
INTRODUCTION
Colorectal cancer was one of the top three cancer types worldwide according to the Global Cancer Statistics in 2012 [1], which has become a heavy health burden. It was estimated that 49,190 deaths were caused by colorectal cancer in United States in 2016 [2]. Incidence rate of colorectal cancer in developed countries was several times higher than that in developing countries [1], however, the rising trend in some emerging developing countries due to popularization of westernized life style cannot be ignored [2–5]. With promoted preventive measures and advanced screening technology, colorectal cancer mortality rate was reported to decline in many countries [6, 7]. Nonetheless, 40 to 50 percent of colorectal cancer patients will suffer from recurrence or death after surgical resection, so further adjuvant treatment was needed [8].
Chemotherapy is the major treatment approach after surgery. For example, Fluorouracil plus Leucovorin (FU+LV) is the standard chemotherapy regimen for colon cancer patients [9, 10]. Oxaliplatin (OXP) is the third generation platinum derivatives with a significant role in colorectal management, which can inhibit DNA synthesis and replication and thus to decrease tumor cell growth. Many clinical trials have proved the addition of OXP in standard FU+LV chemotherapy (FOLFOX regimen) presented notable improved treatment efficiency in stage III and stage IV colon cancers, but its benefit in high-risk stage II colon cancers patients is yet to be confirmed [11–16]. However, toxicities are main concerns in use of OXP [17]. Therefore an ideal drug delivery vesicle is required to achieve slow and sustained release of OXP so as to reduce side effects without decreasing efficacy.
Fibrin glue (FG) is a medical hemostat made of fibrinogen and thrombin, which is commonly used in surgical procedures, by formation of fibrin clot thereby promoting hemostasis, tissue adhesion and sealing. FG exhibits several advantages such as good biocompatibility, safety and biodegradability. FG was proved to be a desirable vesicle for antibiotics, drugs, genes and growth factors [18–23]. Moreover, several studies have demonstrated combination of FG and anti-tumor agent as drug delivery system, showing inspiring results in chemotherapy of pancreatic cancer, retinoblastoma, liver cancer, melanoma, gastric cancer, esophageal cancer and glioma [24–30]. In spite of this, few research has reported FG application in colorectal cancer chemotherapy, thus it will be intriguing to combine OXP and FG in colorectal cancer treatment.
In our study, we firstly used FG as sustainable and safe drug delivery system for OXP in both subcutaneous model and abdominal metastasis model of murine colon cancer. Moreover, we further innovatively explored the effect of OXP and FG in immune microenvironment, tumor apoptosis, tumor proliferation and tumor angiogenesis, aiming at providing evidence for novel chemotherapy approach in the management of colorectal cancer.
RESULTS
Inhibitory effect of OXP on colon tumor cells growth
MTT assays were performed in CT26 cells treated with different concentration of OXP for 24h and 48h, respectively, to verify the effect of OXP on colon cancer cells metabolic activity. Results showed that CT26 cells growth was significantly inhibited by OXP in concentration and time dependent manners (Figure 1). MTT assay results determined the inhibitory effect of OXP to colon tumor cells growth.
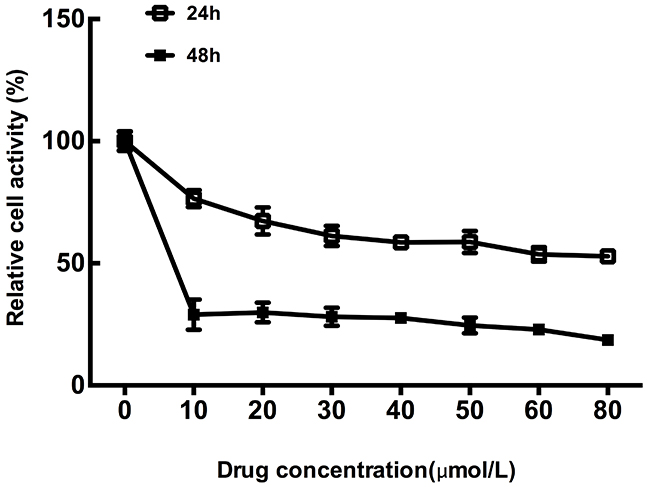
Figure 1: Inhibitory effect of oxaliplatin on colon tumor cells growth. The cell survivals of CT26 cells exposed to different concentrations of Oxaliplatin for 24h and 48h were measured by MTT assay. Each point represented the mean ± standard error of mean (SEM), n=6.
In vivo antitumor activity
Subcutaneous mouse model and abdominal metastasis mouse model were established to evaluate antitumor effect of OXP+FG in colon cancer. Mice bearing colon cancer were randomly allocated into Normal Saline (NS), FG, OXP and OXP in combination with FG (OXP+FG) groups in both models. In subcutaneous model, there was no obvious difference among body weights of NS group, FG group, OXP group and OXP+FG group (Figure 2A). Subcutaneous tumor volumes were dramatically smaller in OXP+FG group compared with those in NS group, FG group and OXP group (Figure 2B). Consistently, subcutaneous tumor weights of OXP+FG group were distinctly suppressed compared with other treatment groups (Figure 2C). Images of subcutaneous tumor were shown (Figure 2D). In abdominal metastasis model, body weights of OXP and FG group were slightly less than body weights of other three groups (Figure 3A). In addition, OXP+FG group showed reduced ascites compared with NS group, FG group and OXP group (data not shown). Tumor weights of OXP+FG group were significantly less versus those of NS group, FG group and OXP group (Figure 3B). Furthermore, there was a significant decrease of nodules amount (<3mm and >3mm) in OXP+FG group compared with that in other groups (Figure 3C and 3D). In summary, all results above proved that OXP+FG treatment showed enhanced anti-tumor activity in both subcutaneous models and abdominal metastasis model of colorectal cancer, in comparison with NS, FG and OXP treatments.
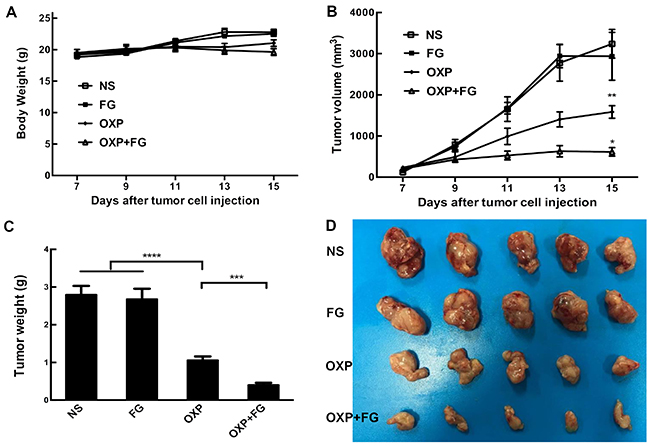
Figure 2: Oxaliplatin and FG combination showed enhanced anti-tumor effect in subcutaneous model of colon cancer. Mice bearing colon tumor were randomly allocated into NS group, FG group, OXP group and OXP+FG group and started to receive treatment on 7th day after inoculation. (A) Mice body weights of different treatment groups (P>0.05, OXP+FG versus NS group, FG group and OXP group). (B) Changes of subcutaneous tumor volumes of different groups (P<0.01, OXP group versus NS and FG groups. P<0.05, OXP+FG group versus OXP group). (C) Tumor weights of different groups (P<0.0001, OXP group versus NS and FG groups. P<0.001, OXP+FG group versus OXP group). (D) Photograph of tumors on the 15th day after implantation. Each bar represented the SEM, n=5.
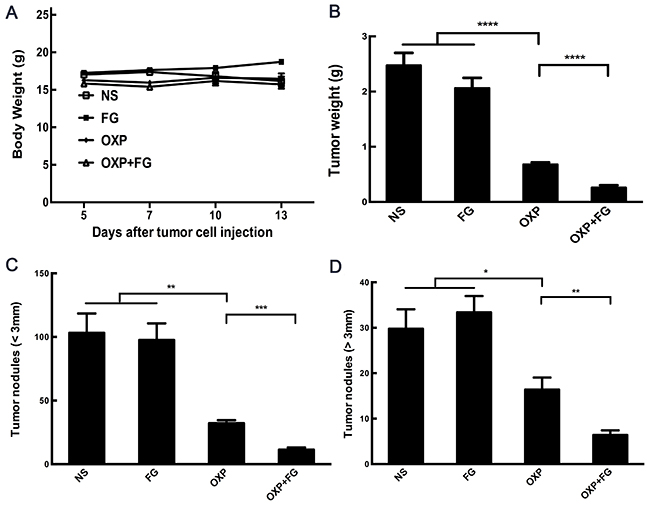
Figure 3: Oxaliplatin and FG combination showed enhanced anti-tumor effect in abdominal metastasis model of colon cancer. Mice bearing colon tumor received different treatments were divided into four groups: NS group, FG group, OXP group and OXP+FG group and started to receive treatment on 5th day after peritoneal injection. (A) Mice body weights of different treatment groups (P<0.05, OXP+FG versus NS group, FG group and OXP group). (B) Tumor weights of different groups (P<0.0001, OXP group versus NS and FG groups. P<0.0001, OXP+FG group versus OXP group). (C) Tumor nodules amount (<3mm) (P<0.01, OXP group versus NS and FG groups. P<0.001, OXP+FG group versus OXP group). (D) Tumor nodules amount (>3mm) (P<0.05, OXP group versus NS and FG groups. P<0.01, OXP+FG group versus OXP group). Each bar represented the SEM, n=5.
OXP+FG increased activated CD8+ T cells and reduced Treg cells in vivo
To explore the anti-tumor mechanism of OXP+FG, analysis of immune microenvironment in mice was carried out in our study. Interestingly, activated CD8+ T cells percentage was found increased while Treg cells percentage was decreased in mice spleens of OXP+FG group, in comparison of NS group, FG group and OXP group (Figure 4A and 4B). The results demonstrated that OXP+FG could increase activated CD8+ T cells and reduce Treg cells in vivo.
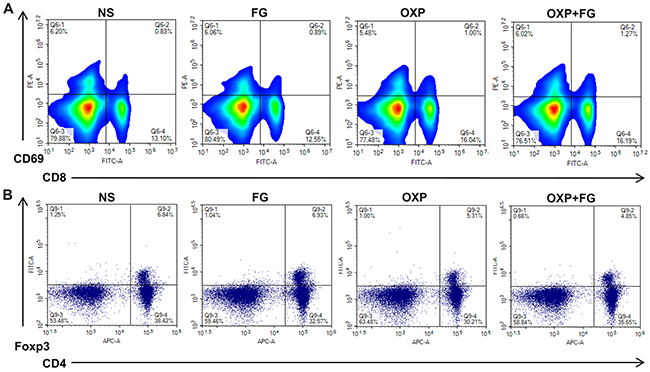
Figure 4: OXP and FG combination promoted activated CD8+ T cells and reduced regulatory T cells. Lymphocytes from mice spleens were stained with CD8, CD69, CD4 and Foxp3 fluorochrome-labeling antibodies. Cells with positive CD8 and CD69 staining indicated activated CD8+ T cells. Cells with positive CD4 and Foxp3 indicated regulatory T cells (Treg) cells. (A) Representative images of flow cytometry result for activated CD8+ T cells in different treatment groups. (B) Representative images of flow cytometry result for Treg cells in different treatment groups.
OXP+FG increased IFN-γ concentration
To determine the quantity of IFN-γ in ascites, an IFN-γ ELISA Kit was applied in our study. Results showed that concentration of IFN-γ in ascites of OXP+FG group was significantly higher than that in ascites of NS group, FG group and OXP group (Figure 5). Therefore, results suggested that OXP+FG evoked more IFN-γ against tumor cells.
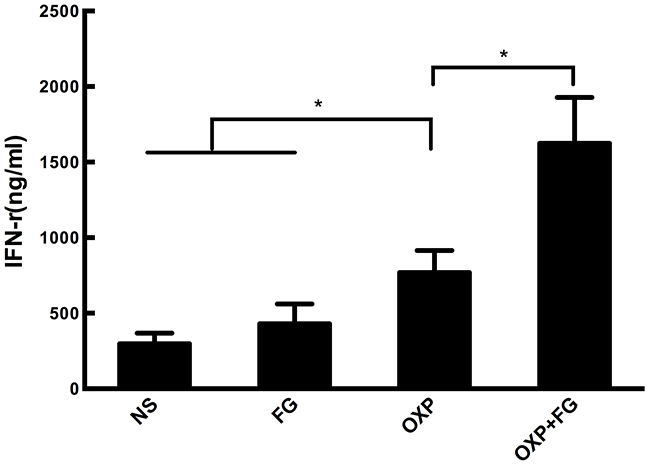
Figure 5: OXP and FG combination significantly increased INF-γ concentration. IFN-γ in ascites of mice was detected with ELISA method. Significantly increased concentration of IFN-γ was observed in OXP group compared with NS group, FG group (P<0.05, OXP group versus NS and FG groups). The concentration of IFN-γ in OXP+FG group was higher than that of OXP group (P<0.05, OXP+FG group versus OXP group). Each bar represented the SEM, n=5.
OXP+FG promoted tumor apoptosis in vivo
TUNEL assay was performed to investigate tumor apoptosis in different treatment groups. Tumor cells with positive TUNEL staining represented apoptotic tumor cells. In OXP+FG group and OXP group, TUNEL staining positive cells were obviously more than those in NS group and FG group. Moreover, TUNEL staining positive cells in OXP+FG group were more than OXP group (Figure 6). Therefore, results suggested OXP+FG treatment promoted more tumor apoptosis compared with NS, FG and OXP treatment alone.
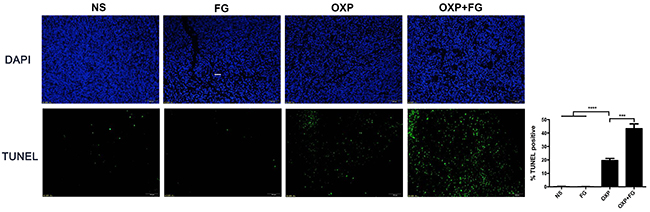
Figure 6: OXP and FG combination promoted tumor apoptosis in vivo. TUNEL assay of tumor sections was performed to analyze tumor apoptosis. Cells with positive TUNEL staining represented apoptotic cell. Results demonstrated increased apoptotic tumor cells in OXP group compared with NS group and FG group (P<0.0001, OXP group versus NS and FG groups). There were more apoptotic tumor cells in OXP+FG group than that in OXP group (P<0.001, OXP+FG group versus OXP group). Representative images were showed above. Each bar represented the SEM, n=5.
OXP+FG decreased tumor proliferation in vivo
Ki67 staining was carried out to detect tumor proliferation in NS group, FG group, OXP group and OXP+FG group. Ki67 staining positive cells indicated cells with high proliferation activity. In OXP+FG group and OXP group, Ki67 staining positive cells were fewer than those in NS group and FG group. Additionally, OXP+FG group showed less Ki67 staining positive cells than OXP group (Figure 7). Results demonstrated OXP+FG exhibited stronger inhibitory effect on tumor proliferation in vivo, compared with NS, FG and OXP treatment alone.
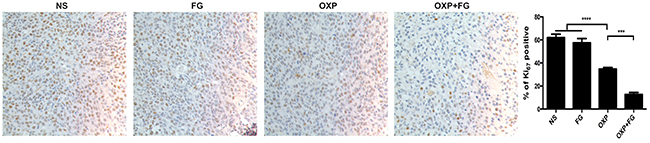
Figure 7: OXP and FG combination attenuated tumor proliferation in vivo. Ki67 staining of tumor sections was applied to explore tumor proliferation. Cells with positive Ki67 staining represented cells with high proliferation activity. Results showed decreased proliferation of tumor cells in OXP group, compared with that in NS group and FG group (P<0.0001, OXP group versus NS and FG groups). In OXP+FG group, there were less tumor cells with high proliferation activity than that in OXP group (P<0.001, OXP+FG group versus OXP group). Representative images of each group were showed above.
OXP+FG inhibited tumor angiogenesis in vivo
As angiogenesis is an important event in tumor development and progression, investigation on microvessel density (MVD) of tumor sections was performed with CD31 staining method. In OXP+FG group and OXP group, immunohistochemical results showed decreased MVD compared with that in NS group and FG group. Moreover, there were less CD31 staining positive cells in OXP+FG group than those in OXP group (Figure 8). Results revealed that OXP+FG might play anti-tumor activity through suppressing angiogenesis of tumor.
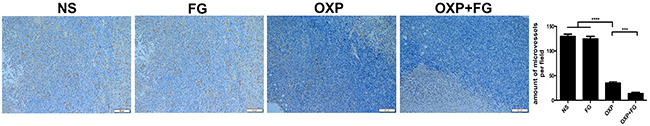
Figure 8: OXP and FG combination repressed tumor angiogenesis. CD31 staining can be used to label microvessel in tumor sections. Results showed reduced microvessel densities (MVD) of tumor cells in OXP group, compared with that in NS group and FG group (P<0.0001, OXP group versus NS group and FG group.). There were less microvessels in OXP+FG group than that in OXP group (P<0.001, OXP+FG group versus OXP group). Representative images of each group were showed above.
Safety assessment
To further evaluate the safety of FG and OXP combination on experiment animals, HE staining and serum biochemical parameters measurement were carried out. There was no visible treatment toxicity to vital organs according to the HE staining of heart, liver, spleen, lung and kidney sections (Figure 9). Test results of key biochemical parameters in serum indicated no differences among four treatment groups (Figure 10). Moreover, monitoring of animal growth status such as body weight, excretions and appearance showed no obvious abnormalities.
Figure 9: OXP and FG combination exhibited no toxicity to vital organs. HE staining images of heart (A), liver (B), spleen (C), lung (D) and kidney (E) were presented. No obvious pathohistological abnormalities were observed in NS group, FG group, OXP group and OXP+FG group.
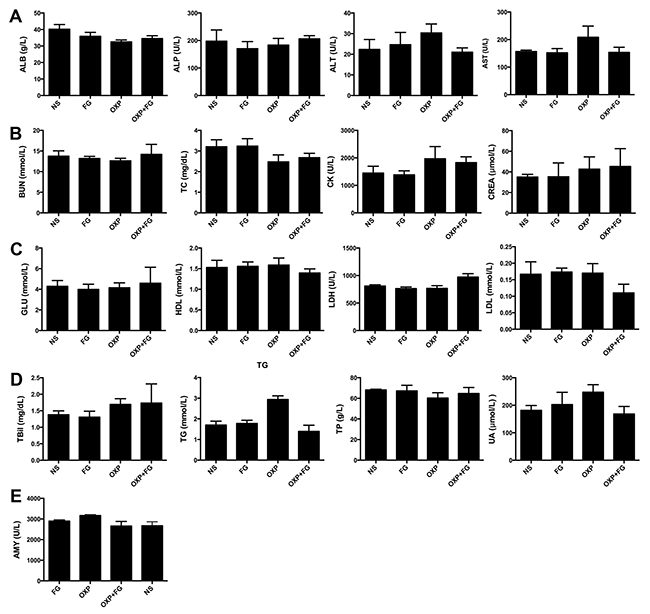
Figure 10: OXP and FG combination exhibited no systemic toxicity in vivo. Blood serums of mice from different treatment groups were assessed for some biochemical parameters (ALB, albumin; ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; BUN, blood urea nitrogen. TC, total cholesterol; CK, creatine kinase; CREA, creatinine; GLU, glucose; HDL, high density lipoprotein-cholesterol; LDH, lactate dehydrogenase; LDL, low density lipoprotein-cholesterol; TBil, total bilirubin; TG, triglycerides; TP, total protein; UA, uric acid; AMY, amylase). (A) ALB, ALP, ALT, AST (P>0.05, P>0.05, P>0.05, P>0.05 respectively, for all groups comparison). (B) BUN, TC, CK, CREA (P>0.05, P>0.05, P>0.05, P>0.05, respectively, for all groups comparison). (C) GLU, HDL, LDH, LDL (P>0.05, P>0.05, P>0.01, P>0.05, respectively, for all groups comparison). (D) TBil, TG, TP, UA (P>0.05, P<0.01, P>0.05, P>0.05, respectively, for all groups comparison). (E) AMY (P>0.05 for all groups comparison). Each bar represented the SEM, n=5.
DISCUSSION
Cancer has been a thorny issue for human health and colorectal cancer is among the most prevalent cancer types all around the world. Although great progresses were achieved on the management of colorectal cancer in recent decades, problems such as local recurrence and distal metastasis are urgent to be overcome. Chemotherapy regimen containing OXP shows pleasant anti-tumor performance but limited by drug adverse effects.
Our study innovatively combined OXP and FG in colon cancer treatment and achieved remarkable anti-tumor activity. Both murine colon cancer subcutaneous model and abdominal metastasis model were established for investigating the therapeutic effects and underlying mechanism. In vitro, OXP showed strong inhibitory effect to CT 26 cells growth. In vivo, OXP+FG presented remarkable enhanced anti-tumor effect in both subcutaneous model and abdominal metastasis model in comparison with NS, FG and OXP alone, through increasing activated CD8+ T cells, reducing Treg cells and evoking more IFN-γ. Moreover, OXP+FG suppressed tumor proliferation and tumor angiogenesis as well as induced tumor cell apoptosis. No obvious toxicity was exhibited in vivo among all treatment groups.
Some studies have reported enhanced anti-tumor performance of FG mixed with chemotherapeutic agents in a few cancers. Kitazawa monitored a sustained release of doxorubin (DOX) from FG containing sodium alginate in rats with liver cancer and reported its benefit in local delivery for higher drug concentration in tumor extracellular fluid than that in blood [27]. In addition, Ogura founded gemcitabine (GEM) and FG was more effective than GEM in inhibiting growth of human pancreatic cancer cells in nude mice [28]. Tanaka founded mixture of mitomycin (MMC) and FG showed twice the effect of MMC in gastric and esophageal cancer [25]. Our result was coincide with previous studies that chemotherapeutic agent OXP mixed in FG was more efficient in anti-tumor activity compared with OXP alone, which was probably due to maintained concentration and sustained release of drug. Moreover, no obvious systemic toxicity was observed in our study, demonstrating FG might be a desirable drug vesicle to reduce drug diffusion into distal normal tissue.
OXP is known to function through blocking the duplication of DNA. Recently, OXP was revealed to induce immunogenic cell death associated with High mobility group box 1(HMGB1) release and calreticulin exposure [31]. When in combination with liver-specific IL-12 or IL-7, OXP increased CD8+/ Treg cell ratio in murine metastatic colon cancer model, reducing immune suppressive response and further enhancing anti-tumor immune response [32–35]. Moreover, it was reported that colorectal cancer patients had increased percentage of Treg cells both in peripheral blood and tumor infiltrating lymphocytes [36]. Many present studies supported that T cells shifted from host-protective to tumor promoting Treg cells, associated with poor prognosis in various cancers [37–40]. Depletion of Treg cells improved anti-tumor immunity in colitis-associated colon cancer and improved the survival of post-surgery colorectal cancer patients [41, 42]. Our result showed suppressed Treg cells in vivo and enhanced anti-tumor activity by OXP and FG combination, which was consistent with their findings.
Activated CD8+ T cells are known as cytotoxic T cells and are very important in adaptive immunity, which can secret cytotoxic granules and induce target cell apoptosis thus to play anti-tumor role. However, CD8+ T cells are often hampered by Treg cells through cell contact and transforming growth factor (TGF)-β [43]. Our study found increased activated CD8+ T cells and reduced Treg cells along with enhanced anti-tumor performance in colorectal cancer and tumor cell apoptosis was promoted. Suzuki demonstrated intratumoral CD8+/Treg cell ratio was positively correlated with both disease-free survival and overall survival in colorectal cancer patients [44]. Our results provided supportive evidence to Suzuk’s viewpoint.
IFN-γ is secreted by T helper (Th) 1 cells, CD8+ cytotoxic T cells, macrophages, NK cells and mucosal epithelial cells, playing versatile anti-tumor roles on cancer cells. IFN-γ participates in anti-proliferative, pro-apoptotic and anti-angiogenesis processes [45–48]. In addition, Ni founded IFN-γ showed synergistic effects with OXP to eliminate both colon cancer initiating label-retaining cells (LRCCs) and non-LRCCs [49]. Overacre-Delgoffe discovered IFN-γ could increase the fragility of Treg cells to heighten anti-tumor immunity [50]. In our study, we found there was a significantly increase of IFN-γ concentration in ascites of OXP+FG group compared with NS group, FG group and OXP group, which could be one of the reasons to enhance anti-tumor performance through promoting tumor cell apoptosis, suppressing angiogenesis and tumor cell proliferation.
In conclusion, our study innovatively proposed the combination of OXP and FG as a novel drug delivery system for colorectal cancer treatment. OXP+FG presented remarkablely enhanced anti-tumor efficacy in colorectal cancer in comparison with OXP alone, mediated by reducing immunosuppressive microenvironment, inhibiting tumor proliferation, attenuating angiogenesis and promoting tumor apoptosis. This is the first study to combine OXP and FG in chemotherapy of colorectal cancer and reveal its potential anti-tumor mechanisms. FG is now commonly used as medical hemostat for patients during surgery, thus it will be very meaningful and promising to realize local drug delivery by FG in chemotherapy of colorectal cancer patients.
MATERIALS AND METHODS
Reagents
Oxaplatin Injection (OXP) was obtained from Jiangsu Hengrui Medicine (China), Fibrin Glue (Human) was purchased from Shanghai RAAS (China). 3-(4, 5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (USA) and dimethyl sulfoxide (DMSO) from KeLong Chemicals (China). Diaminobenzidine (DAB) was purchased from Servicebio (China). 4’, 6-diamidino-2-phenylindole (DAPI) was purchased from Beyotime (China). Antibodies purchased included: CD8, CD4, CD69 from Abcam (USA), goat anti-mouse Ki67 antibody, goat anti-mouse CD31 antibody and rabbit anti-goat horseradish peroxidase (HRP)-conjugated secondary antibody from Servicebio (China). Terminal deoxy-nucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) kit was purchased from Promega (USA). Anti-human forkhead box P3 (Foxp3)/Transcription Factor Staining Buffer Set and mouse IFN-γ ELISA kit were purchased from affymetrix, eBioscience (USA).
Cell lines
Colon carcinoma cell line CT 26 was purchased from American Type Culture Collection (ATCC Number: CRL-2638TM, USA). Cells were cultured in RPMI 1640 basal medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 0.1% Amikacin and incubated in plastic cell culture dishes in humidified incubator at 37°C with 5% CO2.
MTT assay
Cells viability was measured by MTT assay. 3*103 cells/well and 5*103 cells/well were seeded into two 96-well plates, respectively, and incubated overnight. Then the culture medium was replaced by 200μl RPMI 1640 medium containing OXP at concentration of 0-80μg/ml with 6 duplicate wells for each concentration. After 24h and 48h incubation, 20μl MTT solution (5mg/ml) was added to each well and co-cultured with cells in dark humidified incubator at 37°C with 5% CO2 for 3 hours. Next, culture medium was removed and formazan was dissolved by 150μl DMSO for 10 minutes. Optical density (OD) was determined by spectrophotometer at 570nm with a microplate reader (OPTI max, Molecular Dynamics). Cell viability was expressed as: cell viability (%) = (mean OD in test wells) / (mean OD in control wells) ×100.
Animal model establishment and treatment
6-8 weeks female BALB/c mice were obtained from the Animal Center Laboratory of Beijing HFK Bioscience (Beijing, China). All animals are provided with sterilized water and chow and maintained under SPF (specific pathogen free) environment. All study procedures were approved by the Animal Ethics Committee of West China Hospital, Sichuan University. 1*106 CT26 cells and 2*105 CT26 cells were suspended in 100ul RPMI 1640 basal medium and injected subcutaneously and intraperitoneally, respectively, to establish subcutaneous tumor model and abdominal metastasis model. Animals were randomized into four groups before treatment: (A) NS: normal saline. (B) FG: fibrinogen + thrombin. (C) OXP. (D) OXP+FG. For subcutaneous tumor model, OXP 10 mg/kg together with fibrinogen 100μl + thrombin 100μl, fibrinogen 100μl + thrombin 100μl, OXP 10 mg/kg and NS 200μl, were given intratumorally 7 days after CT26 inoculation. For abdominal metastasis model, OXP 5 mg/kg together with fibrinogen 100μl + thrombin 100μl, fibrinogen 100μl + thrombin 100μl, OXP 5 mg/kg and NS 200μl, were given intraperitoneally 5 days after CT26 injection. Mice body weights and subcutaneous nodule sizes (length and width) were measured every 3 days. Tumor volume was calculated as the following formula: Tumor volume (mm3) = length*width2*0.5. Mice were euthanized when they presented cachexia or tumor surface started to ulcerate except that 3 mice from each group were euthanized three days after first treatment for early investigation on immune microenvironment. Ascites were collected by aspiration of a 5ml needle. Both abdominal nodules and subcutaneous tumors were weighted and prepared for histological investigation. Moreover, abdominal nodules were divided into > 3mm and < 3mm group according to diameter and counted by two independent investigators though naked eyes.
Flow cytometry
Spleens were harvested, grinded, filtered and eventually suspended in PBS after red blood cells splitted with lysis buffer. Pellets of cells were dispersed in 100μl cold PBS and stained by 1μl of the following fluorochrome-conjugated antibodies: CD8, CD4, CD69 for 30min in dark and then washed with PBS. Intracellular Foxp3 staining was conducted with Foxp3/Transcription Factor Staining Buffer Set (affymetrix eBioscience) according to the product description. Stained cells were pretreated with cold PBS containing 5% FCS, then centrifuged pellets were dispersed in fixation/permeabilization buffer in 4°C dark atmosphere for 2h. Consequently, cells were washed and resuspended in 200μl permeabilization buffer. Afterward, FOXP3 antibodies were added and incubated in dark for 30 minutes. Cells were acquired and analyzed by NovoCyte flow cytometer (ACEA Biosciences, CA, USA).
Ki67 and CD31 assay
For Ki67 and CD31 staining, 4μm paraffin embedded sections went through antigen retrieval and peroxidase inactivation. Following normal serum (ZSGB Bioscience, China) blockage, primary antibodies (goat anti-mouse Ki67 antibody and goat anti-mouse CD31 antibody, Servicebio, China) were added and incubated overnight at 4°C. Next, HRP-conjugated secondary antibodies (Servicebio, China) were added and incubated for 50minutes at 37°C. DAB was used to render color. A single microvessel was defined as a single cell with positive CD31. Cells with positive Ki67 staining represented cells with high proliferation activity. Specimens were observed in a light microscope (Ki67×200, CD31×100, Olympus, Japan). Ki67 Quantification was performed though calculating Ki67 positive cells percentage in five randomly selected fields at ×200 magnification. CD31 Quantification was performed though calculating CD31 positive cell amounts in five randomly selected fields at ×100 magnification
TUNEL assay
To investigate the effect of OXP and FG on apoptosis of colon tumor, TUNEL staining is conducted in deparaffinized and hydrated tumor sections according to the manufacturer’s instruction (Promega, USA). DAPI was used for nuclear staining. Specimens were observed in a fluorescence microscope (×100, Olympus, Japan). Quantification of apoptosis was performed though calculating TUNEL positive cells percentage in five randomly selected fields at ×100 magnification.
ELISA of ascites
Supernatants of ascites were measured with mouse IFN-γ ELISA kit (affymetrix, eBioscience). All procedures were performed according to the product instruction. Briefly, 100μl/well standards and supernatants were added into capture antibody coated and blockage buffer blocked wells overnight, followed by incubation with detection antibody for 1h and Avidin-HRP for 30 minutes. Besides, washes with PBS containing 0.5% (v/v) Tween-20 solution (PBS-T) were conducted between each step. TMB substrate was added to color rendering in dark for 15minutes, which was finally ended by stop solution. Results were determined by a spectrophotometer at 450nm with a microplate reader. A standard curve was plotted to determine the quantity of IFN-γ in the test samples.
Safety assessment
Organs including kidney, liver, lung, spleen, heart of mice were embedded in paraffin and cut into 4 μm-thick sections. HE staining was carried out as standard procedure dewaxing, hydration, Eosin staining, Haematoxylin staining and sealing. Sections were observed in a light microscope (×200, Olympus, Japan). Blood were collected after eyeball extraction and serums were harvested after overnight still placement at 4°C and measured for a few biochemical parameters (Hitachi, Japan).
Statistical analyses
Graphic was performed with PRISM version 5.0 (GraphPad Software). Statistical analysis of multiple groups was performed with one-way analysis of variance (ANOVA). Statistical analysis between two groups was performed with Student’s t test. Results were represented as mean ± standard error of the mean (SEM). A P-value of less than 0.05 was considered statistically significant for all experiments.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87-108. https://doi.org/10.3322/caac.21262.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7-30. https://doi.org/10.3322/caac.21332.
3. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66: 115-32. https://doi.org/10.3322/caac.21338.
4. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends--an update. Cancer Epidemiol Biomarkers Prev. 2016; 25: 16-27. https://doi.org/10.1158/1055-9965.EPI-15-0578.
5. Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009; 59: 366-78. https://doi.org/10.3322/caac.20038.
6. Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I, Seeff LC, van Ballegooijen M, Goede SL, Ries LA. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010; 116: 544-73. https://doi.org/10.1002/cncr.24760.
7. Bosetti C, Levi F, Rosato V, Bertuccio P, Lucchini F, Negri E, La Vecchia C. Recent trends in colorectal cancer mortality in Europe. Int J Cancer. 2011; 129: 180-91. https://doi.org/10.1002/ijc.25653.
8. Obrand DI, Gordon PH. Incidence and patterns of recurrence following curative resection for colorectal carcinoma. Dis Colon Rectum. 1997; 40: 15-24.
9. Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. International multicentre pooled analysis of colon cancer trials (IMPACT) investigators. Lancet. 1995; 345: 939-44.
10. Sargent D, Sobrero A, Grothey A, O’Connell MJ, Buyse M, Andre T, Zheng Y, Green E, Labianca R, O’Callaghan C, Seitz JF, Francini G, Haller D, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009; 27: 872-7. https://doi.org/10.1200/JCO.2008.19.5362.
11. Graham JS, Cassidy J. Adjuvant therapy in colon cancer. Expert Rev Anticancer Ther. 2012; 12: 99-109. https://doi.org/10.1586/era.11.189.
12. Shah MA, Renfro LA, Allegra CJ, Andre T, de Gramont A, Schmoll HJ, Haller DG, Alberts SR, Yothers G, Sargent DJ. Impact of patient factors on recurrence risk and time dependency of oxaliplatin benefit in patients with colon cancer: analysis from modern-era adjuvant studies in the adjuvant colon cancer end points (ACCENT) database. J Clin Oncol. 2016; 34: 843-53. https://doi.org/10.1200/JCO.2015.63.0558.
13. Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera F, de Gramont A. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009; 27: 3109-16. https://doi.org/10.1200/JCO.2008.20.6771.
14. de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000; 18: 2938-47. https://doi.org/10.1200/jco.2000.18.16.2938.
15. Andre T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, Topham C, Zaninelli M, Clingan P, Bridgewater J, Tabah-Fisch I, de Gramont A. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004; 350: 2343-51. https://doi.org/10.1056/NEJMoa032709.
16. Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004; 22: 23-30. https://doi.org/10.1200/JCO.2004.09.046.
17. Capdevila J, Elez E, Peralta S, Macarulla T, Ramos FJ, Tabernero J. Oxaliplatin-based chemotherapy in the management of colorectal cancer. Expert Rev Anticancer Ther. 2008; 8: 1223-36. https://doi.org/10.1586/14737140.8.8.1223.
18. Kidd ME, Shin S, Shea LD. Fibrin hydrogels for lentiviral gene delivery in vitro and in vivo. J Control Release. 2012; 157: 80-5. https://doi.org/10.1016/j.jconrel.2011.08.036.
19. Wong C, Inman E, Spaethe R, Helgerson S. Fibrin-based biomaterials to deliver human growth factors. Thromb Haemost. 2003; 89: 573-82.
20. Cruysberg LP, Nuijts RM, Gilbert JA, Geroski DH, Hendrikse F, Edelhauser HF. In vitro sustained human transscleral drug delivery of fluorescein-labeled dexamethasone and methotrexate with fibrin sealant. Curr Eye Res. 2005; 30: 653-60. https://doi.org/10.1080/02713680590968600.
21. Wang SS, Yang MC, Chung TW. Liposomes/chitosan scaffold/human fibrin gel composite systems for delivering hydrophilic drugs--release behaviors of tirofiban in vitro. Drug Deliv. 2008; 15: 149-57. https://doi.org/10.1080/10717540801952456.
22. Fujimoto K, Yamamura K, Osada T, Hayashi T, Nabeshima T, Matsushita M, Nishikimi N, Sakurai T, Nimura Y. Subcutaneous tissue distribution of vancomycin from a fibrin glue/Dacron graft carrier. J Biomed Mater Res. 1997; 36: 564-7.
23. Woolverton CJ, Fulton JA, Salstrom SJ, Hayslip J, Haller NA, Wildroudt ML, MacPhee M. Tetracycline delivery from fibrin controls peritoneal infection without measurable systemic antibiotic. J Antimicrob Chemother. 2001; 48: 861-7.
24. Anai S, Hide T, Takezaki T, Kuroda J, Shinojima N, Makino K, Nakamura H, Yano S, Kuratsu J. Antitumor effect of fibrin glue containing temozolomide against malignant glioma. Cancer Sci. 2014; 105: 583-91. https://doi.org/10.1111/cas.12397.
25. Tanaka T, Yamana H, Fujita H, Ono T, Tou Y, Fujii T, Uhi T, Shirouzu K. [An experimental study on antitumor effect of MMC-fibrin glue mixture]. [Article in Japanese]. Gan To Kagaku Ryoho. 1996; 23: 1400-2.
26. Viale M, Rossi M, Russo E, Cilli M, Aprile A, Profumo A, Santi P, Fenoglio C, Cafaggi S, Rocco M. Fibrin gels loaded with cisplatin and cisplatin-hyaluronate complexes tested in a subcutaneous human melanoma model. Invest New Drugs. 2015; 33: 1151-61. https://doi.org/10.1007/s10637-015-0291-x.
27. Kitazawa H, Sato H, Adachi I, Masuko Y, Horikoshi I. Microdialysis assessment of fibrin glue containing sodium alginate for local delivery of doxorubicin in tumor-bearing rats. Biol Pharm Bull. 1997; 20: 278-81.
28. Ogura Y, Mizumoto K, Tanaka M, Ohuchida K, Murakami M, Yamada D, Ishikawa N, Nagai E. Strategy for prevention of local recurrence of pancreatic cancer after pancreatectomy: antitumor effect of gemcitabine mixed with fibrin glue in an orthotopic nude mouse model. Surgery. 2006; 140: 66-71. https://doi.org/10.1016/j.surg.2005.11.012.
29. Van Quill KR, Dioguardi PK, Tong CT, Gilbert JA, Aaberg TM Jr, Grossniklaus HE, Edelhauser HF, O’Brien JM. Subconjunctival carboplatin in fibrin sealant in the treatment of transgenic murine retinoblastoma. Ophthalmology. 2005; 112: 1151-8. https://doi.org/10.1016/j.ophtha.2004.11.060.
30. Stendel R, Scheurer L, Schlatterer K, Gminski R, Mohler H. Taurolidine-Fibrin-Sealant-Matrix using spray application for local treatment of brain tumors. Anticancer Res. 2004; 24: 631-8.
31. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010; 29: 482-91. https://doi.org/10.1038/onc.2009.356.
32. Gou HF, Huang J, Shi HS, Chen XC, Wang YS. Chemo-immunotherapy with oxaliplatin and interleukin-7 inhibits colon cancer metastasis in mice. PLoS One. 2014; 9: e85789. https://doi.org/10.1371/journal.pone.0085789.
33. Hernandez-Alcoceba R, Berraondo P. Immunochemotherapy against colon cancer by gene transfer of interleukin-12 in combination with oxaliplatin. Oncoimmunology. 2012; 1: 97-9. https://doi.org/10.4161/onci.1.1.17930.
34. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005; 102: 18538-43. https://doi.org/10.1073/pnas.0509182102.
35. Gonzalez-Aparicio M, Alzuguren P, Mauleon I, Medina-Echeverz J, Hervas-Stubbs S, Mancheno U, Berraondo P, Crettaz J, Gonzalez-Aseguinolaza G, Prieto J, Hernandez-Alcoceba R. Oxaliplatin in combination with liver-specific expression of interleukin 12 reduces the immunosuppressive microenvironment of tumours and eradicates metastatic colorectal cancer in mice. Gut. 2011; 60: 341-9. https://doi.org/10.1136/gut.2010.211722.
36. Ling KL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD, Warren BF, Piris J, Roncador G, Fox SB, Banham AH, Cerundolo V. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun. 2007; 7: 7.
37. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007; 25: 2586-93. https://doi.org/10.1200/jco.2006.09.4565.
38. Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Housseau F. Regulatory T-cell response to enterotoxigenic bacteroides fragilis colonization triggers IL17-dependent colon carcinogenesis. Cancer Discov. 2015; 5: 1098-109. https://doi.org/10.1158/2159-8290.CD-15-0447.
39. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004; 10: 942-9. https://doi.org/10.1038/nm1093.
40. Gounaris E, Blatner NR, Dennis K, Magnusson F, Gurish MF, Strom TB, Beckhove P, Gounari F, Khazaie K. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 2009; 69: 5490-7. https://doi.org/10.1158/0008-5472.CAN-09-0304.
41. Scurr M, Gallimore A, Godkin A. T cell subsets and colorectal cancer: discerning the good from the bad. Cell Immunol. 2012; 279: 21-4. https://doi.org/10.1016/j.cellimm.2012.08.004.
42. Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M, von Smolinski D, Kasper S, Sparwasser T, Gruber AD, Schuler M, Sakaguchi S, Roers A, et al. Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer Res. 2014; 74: 4258-69. https://doi.org/10.1158/0008-5472.CAN-13-3065.
43. Khazaie K, von Boehmer H. The impact of CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin Cancer Biol. 2006; 16: 124-36. https://doi.org/10.1016/j.semcancer.2005.11.006.
44. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009; 27: 186-92. https://doi.org/10.1200/JCO.2008.18.7229.
45. Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003; 8: 237-49.
46. Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ. Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat Med. 1998; 4: 408-14.
47. Brown TJ, Lioubin MN, Marquardt H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and oncostatin M for human melanoma cells. J Immunol. 1987; 139: 2977-83.
48. Coughlin CM, Salhany KE, Gee MS, LaTemple DC, Kotenko S, Ma X, Gri G, Wysocka M, Kim JE, Liu L, Liao F, Farber JM, Pestka S, et al. Tumor cell responses to IFN gamma affect tumorigenicity and response to IL-12 therapy and antiangiogenesis. Immunity. 1998; 9: 25-34.
49. Ni C, Wu P, Zhu X, Ye J, Zhang Z, Chen Z, Zhang T, Zhang T, Wang K, Wu D, Qiu F, Huang J. IFN-gamma selectively exerts pro-apoptotic effects on tumor-initiating label-retaining colon cancer cells. Cancer Lett. 2013; 336: 174-84. https://doi.org/10.1016/j.canlet.2013.04.029.
50. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, Shuai Y, Normolle DP, Kirkwood JM, et al. Interferon-gamma drives treg fragility to promote anti-tumor immunity. Cell. 2017; 169: 1130-41 e11. https://doi.org/10.1016/j.cell.2017.05.005.