INTRODUCTION
Streptococcus iniae, a Gram-positive bacterium, was initially isolated from freshwater Amazon dolphin (Inia geoffrensis) in the 1970s [1]. To date, S. iniae has been characterized as a global zoonotic pathogen that mainly infects a broad range of marine and freshwater fish, including bream, trout, tilapia, salmon, barramundi, yellowtail, Japanese flounder, hybrid striped bass, channel catfish and Amazon dolphin [2, 3]. The common histopathologic characteristics of infected fish are septicaemia and meningitis, resulting in high mortality in farmed fish and enormous economic losses in aquaculture [3]. The pathogen could also opportunistically infect elderly humans associated with the handling and preparation of infected fish [4]. While several studies have been conducted on S. iniae, attention to date has mainly focused on the so-called virulence factors SiM protein [2,5]. interleukin-8 protease [6], streptolysin S [7]. C5a peptidase [8], capsule [1, 9–11]. phosphoglucomutase [12], exopolysaccharide [13] and α-enolase [14, 15], which are potentially involved in the infection process. A number of candidate S. iniae vaccines have been developed based on these factors [2]. However, subsequent reports demonstrate that vaccines, such as those based on the well studied M-protein, SiMA, are not effective in farmed fish [16]. Further research is therefore required to clarify the fundamental pathogenic mechanisms of S. iniae.
High-throughput and next-generation sequencing techniques have facilitated significant advances in bacteriology and enhanced our understanding of the biology, diversity and evolution of bacteria [17]. Sequence-based analyses have provided unexpected insights into bacterial diversity and allowed us to monitor the spread of infection and devise new drugs and vaccines [18]. By 2015, eight available genomes of S. iniae were recorded on the NCBI database: YSFST01-82, SF1, 9117, CAIM 527, ISET0901, IUSA1, KCTC 11634BP and ISNO (Table 1). While the complete genome sequence of S. iniae SF1 has been reported [19], comparative genomic analysis of closely related S. iniae species is yet to be conducted. In this study, we have reported the complete genome sequence of S. iniae DX09, a strain isolated from cultured catfish (Ictalurus punctatus) (Supplementary Figure 1) in the Guangxi region of China, for the first time and compared it to all known S. iniae genomes available in the NCBI database. Our collective data provide novel insights into the genomic features as well as adaptation and evolutionary mechanisms of S. iniae.
Table 1: Overview of the sequenced genomes of Streptococcus iniae
Strain |
GenBank accession |
Size (Mb) |
GC% |
Gene |
Host |
Geographical distribution |
|---|---|---|---|---|---|---|
DX09 (this study) |
GCA_001654755 |
1.98 |
36.60 |
1878 |
Ictalurus punctatus |
China |
YSFST01-82 [55] |
GCA_000831485 |
2.09 |
36.80 |
2029 |
Paralichthys olivaceus |
South Korea |
SF1 [19] |
GCA_000403625 |
2.15 |
36.70 |
2196 |
Paralichthys olivaceus |
China |
9117 [56] |
GCA_000300915 |
2.08 |
36.80 |
2034 |
Homo sapiens |
USA |
CAIM 527 [57] |
GCA_001595425 |
2.10 |
36.50 |
1991 |
Inia geoffrensis |
USA |
ISET0901 [58] |
GCA_000648525 |
2.07 |
36.80 |
1997 |
Oreochromis niloticus |
Israel |
IUSA1 [59] |
GCA_000331915 |
2.22 |
36.30 |
2156 |
Sparus aurata |
Spain |
KCTC 11634BP [60] |
GCA_000320965 |
2.06 |
37.00 |
1903 |
Paralichthys olivaceus |
South Korea |
ISNO [53] |
GCA_000648555 |
2.07 |
36.80 |
1996 |
Oreochromis niloticus |
USA |
RESULTS AND DISCUSSION
Overview of the streptococcus iniae DX09 genome sequence
S. iniae DGX07 (recently re-named DX09) was initially isolated from the spleen of diseased catfish in our laboratory. Fish infected with DX09 exhibited serious septicaemia. Intact bacterial cells were observed in macrophages of the spleen (Figure 1). Previous translocation assays in vitro demonstrated that S. iniae could successfully invade skin epithelial cells and exist freely in the cytoplasm [20]. The ability to survive intracellularly after invasion of fish cells is important for pathogenic S. iniae.
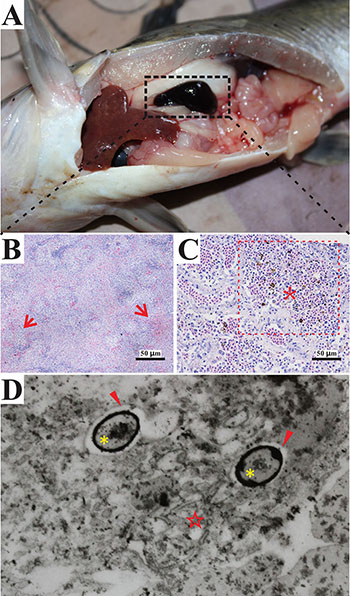
Figure 1: Main symptoms and histopathological changes of infected Ictalurus punctatus. (A) Hyperemia swelling in spleen of Ictalurus punctatus infected with Streptococcus iniae; (B) Pathological changes in spleen were observed splenic sinus congestion (arrows); (C) Melano-macrophage center (*) in spleen; (D) Transmission electron micrographs (TEM × 17000) of primary I. punctatus spleen macrophages fixed after infection with S. iniae. Detailed analysis of S. iniae within vacuoles (arrowheads); bacteria (*:) were sequestered in vacuoles. (☆:) endoplasmic reticulum. Images were finally generated using CorelDRAW X6 software. Copyright (c) 2015–2016 [Tao Liu] and CorelDRAW X6. All rights reserved.
Next, we used the Illumina Miseq Sequencing platform to yield 22131 Mb paired-end reads that were assembled into 144 contigs and 122 scaffolds, giving 1211-fold coverage of the S. iniae DX09 genome (Supplementary Table 1). Our results disclosed a 1.98 Mb chromosome with an average GC content of 36.6%, which represents the smallest genome among all sequenced Streptococcus iniae so far. The gene region accounted for 87% of the genome and was composed of 1893 coding sequences (Supplementary Table 2). In total, 34 tRNA genes (Supplementary Table 3) representing 18 amino acids and 4 rRNA genes (Supplementary Table 4) were identified. Functional analysis based on Clusters of Orthologous Groups (COG) classification (Supplementary Figure 2) revealed that the three most abundant functional categories within the DX09 genome were ‘Amino acid transport and metabolism (E)’, ‘Carbohydrate transport and metabolism (G)’ and ‘Translation, ribosomal structure and biogenesis (J)’. The large number of coding sequences (CDS) identified suggests that the DX09 lifestyle requires the efficient uptake of nutrients from the host (fish) or environment (water).
A Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) locus identified within the genome of DX09 (contig 1: 92,487 bp to 100,254 bp) exhibited a high level of sequence identity to those from other S.iniae (data not shown). This finding was in agreement with data from a previous study on the CRISPR/Cas systems of S.iniae SF1 [19].
The general genomic features of all sequenced Streptococcus iniae are summarized in Table 1 and the phylogenetic relationship is showed in Supplementary Figure 3.
The PZ-I region with an ess locus is unique to S. iniae strains DX09, ISET0901, ISNO, CAIM 527, IUSA1 and SF1
A complete nucleotide sequence-based pangenome was created from nine genomes (Table 1) with Gegenees and subsequently used as a reference for comparison with nine other S. iniae genomes (Table 1) using the BLAST Ring Image Generator (BRIG)28 (Figure 2A). The data revealed extremely high conservation amongst the lineages, with mobile genetic elements accounting for the majority of differences. We detected one specific region of genome plasticity in DX09, ISET0901, ISNO, CAIM 527, IUSA1 and SF1 strains, designated plasticity zone I (PZ-I), which was >18 kb in size and absent from YSFST01-82, KCTC 11634BP and 9117 (Figure 2A). Further investigation of the DX09 genome revealed that some genes (A7N10_RS03660– A7N10_RS03685, overlapping region between the red line and blue box in Figure 2A) specifically present in PZ-I are partially related to the Ess/type VII secretion system (T7SS). Additionally, a four-gene putative cluster (encoded by A7N10_RS03635-A7N10_RS03650), O-glycosy[1] hydrolase (encoded by A7N10_RS03655) and several hypothetical proteins (encoded by A7N10_RS03610-A7N10_RS03630) were detected in the PZ-I region (Figure 2A, red line). Genes encoding DX09-T7SS were identified at the ess locus (Figure 2A, blue box), potentially representing all the T7SS genes in Firmicutes bacteria [21]. Organization and comparison of the ess loci within all sequenced S. iniae strains are presented in Figure 2B. The core regions of ess locus mostly retained gene synteny between DX09 and other related strains. T7SS, specifically present in Gram-positive bacteria [21], is proposed to be involved in translocation of proteins (generally virulence factors) to the extracellular environment [22]. T7SS was initially described and termed the ‘ESX protein secretion system’ in pathogenic mycobacteria [23] belonging to the class Actinobacteria with high GC content. Subsequently, a related secretion system (ESS protein secretion) was detected in some Firmicutes bacteria, including Staphylococcus aureus [24–29], Listeria monocytogenes [21, 30], Bacillus subtilis [21, 30] and Streptococcus agalactiae [21, 30]. We have identified an ESS secretion system in S. iniae, which also belongs to the low-GC Firmicutes bacterial group. Pathogenic mycobacteria generally possess up to five different T7SS (ESX-1, ESX-2, ESX-3, ESX-4 and ESX-5), which have possibly evolved via gene duplication [31]. Several studies suggest that ESX-1, the first T7SS identified, strongly influences host–pathogen interactions, in particular, the ability of bacteria to escape the phago-vacuole of infected cells (macrophages and dendritic cells) [23]. ESX-1 is partially missing in the vaccine strain Mycobacterium bovis Bacille Calmette-Guérin [32], indicating a contribution to virulence. Previous investigations have demonstrated a crucial role of T7SS in bacterial survival within the host [21, 23, 31, 32], consistent with transmission electron micrographs of interactions between S. iniae DX09 and spleen cells from diseased catfish. Moreover, considerable evidence is available supporting essential roles of the actinobacterial ESS protein secretion system in virulence [26–29].
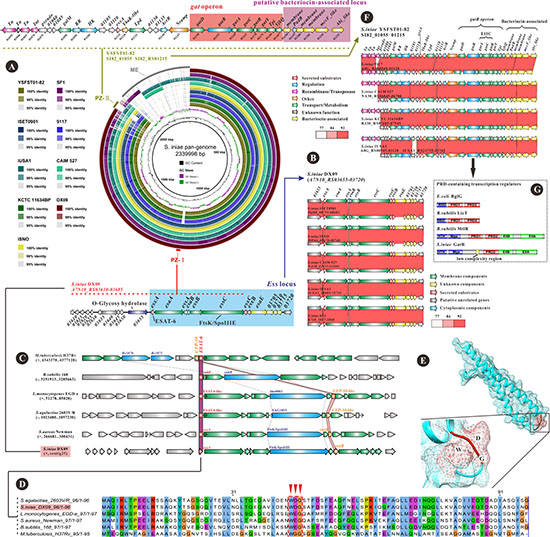
Figure 2: BLASTN-based sequence comparison of nine Streptococcus iniae strains against the S. iniae pangenome reference generated using Gegenees. (A) Circular representation of comparison between the pan-genome and the nine S. iniae genomes obtained with BRIG [37]. The image shows similarities between a central pan-genome and nine S. iniae genome sequences as a set of concentric rings, where BLAST matches are coloured on a sliding scale indicating a defined percentage identity (thresholds set at default 98% and 95%). The innermost rings represent GC content (black) and GC skew (purple/green) of the pan-genome. The positions of the plasticity regions are marked in the outer ring. Plasticity zone(PZ)- I with some gene clusters (using S. iniae DX09 as an example) is shown on the dashed line. The Ess locus is in the blue box (below). PZ-I was unique to S. iniae strains DX09, ISET0901, ISNO, CAIM 527, IUSA1 and SF1, and absent from the strains YSFST01-82, 9117 and KCTC 11634BP. PZ-II with a gat operon (red box) and putative bacteriocin-associated locus (purple box) is presented at the top of the olive dashed line (S. iniae YSFST01-82). PZ-II was unique to S. iniae strains YSFST01-82, 9117, CAIM 527, KCTC 11634BP and IUSA1, and absent from the genomes of DX09, ISET0901, ISNO and SF1. ME, mobile elements including prophages, integrative and conjugative elements and insert sequences. (B) Detailed comparison of the Ess locus between DX09 (blue box in Figure 2A) and other S. iniae possessing the Ess locus in Figure 2A. Regions with nucleotide identity >77% are connected by red windows using a colour intensity gradient based on the identity scores of BLASTN comparisons in WebACT. The Ess locus from the start of esxA to the end of a unknown component (such as A7N10_RS03715 in DX09) covers ~14 kb and is almost 100% identical between the DX09, ISET0901, ISNO, CAIM 527, IUSA1 and SF1 strains (only two differences over this region). Hallmark genes of the Ess locus are indicated in the top panel. Accession numbers of ISET0901, ISNO, CAIM 527, IUSA1 and SF1 are presented on the left. Accession numbers of DX09 are given on the top and omitted in subsequent comparisons. Arrows of the same colour indicate homologous genes among bacteria. Genes encoding secreted substrates are coloured in red, membrane components in green, cytoplasmic components in blue, and unknown components in yellow. Putative unrelated genes are shaded in grey. (C) Comparison of different type VII secretion systems (T7SS, encoded by the Ess locus) between DX09 with Ess-containing Gram-positive bacteria, including Actinobacteria (Mycobacterium tuberculosis strain H37Rv) and Firmicutes (Bacillus subtilis strain 168, Listeria monocytogenes strain EGD-e, Streptococcus agalactiae strain 2603V/R and Staphylococcus aureus strain Newman). Colour codes are presented in the key. Type VII secretion systems are indicated and all region-specific genes shaded in grey. Members of the FtsK/SpoIIIE and ESAT-6/WXG100 families are both present in all the different type VII secretion systems. The dashed line represents homology of FtsK/SpoIIIE (blue arrow) and purple/pink region homology of the ESAT-6/WXG100 family. Other T7SS-related genes in each bacterial specimen are coloured in green. Putative unrelated genes are shaded in grey. (D) Protein sequence alignment of DX09 EsxA with ESAT-6/WXG100 proteins from other Gram-positive bacteria. All six proteins contain the tryptophan-variable-glycine (WXG) motif (red inverted triangle). (E) Predicted structure of the EsxA protein in DX09. The WXG motif is indicated in red. (F) Detailed comparison of the PZ-II loci between S. iniae YSFST01-82 and other S. iniae strains possessing PZ-II in Figure 2A. Regions with nucleotide identity > 77% are connected by red windows using a colour intensity gradient based on identity scores of BLASTN comparisons in WebACT. The Ess locus was almost 100% identical between the YSFST01-82, 9117, CAIM 527, KCTC 11634BP and IUSA1 strains (only mobile elements with some transposase sequences are different over this region). Hallmark genes of PZ-II are indicated in the top panel. Accession numbers of 9117, CAIM 527, KCTC 11634BP and IUSA1 are presented on the left. Accession numbers of YSFST01-82 are presented at the top and omitted in subsequent comparisons. (G) PRD-containing regulators as multidomain proteins potentially containing EIIB-like and EIIA-like domains. Antiterminators BglG (from Escherichia coli) and licT (from B. subtilis) share similar domain structures, both containing the RNA binding domain (RBD) that can mediate antitermination activity. The PRD-containing transcriptional activator MtlR (from B. subtilis) and putative PRD-containing transcription activator GatR (from S. iniae) share similar domain structures. Both contain two DNA-binding domains, HTH and Mga, and EIIB-like and EIIA-like domains. Images were generated with the software CorelDRAW X6. Copyright (c) 2015–2016 [Tao Liu] and CorelDRAW X6. All rights reserved.
Analysis of genes within the ess locus in DX09 validated their importance in substrate secretion. esxA (A7N10_RS03660), the first gene of the ess locus, may encode a virulence factor and is potentially secreted into the extracellular milieu. esxB (A7N10_RS03695) and esxC (A7N10_RS03690) possibly encode two other virulence factors (EsxB and EsxC) located in the ess locus. esxA,esxB and esxC are clustered with nine other genes, some of which may play essential roles in secretion of the translated EsxA, EsxB and EsxC proteins. Among these, esaA, essA, essB and essC (encoded by A7N10_RS03665, A7N10_RS03670, A7N10_RS03680 and A7N10_RS03685, respectively) are membrane-related components with transmembrane domains that have potential functions in secretion (Figure 2A, 2B; green arrows in the gene clusters). These results are in agreement with data obtained for T7SS in Staphylococcus aureus [26]. However, the functions of other genes within this locus remain to be established (Figure 2A, 2B; yellow arrows in the gene clusters).
Comparison of different gene clusters encoding T7SS between Actinobacteria and Firmicutes
Protein secretion has been most extensively investigated in Gram-negative bacteria, which possess six different specialized secretion systems, designated types I–VI [33]. In contrast, Gram-positive bacteria have developed T7SSs due to their complex cell envelope [21, 23, 32]. Various T7SSs appear to be present in both high-GC Gram-positive (Actinobacteria) and low-GC Gram-positive (Firmicutes) bacteria [21, 26]. To further determine the relationship between T7SS from actinobacteria and firmicutes bacteria, we compared essDX09 with T7SS from actinobacteria Mycobacterium tuberculosis H37Rv (containing the ESX-1 secretion system) and firmicutes B. subtilis 168, L. monocytogenes EGD-e, S. agalactiae 2603V/R and S. aureus Newman. The results illustrate the conserved core regions (Figure 1C). The common features between actinobacteria ESX-1 and firmicutes ESS secretion systems appear limited, with only two conserved components. One is an ATPase (Figure 2C, blue arrows) with a FtsK/SpoIIIE domain encoded by essC in DX09 (dashed line represents homology) and the second is secreted proteins of the ESAT-6/CFP-10 family encoded by esxA/esxB in DX09 (Figure 2C, red/orange arrows; purple/pink region represents homology). Our results were in agreement with data from a previous study suggesting that T7SS of firmicutes belongs to a specific and distant subfamily [21, 23, 24, 26, 32]. Notably, B.subtilis lacked the CFP-10-like protein in its T7SS (Figure 2C).
Moreover, the essDX09 locus shares extensive similarity in terms of gene content and arrangement with T7SS-ess from L. monocytogenes and S. aureus. However, T7SS encoded by the ess locus has only been well documented in S. aureus to date [24–29, 34, 35]. In the human pathogen, S. aureus, a small EsxA belongs to a ESAT-6-like protein [27]. The small protein, ESAT-6, was initially identified as a virulence factor secreted by T7SS-ESX of M. tuberculosis [21]. These protein members were characterised by a central tryptophan-variable-glycine (WXG) motif [36]. To determine whether esxA from S.iniae DX09 contains this motif, we compared the protein sequences between actinobacteria and firmicutes bacteria (Figure 2C). As expected, the conserved WXG motif (Figure 2D, red inverted triangle) was identified in the centre of the sequence. Examination of the structure of esxADX09 revealed a helical hairpin with the WXG motif localized between the two α-helices (Figure 2E, red region), consistent with previous reports on the ESAT-6-like protein structure [21, 36].
Protein secretion is critical for both pathogenic and nonpathogenic organisms to exploit nutrient resources within the host or specific niches and escape the immune system. Here, we have identified a T7SS encoded by the ess locus in S. iniae at the complete genome level for the first time. Based on the collective findings, we conclude that heterogeneity of T7SS-ess in different S. iniae strains is responsible for specific niches infect different hosts.
The PZ-II region with a phosphoenolpyruvate-carbohydrate phosphotransferase system (PTS) is unique to S. iniae strains YSFST01-82, 9117, CAIM 527, KCTC 11634BP and IUSA1
BRIG is a user-friendly program that generates genome comparisons for multiple prokaryotes [37]. Previous BRIG-based analyses only used target bacteria as the reference genome to compare closely related organisms [38, 39]. This comparative analysis has been effectively applied to identify all the regions contained by the target bacterial genome. However, the method sometimes overlooks specific zones harboured in closely related organisms that may be lost in the target bacterial genome. Therefore, in the current study, a pangenome created from nine genomes was used as the reference for comparison with nine S. iniae genomes to assess whether DX09 or closely related strains lack specific genomic regions. Consequently, we identified a 28 kb plasticity zone II (PZ-II) as a strain-specific region within YSFST01-82, 9117, CAIM 527, KCTC 11634BP and IUSA1, which was absent from DX09, ISET0901, ISNO and SF1 (Figure 2A). Four mobile elements exist in PZ-II of YSFST01-82, three of which are insertion sequences (IS) with the primary transposase sequence of each element encoded by SI82_01055, SI82_01060 and SI82_01065, and one encoding an integrase (SI82_01065) belonging to the rve superfamily (Figure 2A). This finding suggests that PZ-II is a horizontal gene transfer element that is unlikely to have been transferred in a single event and possibly incorporated into the host chromosome via a series of integration events. Comparative analysis of PZ-IIYSFST01-82 with PZ-II regions from different S. iniae strains is presented in Figure 2F, which illustrates the remarkable conservation of the core regions. With the exception of the mobile elements, PZ-IIYSFST01-82 shared extensive similarity in DNA sequence (> 99%), gene arrangement and content with PZ-II9117, PZ-IICAIM 527, PZ-IIKCTC 11634BP and PZ-IIIUSA1, consistent with the gene synteny observed in Figure 2A. Analysis of genes within the PZ-II core, such as the gat operon, revealed critical roles in nutrient transport and metabolism. In enteric bacteria, the gat operon is involved in galactitol (Gat) transport and metabolism [40]. This process is often associated with the phosphoenolpyruvate-carbohydrate phosphotransferase system (PTS), which takes up and phosphorylates carbohydrates and plays a key role in utilization of energy-efficient carbon sources in most bacteria [41]. The PTS is generally composed of three proteins, specifically, enzyme 1 (EI), histidine protein (HPr) and enzyme 2 complexes (EII) [41, 42]. During uptake of PTS carbohydrates, a phosphoryl group is transferred from phosphoenolpyruvate (PEP) via the EI and HPr to sugar-specific EII, which then phosphorylates the carbohydrate [41, 42]. Hence, different bacteria often possess variable EIIs usually consisting of one hydrophobic integral membrane domain (EIIC) and two hydrophilic domains (EIIA and EIIB) [42]. Within PZ-II, gatA, gatC and gatB encode EIIAGat, EIICGat and EIIBGat, respectively, which are specifically responsible for galactitol uptake and phosphorylation in S. iniae (Figure 2A, 2F). Importantly, gatR encodes a PRD-containing transcription regulator, denoted GatR (Figure 2G), which may act on the gat operon (discussed below). Notably, a tagatose-1,6-bisphosphate-aldolase encoded by gatY was identified in the gat operon in PZ-II, suggesting that phosphoryl-Gat is further metabolized in the tagatose 6-phosphate pathway [40], the sole route of lactose and D-galactose metabolism in S. aureus. Four other genes encoded by galM, gatD, Tpi and YjbQ from PZ-II additionally belong to this metabolic pathway (Figure 2A, 2F).
PZ-II carries additional genes capable of conferring survival benefits in niche-specific adaptations, including mccF_like and Abi_like involved in bacteriocin self-immunity. Bacteriocins are ribosomally synthesized antimicrobial peptides active against other closely related bacteria [43]. The mccF_like gene belongs to the Peptidase_S66 superfamily and encodes a microcin C7 self-immunity protein resistant to microcin C7 (MccF) [44]. The Abi _like protein belongs to the Abi family (also known as the CAAX prenyl protease family) consisting of putative membrane-bound metalloproteases [45]. In general, Abi is located downstream of bacteriocin structural genes, where it may be involved in self-immunity [45]. Within PZ-II, a number of membrane-related proteins containing several transmembrane regions and a PadR family transcriptional regulator were identified upstream of mccF_like and Abi_like proteins. A novel bacteriocin-like locus was identified in this region of PZ-II. Bacteriocin-associated genes can be effectively utilized as a means to determine unknown bacteriocins in sequenced bacterial genomes [45]. Other genes located in PZ-II, such as YbaK-like and Nramp, encode bacterial prolyl-tRNA synthetase and natural resistance-associated macrophage protein, respectively, both of which are potential contributors to bacterial survival.
Interestingly, esxA was located both in PZ-I and PZ-II. esxAPZ-I shared 86.6% DNA sequence identity with esxAPZ-II, both of which contained the WXG motif at the amino acid level (Supplementary Figure 4). Our results support a critical role of esxA in all strains of S. iniae.
Proposed mechanism of regulation of PTS regulatory domain (PRD)-containing transcriptional activators during uptake of galactitol in S. iniae
The gat operon encoding a PTS occupies a major region of PZ-II in the S. iniae strains discussed above. Therefore, Gat uptake and metabolism appear essential for YSFST01-82, 9117, CAIM 527, KCTC 11634BP and IUSA1 strains. Previous studies indicate that PTS can regulate usage of various carbon sources, which are involved in carbon catabolite repression and self-regulation of PTS [41, 42]. These regulatory processes allow bacteria make the most out of nutrients. Several operons participating in catabolism of PTS sugars are controlled by regulators that contain a PRD [46]. PRD-mediated regulation has been extensively studied for the Escherichia coli antiterminator BglG [47], B. subtilis antiterminator licT [48, 49] and B. subtilis transcriptional regulator MtlR [50, 51]. To assess the functional relationships between the PRD-containing transcriptional activator of S.iniae, GatR, and the above regulators from E. coli and B. subtilis, we conducted a domain correlation analysis (Figure 2G). The antiterminator BglG regulates the bgl (β-glucoside) operon, which is involved in the uptake and utilization of β-glucosides [46]. BglG controls the bgl operon by inactivating terminators that frame this region, thereby allowing transcriptional elongation [41, 42]. The antiterminator licT modulates expression of the bglPH operon for uptake and utilisation of β-glucoside [49]. In fact, BglG and licT share similar mechanisms of regulation and present the same modular structures comprising three domains (N-terminal RNA-binding and two PTS regulatory domains, PRD1 and PRD2) (Figure 2G). In the absence of inducer, BglG and licT are inactivated via membrane sequestration and subsequent phosphorylation of PRD1 mediated by phosphorylated EIIB (P-EIIB) [46, 49, 52]. In the presence of inducer (β-glucoside), the incoming inducer becomes phosphorylated and P-EIIB is dephosphorylated [42, 52]. Consequently, BglG and licT are also dephosphorylated at PRD1, released into the cytoplasm and activated [42, 52]. BglG and licT additionally require phosphorylation of PRD2 for activation [42]. However, GatR of S. iniae is distinct from these proteins, presenting a different modular structure. Interestingly, we observed that the B. subtilis transcriptional regulator (MtlR) shares a similar domain structure with S. iniae GatR (Figure 2G). Both structures contain a HTH DNA-binding and Mga DNA-binding domain, clearly indicative of DNA binding rather than antiterminator activity. The DNA-binding domain is followed by PRD as well as EIIB-like and EIIA-like domains, which are further absent in BglG and licT. One major difference is that MtlR possesses two PRDs (PRD1 and PRD2) while GatR only contains PRD2. In contrast to BglG and licT, MtlR from B. subtilis is activated via bacterial membrane sequestration mediated by its cognate membrane EIIBMtl [50–52]. B. subtilis MtlR controls expression of the mtlAFD operon, which mediates the utilization of mannitol [51]. In the absence of mannitol, intracellular P-EIIBMtl prevails, which does not interact with MtlR [52]. Consequently, MtlR is inactive and released into the cytoplasm [52]. In the presence of mannitol, P-EIIBMtl is dephosphorylated and sequesters MtlR to the bacterial membrane [52]. Interactions with the bacterial membrane rather than EIIBMtl appear important for MtlR activation. Based on these results, together with previous findings, we propose that GatR activation is regulated by Gat uptake and further metabolism occurs via a modified glycolytic pathway, known as the tagatose-6-phosphate pathway (Supplementary Figure 5). In S.iniae, EIIBGat may be phosphorylated in the absence of galactitol and thus unable to sequester GatR, which remains inactive in the cytoplasm. If galactitol is present, the compound is phosphorylated and taken up by EIIGat, resulting in dephosphorylation. EIIGat may subsequently interact with the cognate EIIFru-like or EIIBgl-like domain of GatR, leading to its sequestration into the bacterial membrane. Furthermore, PRD2 of GatR is generally phosphorylated by PTS-PEP, PTS-EI and PTS-Hpr for activation. Two conditions therefore need to be fulfilled to render GatR active: (a) phosphorylation at PRD2 by PTS and (b) interaction with unphosphorylated EIIBGat, which sequesters it into the bacterial membrane. In this way, in the presence of galactitol, the gat operon could be effectively expressed for galactitol uptake and further utilization. Therefore, S. iniae YSFST01-82, 9117, CAIM 527, KCTC 11634BP and IUSA1 appear to have evolved a strategy to utilize galactitol sequentially, which may sustain a higher growth rate for these strains in a specific niche.
PZ-I and PZ-II contribute to adaptation in different host niches
We have illustrated the major functions encoded by genes within the PZ-I and PZ-II regions at the complete genome level. Briefly, PZ-I encodes a T7SS-ESS that allows the secretion of specific bacterial proteins into the environment while PZ-II potentially encodes PTSGat that may help bacteria make the most efficient use of galactitol and other available carbon sources. In this study on S. iniae, a critical question is why some strains of S. iniae have evolved PZ-I while others possess PZ-II. Here, we mainly focused on the different geographical distributions and hosts of the nine S. iniae strains under study. Four of these strains were from Asia, two from Europe and the Middle East and three from America (Table 1; Figure 3A, strains with PZ-I are coloured red, strains with PZ-II are blue, strains with both PZ-I and PZ-II are black). S. iniae ISNO is an attenuated vaccine strain selected using novobiocin [53]. Other than ISNO, the geographical distribution of the remaining strains does not appear to be associated with the existence of PZ-I and PZ-II. Interestingly, strains from different hosts contained different plasticity zones. DX09 isolated from Ietalurus Punetaus and ISET0901 (also including ISNO) from Nile tilapia (Oreochromis niloticus) only carried PZ-I. In contrast, 9117, YSFST01-82 and KCTC 11634BP were respectively isolated from Homo sapiens and olive flounder (Paralichthys olivaceus) and only carried PZ-II in their genome. CAIM 527 and IUSA1 contained both PZ-I and PZ-II and were isolated from Inia geoffrensis and Sparus aurata,respectively. Unexpectedly, the strain SF1 isolated from Paralichthys olivaceus carried PZ-I but lacked PZ-II (Figure 3B; the red circle represents the PZ-I-containing strains, blue circle represents the PZ-II-containing strains, and the overlapping part represents strains with both PZ-I and PZ-II). Considering this situation, we further used the complete genome of SF1 as the reference and that of DX09 as the background to assess whether genomic changes exist in SF1. We identified two prophages harbored in SF1, designated Prophage zones 1 and 2, respectively, which were absent in DX09 (Figure 3C), in agreement with a previous complete genome analysis of SF1. Importantly, we identified a specific PTS locus located downstream of Prophage zone 2 within the SF1 genome (Figure 3D). This PTS appears to contribute to trehalose uptake and includes a PRD-containing transcriptional activator. It is therefore conceivable that transfer of Prophage zone 2 into the genome of SF1 affects the expression of specific PTS, resulting in SF1 gaining access to the potential trehalose in its host. In fact, several mobile elements are known to affect gene expression in prokaryotic genomes, including IS, repeat sequence, horizontal gene transfer and integrative conjugative elements. Therefore, SF1 without PZ-II may be able to infect Paralichthys olivaceus as a result of PTS-related specific sugar uptake. Taken together, our results clearly demonstrate that PZ-I mediated T7SS and PTS provide competitive advantages in bacterial evolution and influence the adaptation of bacterial populations or virulence of pathogenic strains.
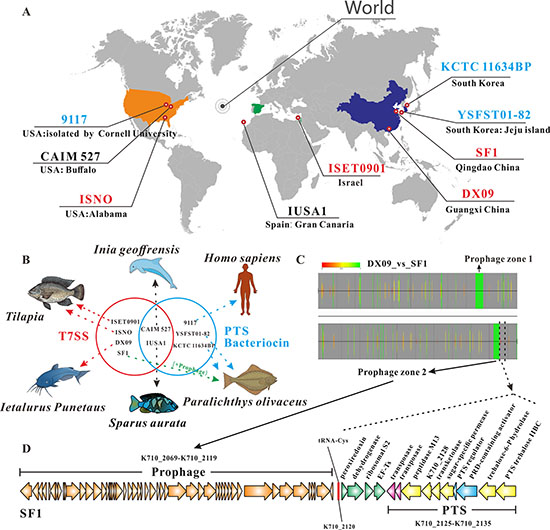
Figure 3: Plastcity zone (PZ)-I and PZ-II contribute to adaptation in different host niches. (A) Geographical distribution of all sequenced Streptococcus iniae. Strains with PZ-I are coloured red; strains with PZ-II are coloured blue; strains with both PZ-I and PZ-II are coloured black. (B) Strains isolated from different hosts present different genome features. PZ-I-containing strains with T7SS are in the red circle; PZ-II-containing strains with PTS and bacteriocin-related locus are in the blue circle; the overlapping region represents strains with both PZ-I and PZ-II. The arrows indicate different hosts for the above strains. (C) Signature analysis of S. iniae SF1 with Gegenees software [54]. A fragmented alignment was performed at 500/500 settings with BLASTN (BLAST+) using S. iniae DX09 as the target genome group and SF1 as the background. The ‘maximum background/average target’ setting was used for biomarker score calculation. Annotations are derived from S.iniae SF1 (GenBank: CP005941). Prophage zones 1 and 2 were identified in SF1. (D) Gene organization in Prophage zone 2 from SF1 and gene cluster with trehalose-PTS located downstream of Prophage zone 2. Images were generated with CorelDRAW X6 software. The base maps in Figure 3B were created using ChemDraw2010 and CorelDRAW X6. Copyright (c) 2015–2016 [Tao Liu], CorelDRAW X6 and ChemDraw2010. All rights reserved.
MATERIALS AND METHODS
Ethics statement
All animal experiments were reviewed and approved by the Committee of Ethics on Animal Care and Experiments at Sichuan Agricultural University. Experimental procedures were perfomed in accordance with the Guidelines for Experimental Animals maintained by the Chinese Ministry of Science and Technology.
Histopathological and transmission electron microscopy analyses of diseased fish infected with S. iniae DX09
Spleen tissue was sampled from naturally infected catfish in Guangxi, China, and prepared for histological examination. Sections of spleen were fixed in 10% formaldehyde and embedded in paraffin. Sections (4 mm in thickness) were stained with Harris haematoxylin-eosin (H&E) and examined under an Olympus light microscope (Japan). To characterize the intracellular presence of S. iniae within primary catfish macrophages, spleen tissue from infected catfish was further examined under a Jeol EX II transmission electron microscope (Jeol, Tokyo, Japan) at 80 kV.
S. iniae DX09 DNA extraction and sequencing
S. iniae DX09 was isolated from diseased I. punctatus in a reservoir cage culture farm (Guangxi, China) and routinely cultured in brain heart infusion (BHI) broth (Oxoid, Basingstoke, UK) at 28°C. Genomic DNA was isolated from 10 mL overnight culture using the TIANamp Bacteria DNA Kit (TIANGEN Biotech, Beijing, China). DNA was dissolved in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and 400 bp genomic DNA libraries subsequently constructed using the Illumina Miseq platform according to the manufacturer’s instructions. Genome sequencing was performed by Novogene (Beijing, China).
Assembly of S. iniae DX09
Low-quality Illumina reads were filtered and high-quality reads used for de novo assembly. Short reads were assembled using the SOAPdenovo alignment tool (version 2.04), a genome assembler developed specifically for next-generation short-read sequences. The SOAP GapCloser was additionally applied to close gaps where possible after assembly.
Sequence analysis and annotation of S. iniae DX09
Protein-coding genes were predicted using Glimmer 3.02, and tRNAscan-SE and RNAmmer used to identify tRNA and rRNA, respectively. The genome sequence was also uploaded onto the Rapid Annotation using Subsystem Technology (RAST) server to check annotated sequences. The functions of predicted protein-coding genes were annotated through comparison with the NCBI-NR, COG, and GO databases. The evolutionary history was inferred using the Neighbor-Joining method based on DNA gyrase (gyrB gene).Evolutionary analyses were conducted in MEGA7.
Pan-genome assembly and genome comparison analysis
To facilitate comparative genomic analysis of all specific regions within all the nine S. iniae genomes, a pangenome was assembled using Gegenees from four complete genomes (YSFST01-82, SF1, ISET0901 and ISNO) and five draft genomes (DX09, this study; 9117, KCTC 11634BP, IUSA1 and CAIM 527). The resulting pangenome was employed as a reference for complete multiple genome BLAST comparisons and viewed using BRIG. Based on Gegenees, we used BRIG to map the signatures of PZ-I, PZ-II and mobile elements to the pangenome for further analysis.
Bioinformatic analysis of PZ-I and PZ-II in genomes
We employed BRIG [37] to map the signatures of PZ-I and PZ-II in DX09 and YSFST01-82 for comparison with other S. iniae genomes. The PZ-I and PZ-II homolog regions in other bacterial genomes were detected with MegaBlast. Individual hits were retrieved and manually searched for the nearby presence of PZ-I/PZ-II hallmark genes, such as integrase, and a gene for O-glycosy[1] hydrolase located near the other end of the plasticity zones. The PZ-I/PZ-II regions of DX09 and YSFST01-82 were isolated in silico from the host genome and pair-wise compared with other S. iniae PZ-I/PZ-II sequences using WebACT (http://www.webact.org/WebACT/home). Regions with nucleotide sequence similarity >77% were exported and displayed on the local gene map using CloningVectors software. The final display was edited for clarity in CorelDRAW X6 software. The ess locus from DX09 was further compared with the homologous T7SS regions from B. subtilis 168, L. monocytogenes EGD-e, S. agalactiae 2603V/R and S. aureus Newman using MicrobesOnline (http://www.microbesonline.org/). Editing and visualization of sequence alignments between DX09-Ess and other T7SS sequences was conducted with Jalview. Three-dimensional structure prediction of EsxA-DX09 was performed with SWISS-MODEL (https://www.swissmodel.expasy.org/) and the data visualized with Chimera 1.9.
Macrosynteny between S. iniae DX09 and S. iniae SF1
A software tool, Gegenees [54], that uses a fragmented alignment approach to facilitate the comparative analysis of hundreds of microbial genomes was employed to compare S. iniae DX09 and S. iniae SF1 genomes based on 200 bp fragment length and step-size of 100 as standard values. The genomes were fragmented and compared, all against all, via a multithreaded BLAST control engine in Gegenees. Gegenees additionally provided a phylogenomic overview of the genomes. The alignment was subsequently mined for genomic regions with conservation patterns matching a defined target group and absent from the background group. SF1 was used as the background in this case. The ‘maximum background/average target’ setting was used for biomarker score calculation. The resulting conservation pattern signatures, such as Prophage zone 1 and Prophage zone 2, could then be viewed and explored graphically.
Author contributions
Yajun Wang and Tao Liu conducted data analysis and wrote the manuscript; Kaiyu Wang provided the conceptual framework and experimental design, and helped with writing of the manuscript; Yajun Wang and Tao Liu performed laboratory work associated with genome sequencing; Tao Liu conducted data analysis and genome assembly as well as histopathology experiments in diseased Ictalurus punctatus. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
FUNDING
This work was supported jointly by Sichuan Technology Support Planning at Sichuan Agricultural University (No. IRT0848) and the Science and Technology Support Program by the Science and Technology Department of Sichuan Province (No. 2014NZ0003).
REFERENCES
1. Locke JB, Colvin KM, Datta AK, Patel SK, Naidu NN, Neely MN, Nizet V, Buchanan JT. Streptococcus iniae capsule impairs phagocytic clearance and contributes to virulence in fish. J bacteriol. 2007; 189:1279–1287.
2. Locke JB, Aziz RK, Vicknair MR, Nizet V, Buchanan JT. Streptococcus iniae M-like protein contributes to virulence in fish and is a target for live attenuated vaccine development. Plos One. 2008; 3:e2824.
3. Chen D, Wang K, Geng Y, Wang J, Huang X, He M. Pathological changes in cultured channel catfish Ictalurus punctatus spontaneously infected with Streptococcus iniae. Dis aquat organ. 2011; 95:203–208.
4. Baiano JC, Barnes AC. Towards control of Streptococcus iniae. Emerg infect dis. 2009; 15:1891.
5. Baiano JC, Tumbol RA, Umapathy A, Barnes AC. Identification and molecular characterisation of a fibrinogen binding protein from Streptococcus iniae. Bmc microbiol. 2008; 8:1.
6. Zinkernagel AS, Timmer AM, Pence MA, Locke JB, Buchanan JT, Turner CE, Mishalian I, Sriskandan S, Hanski E, Nizet V. The IL-8 protease SpyCEP/ScpC of group A Streptococcus promotes resistance to neutrophil killing. Cell host microbe. 2008; 4:170–178.
7. Fuller JD, Camus AC, Duncan CL, Nizet V, Bast DJ, Thune RL, Low DE, De Azavedo JC. Identification of a streptolysin S-associated gene cluster and its role in the pathogenesis of Streptococcus iniae disease. Infect immun. 2002; 70:5730–5739.
8. Purushothaman SS, Hae-Sun P, Cleary PP. Promotion of fibronectin independent invasion by C5a peptidase into epithelial cells in group A Streptococcus. Indian j med res. 2004; 119:44.
9. Heath C, Gillen CM, Chrysanthopoulos P, Walker MJ, Barnes AC. Streptococcus iniae cpsG alters capsular carbohydrate composition and is a cause of serotype switching in vaccinated fish. Vet microbiol. 2016; 193:116–124.
10. Zeng Y, He Y, Wang K, Wang J, Zeng Y, Chen Y, Chen D, Geng Y, OuYang P. cpsJ gene of Streptococcus iniae is involved in capsular polysaccharide synthesis and virulence. Antonie van Leeuwenhoek. 2016; 109:1483–1492.
11. Lowe BA, Miller JD, Neely MN. Analysis of the polysaccharide capsule of the systemic pathogen Streptococcus iniae and its implications in virulence. Infect immun. 2007; 75:1255–1264.
12. Buchanan JT, Stannard JA, Lauth X, Ostland VE, Powell HC, Westerman ME, Nizet V. Streptococcus iniae phosphoglucomutase is a virulence factor and a target for vaccine development. Infect immun. 2005; 73:6935–6944.
13. Eyngor M, Tekoah Y, Shapira R, Hurvitz A, Zlotkin A, Lublin A, Eldar A. Emergence of novel Streptococcus iniae exopolysaccharide-producing strains following vaccination with nonproducing strains. Appl environ microb. 2008; 74:6892–6897.
14. Kim MS, Choi SH, Lee EH, Nam YK, Kim SK, Kim KH. α-enolase, a plasmin (ogen) binding and cell wall associating protein from a fish pathogenic Streptococcus iniae strain. Aquaculture. 2007; 265:55–60.
15. Pancholi V, Fischetti VA. α-Enolase, a novel strong plasmin (ogen) binding protein on the surface of pathogenic streptococci. J biol chem. 1998; 273:14503–14515.
16. Aviles F, Zhang MM, Chan J, Delamare-Deboutteville J, Green TJ, Dang C, Barnes AC. The conserved surface M-protein SiMA of Streptococcus iniae is not effective as a cross-protective vaccine against differing capsular serotypes in farmed fish. Vet microbiol. 2013; 162:151–159.
17. Loman NJ, Pallen MJ. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 2015; 13:787–94.
18. Fricke WF, Rasko DA. Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat rev genet. 2014; 15:49–55.
19. Zhang B, Zhang J, Sun L. Streptococcus iniae SF1: complete genome sequence, proteomic profile, and immunoprotective antigens. Plos one. 2014; 9:e91324.
20. Eyngor M, Chilmonczyk S, Zlotkin A, Manuali E, Lahav D, Ghittino C, Shapira R, Hurvitz A, Eldar A. Transcytosis of Streptococcus iniae through skin epithelial barriers: an in vitro study. Fems microbiol lett. 2007; 277:238–248.
21. Abdallah AM, Nc GVP, Champion PA, Cox J, Luirink J, Vandenbrouckegrauls CM, Appelmelk BJ, Bitter W. Type VII secretion--mycobacteria show the way. Nat rev microbiol. 2007; 5:883–891.
22. Rosenberg OS, Dovala D, Li X, Connolly L, Bendebury A, Finer-Moore J, Holton J, Cheng Y, Stroud RM, Cox JS. Substrates Control Multimerization and Activation of the Multi-Domain ATPase Motor of Type VII Secretion. Cell. 2015; 161:501–512.
23. Houben EN, Korotkov KV, Bitter W. Take five — Type VII secretion systems of Mycobacteria. Biochimica Et Biophysica Acta. 2014; 1843:1707–1716.
24. Warne B, Harkins CP, Harris SR, Vatsiou A, Stanley-Wall N, Parkhill J, Peacock SJ, Palmer T, Holden MT. The Ess/Type VII secretion system ofStaphylococcus aureusshows unexpected genetic diversity. Bmc genomics. 2016; 17:1–13.
25. Anderson M, Aly KA, Chen YH, Missiakas D. Secretion of atypical protein substrates by the ESAT-6 Secretion System of S taphylococcus aureus. Mol microbiol. 2013; 90:734–743.
26. Kneuper H, Cao ZP, Twomey KB, Zoltner M, Jäger F, Cargill JS, Chalmers J, van der Kooi-Pol MM, van Dijl JM, Ryan RP, Hunter WN, Palmer T. Heterogeneity in ess transcriptional organization and variable contribution of the Ess/Type VII protein secretion system to virulence across closely related Staphylocccus aureus strains. Mol microbiol. 2014; 93:928–943.
27. Burts ML, Haselkorn R. EsxA, EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. P natl acad sci usa. 2005; 102:1169–1174.
28. Anderson M, Chen YH, Butler EK, Missiakas DM. EsaD, a Secretion Factor for the Ess Pathway in Staphylococcus aureus. J bacteriol. 2011; 193:1583–1589.
29. Burts ML, Dedent AC, Missiakas DM. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol microbiol. 2008; 69:736–746.
30. Pallen MJ. The ESAT-6/WXG100 superfamily – and a new Gram-positive secretion system? Trends microbiol. 2002; 10:209–212.
31. Houben EN, Bestebroer J, Ummels R, Wilson L, Piersma SR, Jiménez CR, Ottenhoff TH, Luirink J, Bitter W. Composition of the type VII secretion system membrane complex. Mol microbiol. 2012; 86:472–484.
32. Simeone R, Bottai D, Frigui W, Majlessi L, Brosch R. ESX/type VII secretion systems of mycobacteria: Insights into evolution, pathogenicity and protection. Tuberculosis. 2015; 95:S150-S154.
33. Desvaux M, Hébraud M, Talon R, Henderson IR. Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends microbiol. 2009; 17:139–145.
34. Pallen MJ. The ESAT-6/WXG100 superfamily – and a new Gram-positive secretion system? Trends microbiol. 2002; 10:209–212.
35. Bae T, Haselkorn R. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proceedings of the National Academy of Sciences. 2004; 101:12312–12317.
36. Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. Embo j. 2005; 24:2491–2498.
37. Alikhan NF, Petty NK, Zakour NLB, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. Bmc genomics. 2011; 12:402.
38. Garzetti D, Bouabe H, Heesemann J, Rakin A. Tracing genomic variations in two highly virulent Yersinia enterocolitica strains with unequal ability to compete for host colonization. Bmc genomics. 2012; 13:1–15.
39. Miyazaki R, Bertelli C, Benaglio P, Canton J, De Coi N, Gharib WH, Gjoksi B, Goesmann A, Greub G, Harshman K. Comparative genome analysis of Pseudomonas knackmussii B13, the first bacterium known to degrade chloroaromatic compounds. Environ microbiol. 2015; 17:91–104.
40. Nobelmann B, Lengeler JW. Molecular analysis of the gat genes from Escherichia coli and of their roles in galactitol transport and metabolism. J bacteriol. 1996; 178:6790–6795.
41. Deutscher J, Francke C, Postma PW. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiology and Molecular Biology Reviews. 2006; 70:939–1031.
42. Görke B, Stülke J. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat rev microbiol. 2008; 6:613–624.
43. Dobson A, Cotter PD, Ross RP, Hill C. Bacteriocin production: a probiotic trait? Applied and Environmental Microbiology. 2012; 78:1–6.
44. Rebecca FR, Elizabeth MN, Frank Löhr A, Christopher TW. Maturation of an Escherichia coli Ribosomal Peptide Antibiotic by ATP-Consuming N−P Bond Formation in Microcin C7. J am chem soc. 2008; 130:3603–3609.
45. Kjos M, Snipen L, Salehian Z, Nes IF, Diep DB. The abi proteins and their involvement in bacteriocin self-immunity. J bacteriol. 2010; 192:2068–2076.
46. Rothe FM, Bahr T, Stülke J, Rak B, Görke B. Activation of Escherichia coli antiterminator BglG requires its phosphorylation. Proceedings of the National Academy of Sciences. 2012; 109:15906–15911.
47. Amster-Choder O. The bgl sensory system: a transmembrane signaling pathway controlling transcriptional antitermination. Curr opin microbiol. 2005; 8:127–134.
48. Tortosa P, Declerck N, Dutartre H, Lindner C, Deutscher J, Le CD. Sites of positive and negative regulation in the Bacillus subtilis antiterminators LicT, SacY. Mol microbiol. 2001; 41:1381–1393.
49. Lindner C, Hecker M, Le CD, Deutscher J. Bacillus subtilis mutant LicT antiterminators exhibiting enzyme I- and HPr-independent antitermination affect catabolite repression of the bglPH operon. J bacteriol. 2002; 184:4819–4828.
50. Henstra SA, Duurkens RH, Robillard GT. Multiple phosphorylation events regulate the activity of the mannitol transcriptional regulator MtlR of the Bacillus stearothermophilus phosphoenolpyruvate-dependent mannitol phosphotransferase system. J biol chem. 2000; 275:7037–7044.
51. Joyet P, Derkaoui MS, Deutscher J. Control of Bacillus subtilis mtl operon expression by complex phosphorylation-dependent regulation of the transcriptional activator MtlR. Mol microbiol. 2010; 76:1279–1294.
52. Joyet P, Bouraoui H, Aké FMD, Derkaoui M, Zébré AC, Cao TN, Ventroux M, Nessler S, Noirot-Gros MF, Deutscher J. Transcription regulators controlled by interaction with enzyme IIB components of the phosphoenolpyruvate:sugar phosphotransferase system. Biochimica Et Biophysica Acta. 2013; 1834:1415–1424.
53. Pridgeon JW, Zhang D, Zhang L. Complete Genome Sequence of the Attenuated Novobiocin-Resistant Streptococcus iniae Vaccine Strain ISNO. Genome Announcements. 2014; 2.
54. Agren J, Sundström A, Håfström T, Segerman B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. Plos one. 2012; 7:e39107.
55. Rajoo S, Jeon W, Park K, Yoo S, Yoon I, Lee H, Ahn J. Complete Genome Sequence of Streptococcus iniae YSFST01–82, Isolated from Olive Flounder in Jeju, South Korea. Genome Announcements. 2015; 3.
56. Richards VP, Palmer SR, Pavinski Bitar PD, Qin X, Weinstock GM, Highlander SK, Town CD, Burne RA, Stanhope MJ. Phylogenomics and the Dynamic Genome Evolution of the Genus Streptococcus. Genome biol evol. 2014; 6:741–753.
57. Pier GB, Madin SH. Streptococcus iniae sp. nov. , a Beta-Hemolytic Streptococcus Isolated from an Amazon Freshwater Dolphin, Inia geo ffrens is. International Journal of Systematic Bacteriology. 1976; 26:545–553.
58. Pridgeon JW, Zhang D, Zhang L. Complete Genome Sequence of a Virulent Strain, Streptococcus iniae ISET0901, Isolated from Diseased Tilapia. Genome Announcements. 2014; 2.
59. El AF, Acosta F, Real F, Padilla D. Whole-Genome Sequence of the Fish Virulent Strain Streptococcus iniae IUSA-1, Isolated from Gilthead Sea Bream (Sparus aurata) and Red Porgy (Pagrus pagrus). Genome Announcements. 2013; 1.
60. Choi HS, Kwon MG, Kim MS, Park MA, Kim DW, Park JY, Kim JS, Na YJ, Kim MY, Kim DS. Draft Genome Sequence of Beta-Hemolytic Streptococcus iniae KCTC 11634. Genome Announcements. 2013; 1.