutation, another common mutation found in JMML patients, developed stress erythropoiesis in the spleen [14]. Perhaps one reason JMML patients develop extreme splenomegaly is compensatory splenic erythropoiesis, as observed in the Dnmt3a+/−;D61Y mice in this study.
Because DNMT3A and PTPN11 mutations are found in combination in AML and JMML patients, our double mutant mice provide a novel clinically relevant model for developing and evaluating therapies for myeloid leukemia. In AML, the presence of a DNMT3A mutation led to significantly shortened overall survival [1]. For JMML, a disease with poor prognosis, there is currently no established decision-making process for determining which patients would be candidates for experimental therapies. However, it is known that the number of mutations present at diagnosis is strongly correlated with survival when comparing patients who have 0–1 mutations to those with 2 or more mutations [12]. Further work must be done to find the importance of having specifically the DNMT3A and PTPN11 genes mutated together, but these are two commonly mutated genes in myeloid leukemia that we have now shown leads to accelerated disease.
The mechanism of how Dnmt3a loss cooperates with driver mutations to accelerate disease progression is still unknown, but our Dnmt3a+/−;D61Y mouse model will be useful to explore this question. We speculate that the changes in methylation caused by reduced DNMT3A activity may lead to epigenetic changes that alter the normal transcription of tumor suppressor genes needed to dampen Shp2 signaling. Future work doing a genome-wide transcriptional analysis would help to elucidate the mechanism of enhanced myeloid leukemia observed in the double mutant mice.
METHODS
Animal husbandry
Mice with a conditional mutant Shp2 allele, LSL-Shp2D61Y/+, have been previously described [9]. Mice with a conditional knockout Dnmt3a allele have been previously described [13]. Expression of the D61Y mutation, the Dnmt3a mutation, and Mx1-cre were confirmed by genotyping. Three intraperitoneal injections of 300ug polyI:polyC were administered concurrently to mice of each cohort. Mice were housed and bred in accordance with the Institutional Animal Care and Use Committee of the Indiana University School of Medicine.
Flow cytometry
Cell suspensions were incubated for 5 minutes with 10% rat serum (MP Biomedicals) and 0.2% BSA (Roche) in PBS, then stained for 30 minutes at 4°C with biotinylated lineage markers (Mac1, Gr1, CD4, CD8, B220, Ter119, IL7Rα, CD19, and CD3 with streptavidin PerCPcy5.5 as secondary), anti-Sca1-PE, anti-cKit-APC, anti-CD34-FITC, anti-CD16/32-PEcy7, anti-Gr1-FITC, anti-Mac1-APC, biotinylated anti-CD4 and anti-CD8 (with streptavidin APC as secondary), anti-B220-PE, anti-CD71-PE, and/or anti-CD36-APC (eBioscience and BD Biosciences). The analyzer used for flow cytometry was the BD LSR II and data was analyzed using CellQuest.
Complete blood counts
Peripheral blood was collected from the saphenous vein of mice and complete blood counts were measured using a Hemavet 950 (Drew Scientific Group).
Statistical calculations
GraphPad was used to perform the unpaired, two-tailed, Student’s t-tests.
Abbreviations
DNMT3A: DNA methyltransferase 3A; AML: acute myeloid leukemia; MDS: myelodysplastic syndrome; JMML: juvenile myelomonocytic leukemia; MPN: myeloproliferative neoplasm; GMP: granulocyte monocyte progenitor; CMP: common myeloid progenitor; MEP: megakaryocyte erythrocyte progenitor; EP: erythroid progenitor.
Author contributions
LD designed and performed experiments, analyzed data, created figures, and wrote the manuscript. BMR, ELV, VJA, and RJC performed experiments. RK and RJC designed experiments, interpreted data, and wrote the manuscript.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the administrative assistance of Tracy Winkle.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
FUNDING
This work was supported by the U.S. National Institutes of Health (F30 CA210518 to LD and R21 CA202296 to RJC).
REFERENCES
1. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010; 363:2424–33.
2. Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O'Laughlin M, Kandoth C, Baty J, Westervelt P, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011; 25:1153–8.
3. Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013; 122:4086–9.
4. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014; 20:1472–8.
5. Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016; 7:12484.
6. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371:2488–98.
7. Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, Challen GA, Li W, Wheeler D, Rebel VI, Goodell MA. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015; 125:629–38.
8. Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, Helton NM, Guo M, Klco JM, O’Laughlin S, Fronick C, Fulton R, Chang GS, et al. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J Clin Invest. 2017; 127:3657–74.
9. Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, Neel BG. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009; 113:4414–24.
10. Chen L, Chen W, Mysliwski M, Serio J, Ropa J, Abulwerdi FA, Chan RJ, Patel JP, Tallman MS, Paietta E, Melnick A, Levine RL, Abdel-Wahab O, et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia. 2015; 29:1290–300.
11. Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012; 119:559–68.
12. Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, Esquivel E, Yu A, Seepo S, Olsen S, Rosenberg M, Archambeault SL, Abusin G, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. 2015; 47:1326–33.
13. Yu Q, Zhou B, Zhang Y, Nguyen ET, Du J, Glosson NL, Kaplan MH. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. Proc Natl Acad Sci USA. 2012; 109:541–6.
14. Chang YI, You X, Kong G, Ranheim EA, Wang J, Du J, Liu Y, Zhou Y, Ryu MJ, Zhang J. Loss of Dnmt3a and endogenous Kras(G12D/+) cooperate to regulate hematopoietic stem and progenitor cell functions in leukemogenesis. Leukemia. 2015; 29:1847–56.