
INTRODUCTION
Glioblastoma (GBM), the most commonly diagnosed malignant brain tumor in adults [1], has a poor prognosis, with a median survival of only 12-15 months and a high rate of recurrence [2]. Poor prognosis and overall survival (OS) is attributable to the marked inter- and intra-tumoral genetic heterogeneity of GBM tumors [3–6]. Magnetic resonance imaging (MRI) holds great potential for characterizing the phenotypic heterogeneity of GBMs by inferring this from textural information and intensity variations in radiological images. Common techniques such as gadolinium contrast-enhanced T1-weighted imaging highlight perfusion variations in tumor images and advanced image-texture analysis may be able to characterize signal intensity variations within tumors. Texture analysis has many applications in medical image processing and provides one approach to quantify the distribution of gray-level patterns such as homogeneity, entropy, etc., within a set of imaging data [7]. Multiple methods for assessing imaging features and characterizing pixel intensity distributions by quantifying gray levels have been described [8–11]. These methods allow for rigorous and reproducible derivation of detailed, pertinent information, and have been used to analyze MRI features, such as apparent diffusion coefficient, 2-dimensional (2D) spatial habitats [12], and texture features. These characteristics correlate with the grade of disease, patient survival, response to chemotherapy, and genetic and epigenetic status [12–18]. With recent advances in radiomics and radiogenomics (or imaging-genomcis), molecular and genetic heterogeneity can be inferred from MRI features by correlating imaging datasets with corresponding molecular and clinical information.
Several studies have correlated specific features seen in MRI of GBM with patient survival and molecular subtype. However, these investigations have been restricted to 1 or 2 particular MRI sequences and typically considered only features from a single 2D slice [12, 16, 17]. In contrast to these single-sequence, slice-by-slice analyses, radiologists review all acquired MRI sequences in their assessments. Additional research is needed to develop methods for extracting computational radiological features from full multiparametric MR imaging sets. To this end, this study focuses on using four MR sequences to understand the heterogeneity of a tumor region.
We hypothesized that by performing a 3D volumetric analysis of commonly available MRI sequences, we could identify particular imaging habitats [17] correlated with both patient overall survival status and identify key genetic pathways associated with such habitats in GBM. Comprehensive analysis of imaging data could improve the predictive power of this approach and provide novel insights to aid clinical decision-making.
RESULTS
Identifying imaging habitats
Eighty-five patients with GBM identified in the Cancer Genome Atlas who had imaging, clinical, and genomic data available were included in this study. Table 1 shows the patient and respective tumor characteristics within 85 TCGA-GBM cases. Four pre-operative MR scans were obtained for each case: Pre-contrast T1-weighted (T1) image, post-gadolinium T1 (T1c) image, T2-weighted (T2) image, and T2 fluid-attenuated inversion recovery (FLAIR) image. Following skull stripping, rigid registration, and automated segmentation using Brain Tumor Image Analysis (BraTumIA) software [19], we grouped tumor voxels into high and low signal bins for each of the 4 MRI sequences (FLAIR, T1, T1c, and T2) via Kmeans clustering. Across these 4 MR sequences, this leads to the identification of 16 imaging habitats [17] (i.e 24 combinations) based on unique combinations of these high and low signals. The habitats were assigned labels from 0 to 15, with “0” being low-intensity in all 4 acquisition modalities (i.e., FLAIR=0, T1=0, T1c = 0, T2 = 0) and “15” being high-intensity in all 4 (i.e., FLAIR=1, T1=1, T1c = 1, T2 = 1). For instance, habitat 2 represents low intensity in FLAIR, T1, and T2 with high intensity in T1c. Figure 1 describes the process for obtaining these 16 imaging habitats.
Table 1: Clinical data from 85 TCGA patients with primary GBM
Patient or tumor characteristics |
Number of patients (%) |
|---|---|
Age |
|
Median |
59 years |
Range |
18-84 years |
Sex |
|
Male |
65 (%) |
Female |
35 (%) |
Overall survival median (range) |
11.7 (0.5 – 90.9) months |
Disease-free survival median (range) |
6.64 (0.7 – 57.7) months |
IDH1 mutation |
|
Yes |
2 (2) |
No |
83 (98) |
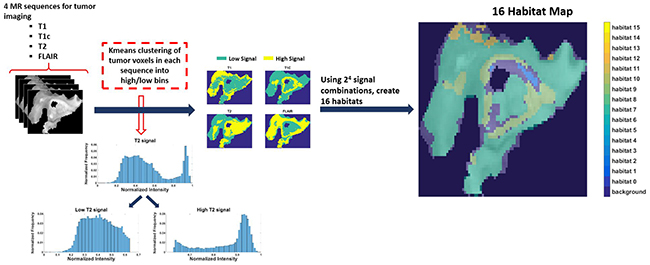
Figure 1: The process of generating 16 spatial habitats. Based on 4 MR sequences (multi-parametric MRI scans), classify each voxel within the tumor volume into high and low categories via kmeans clustering. With 16 (24) signal combinations across the 4 sequences (i.e. 0000-1111), every voxel in the tumor volume can be identified uniquely. The resultant habitat map shows the spatial heterogeneity within tumor.
Identifying important and significant imaging habitats using clinical data
Random survival forest modeling and Cox proportional hazards regression analysis determined imaging habitats 2,7, and 10 to be both important (+ve variable importance) and significant (p-value < 0.05) for determining OS after adjustment for covariates of age, Karnofsky performance score, tumor volume, and IDH1 mutation status (P < 0.05, Supplementary Table 1). These three habitats were thus designated as “relevant” habitats. The process of acquiring these relevant habitats is described in Figure 2. They represent distinct tumor sub-regions of clinical relevance to outcome in GBM, after adjusting for clinical covariates. Their relevance, coupled with the availability of matched genomic data for these patients, enables us to study the molecular mechanisms (pathway activities) associated with the presence of these habitats in GBM.
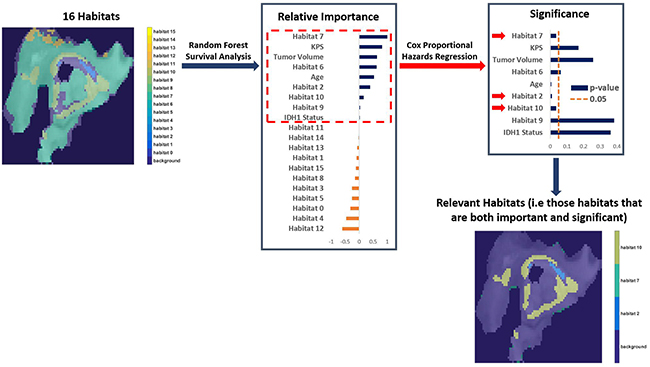
Figure 2: The process of finding important and significant (designated “Relevant”) habitats. After adjusting for clinical covariates (age, Karnofsky performance score, tumor volume, and IDH1 mutation status), we identified important habitats (positive variable importance) via Random Forest survival analysis. Those habitats were then assessed for significance via Cox Proportional Hazards Regression to determine overall survival (OS). Only habitat 2,7, and 10 are both important and significant (i.e “relevant”) in determining OS.
Associating relevant imaging habitats with canonical tumor sub-volumes and genetic pathways
We used the pair-wise Spearman rank test to correlate the amount of each relevant habitat in the tumor with the amount of necrotic, enhancing, non-enhancing, and edema regions within the tumor (Table 2). In particular, habitat 2 was also associated with necrosis (p = 0.0172). For habitat 7 and 10, there was no specific association with canonical tumor sub-volumes, suggesting the need for deeper examination of their physiology and molecular context.
Table 2: Pair-wise Spearman correlation p-values for proportions* of relevant habitats (i.e. those that are important & significant) associated with canonical tumor sub-volumes (edema, necrosis, enhancing, and non-enhancing regions)
Habitat |
Necrosis |
Non-enhancing |
Enhancing |
Edema |
|---|---|---|---|---|
2 |
0.0172 |
0.1003 |
0.2274 |
0.9239 |
7 |
0.1112 |
0.3356 |
0.2670 |
0.1680 |
10 |
0.0937 |
0.8381 |
0.2670 |
0.1680 |
* Habitat proportions are the fraction of tumor pixels belonging to each imaging habitat computed for each patient.
To study the relationships between relevant habitat proportions and molecular pathway data [20], we performed Dirichlet regression analysis. Table 3 lists the key genetic pathways that are most strongly correlated with the relevant habitats. In particular, habitat 2 was positively associated with positive regulation of NFκB transcription-factor activity; while negatively associated with dendrite morphogenesis. Habitat 7 is correlated positively with DNA damage response signal transduction resulting in induction of apoptosis and macrophage activation. Further, habitat 7 was correlated negatively with immune cell activity (monocyte differentiation). Habitat 10 shows positive association with activity of signal transducers and activators of transcription-1 (STAT-1) and Natural killer cell activation, while showing negative correlated with ion channel activity (potassium channel inhibitor activity and voltage gated calcium channel activity).
Table 3: Genetic pathways that are associated (top five in magnitude) with important-significant (i.e “relevant”) imaging habitats
Habitat 2 |
Habitat 7 |
Habitat 10 |
|
|---|---|---|---|
Upregulated Pathways |
Positive regulation of NFκB transcription-factor activity |
DNA damage response signal transduction by p53 class mediator resulting in induction of apoptosis |
Positive regulation of tyrosine phosphorylation of STAT-1 |
Sphinganine 1-phosphate |
Macrophage activation |
Natural killer cell activation |
|
T cell proliferation during immune response |
Neuron projection morphogenesis |
Wnt receptor signaling pathway |
|
Cytokine production during immune response |
Positive regulation of tyrosine phosphorylation of STAT-1 |
Mol beta2 estradiol |
|
T helper 2 cell differentiation |
Natural killer cell activation |
cAMP biosynthetic process |
|
Downregulated Pathways |
Dendrite morphogenesis |
Monocyte differentiation |
Potassium channel inhibitor activity |
Calcineurin A alpha beta B1 |
Response to DNA damage stimulus |
Voltage gated calcium channel activity |
|
Proteasomal ubiquitin dependent protein catabolic process |
T-cell differentiation |
Positive regulation of cyclin dependent protein kinase activity |
|
IGF 1R heterotetramer |
Cell morphogenesis |
Regulation of S phase of mitotic cell cycle |
|
Negative regulation of DNA binding |
Actin cytoskeleton reorganization |
Schwann cell development |
DISCUSSION
Through analysis of multi-parametric MR imaging, we have identified tumor sub-regions with unique combinations of gray-level intensities from each of the four MR modalities included in this study. From these 16 habitats, relevant (i.e. important and significant) habitats were identified based on association with OS status, and further, correlated with morphological and pathological characteristics of tumor, such as leading edge, infiltrating tumor into normal brain, edema, and enhancement around lesion edge. For example, habitat 10 was associated with canonical tumor sub-volumes like edema, peripheral tumor tissue, and enhancement around lesion edge.
Our analysis reveals that the intratumoral abundance of these habitats are associated with outcome. Delineating the extent and location of these aggressive habitats can have implications for delivery of radiotherapy (boosting RT to aggressive habitat areas), surgical resection (an aggressive habitat not impinging on an eloquent area is amenable for possibly complete resection). Additionally, tracking/monitoring the growth of aggressive habitat subvolumes can provide a deeper understanding of disease evolution/recurrence instead of just gross tumor volume. The utility of defining heterogeneity in glioblastoma thus is closely related with prognosis and overall survival of patients [1–4]. Hence, defining such phenotypic heterogeneity is directly relevant for treatment planning, surgical intervention, disease monitoring, and prognosis estimation in the clinic.
Dirichlet regression implicated each relevant habitat with pathway alterations due to unique upstream transcriptional regulators and signaling activity. Imaging-derived habitats also showed some common cellular processes and pathway activity, such as natural killer cell and STAT-1 signaling. These findings are consistent with hallmarks of cancer [21, 22], such as avoiding cell death (natural killer cell) and inducing angiogenesis (tyrosine phosphorylation of STAT-1).
Recent literature supports the relationships inferred between clinical pathologies for habitats (Table 4) and pathway alterations (Table 3). For instance, it is reported that that overexpression of inflammatory cytokines is linked with the leading edge of the gliomas. This is consistent with our findings (positive association of cytokine production) in habitat 2, which is associated with the leading edge of the tumor [23]. Furthermore, it is well known that glioblastoma is infiltrated by diverse immune cells including macrophages [24], which might explain the positive association of macrophage activation and habitat 7. It is also reported that potassium channel inhibitor activity plays a critical role in cell proliferation and cell swelling in neuroblastoma and gliomas [25]. Further, Sforna et al. [26] reported that calcium ions channels play a role in swelling in GBM. These reports are consistent with our clinical pathology interpretation, edema, for habitat 10.
Table 4: Interpretation of important-significant (i.e. relevant) imaging habitats based on multiparametric MR: ‘0’ denotes low in signal intensity and ‘1’ denotes high in signal intensity
Habitat number |
Enhancement combination |
Clinical pathology |
|||
|---|---|---|---|---|---|
FLAIR |
T1 |
T1C |
T2 |
||
2 |
0 |
0 |
1 |
0 |
Leading edge of the tumor |
7 |
0 |
1 |
1 |
1 |
Overall tumor mass including leading edge and infiltrating tumor into normal brain |
10 |
1 |
0 |
1 |
0 |
Edema, peripheral tumor tissue, enhancement around lesion edge |
Our study has some potential limitations. This retrospective study relied on publicly available imaging data acquired across multiple clinical sites. Thus there were some variations in imaging acquisition/sequence parameters (e.g., Relaxation Time and Echo Time) and hardware (e.g., magnetic field strength and receiver coil geometry). In order to mitigate these effects, and in accordance with standard practice, intensity normalization and registration were applied to each image prior to habitat analysis. However, any remaining intensity variations could have potentially affected initial tumor segmentation. A systematic study with standardized image acquisition protocols is needed in order to validate the robustness of these imaging-derived habitat characteristics. Another aspect of uncertainty in this study is derived from the patient outcome data. Because these patients were treated at different institutions, aspects of their treatment regimens may not have been fully standardized, which could potentially affect patient OS duration. Additionally, an inherent challenge with all genomic information derived from tumor specimens is the inability to account for spatial heterogeneity of genomic alterations and tumor cell clones. As with the quality of the imaging data described above, the robustness of the genomic data is also subject to similar uncertainties that can influence the overall generalizability of our conclusions. Also, the presented relationships between habitats and genetic pathway alterations are inferred based on statistical regression methods. These findings could potentially form the basis for targeted perturbation experiments in-vivo to illuminate the mechanistic or causative nature of the relationships between habitats and pathway activity. While outside the scope of this study, research efforts in this direction [27, 28] are essential before these associations can be exploited therapeutically. However, the process of habitat inference and its association mining for prognostic intent can be done quite readily in the current clinical scenario since these 4 MR sequences (T1, T1c, T2, FLAIR) are used routinely.
MATERIALS AND METHODS
Datasets
One hundred patients with primary GBM were identified from The Cancer Genome Atlas (TCGA). Patient image data and corresponding clinical data were extracted from The Cancer Imaging Archive [35]. All patients had undergone routine MRI prior to surgery and treatment. Pre-contrast T1-weighted (T1) images, post-gadolinium T1 (T1c) images, T2-weighted (T2) images, and T2 fluid-attenuated inversion recovery (FLAIR) images were acquired. Patient genomic and molecular data were obtained from cBioPortal [36] and included data on genetic pathway activation (PARADIGM scores [20]). This study abided by the TCGA data use agreement and was Institutional Review Board-exempt.
Of the initial 100 GBM patients identified, 85 patients had a complete set of imaging, clinical, and genomic data available. These patients were separated into 2 survival groups: OS (OS) ≥ 12 months and OS < 12 months. The acquisition parameters for the four MRI sequences used in this study are as follows – T1 (echo time: 15-8.5 ms, relaxation time: 642-400 ms), T1 post contrast (echo time: 15-8.5 ms, relaxation time: 700-400 ms), T2 (echo time: 120-30 ms, relaxation time: 6000-2140 ms) and FLAIR (echo time: 150-90 ms, relaxation time: 11000-6000 ms).
Image processing, image registration and segmentation
For our study, Brain Tumor Image Analysis (BraTumIA) software [19] was used to perform image preprocessing steps (skull stripping and rigid registration), followed by automated segmentation of normal structures (cerebrospinal fluid, gray matter, and white matter) and diseased tissue (necrosis, edema, non-enhancing tissue, and enhancing tissue) [37].
Image habitat analysis
Patient images and the BraTumIA-derived segmentation masks were loaded into MATLAB for habitat analysis. For habitat analysis, we used only the regions of the MR image associated with tumor (i.e. necrosis, edema, non-enhancing tissue, and enhancing tissue). These BraTumIA segmentation labels were combined to create one binary mask of the tumor region for each patient. The masks were applied to each scan to extract intensity values from the tumor. The tumor intensity values were first scaled relative to gray matter and white matter intensity and then subsequently, linearized (scaled from 0 to 1) based on the maximum and minimum in each image. Following Zhou et.al [17] K-means clustering was applied to each MR sequence type (FLAIR, T1, T1c, and T2) across all patients to derive sequence-specific, intensity thresholds that separate high-intensity pixel values from low-intensity pixel values. Each sequence image for each patient was then dichotomized into sub-regions of high or low enhancement (1 or 0, respectively). The combinations of high and low enhancement for each patient resulted in 16 possible imaging habitats, where habitat 0 is represents regions with low-intensity (FLAIR = 0, T1 = 0, T1+C = 0, T2 = 0), habitat 3 represents regions with characteristics (FLAIR = 0, T1 = 0, T1+C = 1, T2 = 1), and habitat 15 represents regions having high-intensity in all acquisitions (FLAIR = 1, T1 = 1, T1+C = 1, T2 = 1). A 3D spatial representation of the imaging habitats was created using these labels. Subsequently, habitat proportions, the fraction of tumor pixels belonging to each imaging habitat, were calculated for each patient. The process of obtaining 16 habitats is described in Figure 1.
Associating imaging habitats with clinical and genomic data
Statistical analysis was performed using R statistical software (R Foundation, Vienna, Austria) with the “survival” (v 2.38-2), “randomForestSRC” (v 2.0.5), and “DirichletReg” (v 0.6-3) packages. Random survival forest (RSF) regression was applied to the imaging habitat proportions to determine if any habitats were associated with OS [16, 38, 39]. The random forest regression identified a subset of habitats that were deemed “important.” Cox proportional hazard regression analysis was then used to determine the association of these (RSF-derived) important habitats with OS (p-values less than 0.05) [40–42], after adjusting for clinical covariates (age, volume, karnofsky score: KPS, IDH1 mutation). Image habitats that were deemed important based on random survival forest regression, and significant from the Cox proportional hazard regression were designated “relevant”. The process of obtaining relevant habitats is depicted in Figure 2. These relevant habitats were then correlated with BraTumIA-derived tumor volume segmentations (edema, necrosis, enhancing regions, and non-enhancing regions) via pair-wise Spearman rank correlation tests. In addition, Dirichlet regression was applied to each relevant habitat to determine if that habitat was associated with pathway activation (PARADIGM) scores. This analysis generated molecular signatures associated with each relevant imaging habitat. The significance of the Dirichlet regression for each pathway was determined by calculating a Bonferroni-adjusted P value, and the strength of association was determined from the absolute value of the Dirichlet regression coefficient.
CONCLUSIONS
This study has elucidated a nuanced relationship between imaging habitats derived from MR images, clinical characteristics, and molecular data in a cohort of GBM patients. The present study implemented an automated methodology that parallels the traditional practice of a neuroradiologist by considering the totality of 3D imaging data (i.e. across multiple MR sequences) from each patient. This technique represents an advance in imaging-genomics. Whereas previous studies derived associated imaging features and genomic data from 2D imaging datasets [12, 29, 30], this study adopted a 3D approach, which is consistent with the progress of the field towards studying 3D image features [10, 31–34] for imaging-genomic analysis. Our approach utilized a multi-parametric representation of 3D image features derived from different imaging sequences overlaid upon each other to generate an imaging phenotype that predicts OS duration, after adjusting for clinical covariates.
This work revealed associations between MRI-derived habitats and oncogenic molecular mechanisms in patients with GBM. The analytical framework and workflow demonstrated in this study are inherently scalable to any number of MR sequences. The four MRI sequences used, FLAIR, T1, T1+C, and T2, are readily available on most modern scanners and used routinely in clinical practice. More advanced sequences such as susceptibility-weighted imaging, diffusion-weighted imaging, or MRI spectroscopy could be incorporated into this framework in a fairly linear manner (with ‘k’ MR sequences, you have 2k habitats). Furthermore, with appropriate imaging and tumor segmentation, this workflow could accommodate data from other tumor types and/or other anatomical sites.
Pathway analysis of the molecular data associated with each habitat showed significantly altered molecular pathways, supporting the hypothesis that each radiographically-distinct tumor habitat is associated with distinct molecular characteristics. Some of these altered pathways pertain to angiogenesis, while others correlate with signaling pathways known to be co-opted by tumor cells to facilitate their avoidance of cell death. The relationships elucidated by this study between imaging habitats and underlying biology may offer additional information regarding the patient’s disease state, complementing inference based on genomics alone. Future work will extend this analysis by considering tumor location, extent of resection, methylguanine-DNA methyltransferase (MGMT) promoter methylation status, as well as extending this work to other tumor types and anatomical sites. A systematic assessment of the impact of acquisition parameter variability on robustness of imaging habitats will also need to be performed before clinical adoption. These results also lay the groundwork for investigations of targeted hypotheses from MRI-guided biopsies [27, 28] in GBM with corresponding genomic analyses to confirm and validate these phenotypic-genomic relationships.
Author contributions
Below is a list of the contribution of all authors towards this study:
Conception and design: Arvind Rao, Katherine Dextraze, Abhijoy Saha
Development of methodology: Arvind Rao, Katherine Dextraze, Abhijoy Saha, Donnie Kim, Shivali Narang, Michael Lehrer
Acquisition of data: Shivali Narang, Anita Rao, Saphal Narang, Arvind Rao
Analysis and Interpretation of Data: Donnie Kim, Shivali Narang, Michael Lehrer, Ganesh Rao, Arvind Rao, Venkatesh Madhugiri, Michelle M. Kim, Sunil Krishnan
Writing, review, and/or revision of manuscript: All authors
Administrative, technical, or material support (i.e., reporting or organizing data, constructing database): All authors
ACKNOWLEDGMENTS
We gratefully acknowledge the support of the R and MATLAB communities.
CONFLICTS OF INTEREST
There are no competing financial interests.
FUNDING
This work was supported by the Cancer Center Support Grant’s Bioinformatics Shared Resource (P30 CA01667 to A.R.), a Research Scholar Grant from American Cancer Society ((RSG-16-005-01) to A.R., a Career Development Award from the MD Anderson Brain Tumor SPORE to A.R., National Institutes of Health grant (K08 NS070928 to G.R.), and a Cancer Prevention and Research Institute of Texas Research training award (No. RP140106 to K.D.).
REFERENCES
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63:11–30.
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
3. Barajas RF Jr, Phillips JJ, Parvataneni R, Molinaro A, Essock-Burns E, Bourne G, Parsa AT, Aghi MK, McDermott MW, Berger MS, Cha S, Chang SM, Nelson SJ. Regional variation in histopathologic features of tumor specimens from treatment-naive glioblastoma correlates with anatomic and physiologic MR imaging. Neuro Oncol. 2012; 14:942–954.
4. Eder K, Kalman B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol Oncol Res. 2014; 20:777–787.
5. Eoli M, Menghi F, Bruzzone MG, De Simone T, Valletta L, Pollo B, Bissola L, Silvani A, Bianchessi D, D’Incerti L, Filippini G, Broggi G, Boiardi A, et al. Methylation of O6-methylguanine DNA methyltransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin Cancer Res. 2007; 13:2606–2613.
6. Shukla S, Pia Patric IR, Thinagararjan S, Srinivasan S, Mondal B, Hegde AS, Chandramouli BA, Santosh V, Arivazhagan A, Somasundaram K. A DNA methylation prognostic signature of glioblastoma: identification of NPTX2-PTEN-NF-kappaB nexus. Cancer Res. 2013; 73:6563–6573.
7. Castellano G, Bonilha L, Li LM, Cendes F. Texture analysis of medical images. Clin Radiol. 2004; 59:1061–1069.
8. Fernandez M, Mavilio A. Texture analysis of medical images using the wavelet transform. AIP Conf Proc. 2002; 630:164–168.
9. Heinonen T, Arola T, Kalliokoski A, Dastidat P, Rossi M, Soimakillio S, Hyttinen J, Eskola H. “Computer aided diagnosis tool for the segmentation and texture analysis of medical images.” In World Congress on Medical Physics and Biomedical Engineering. Springer Berlin Heidelberg. Munich, Germany, pp. 274–276. 2009.
10. Tesar L, Daniel S, Akinobu S, Hidefume K. “3D extension of Haralick texture features for medical image analysis.” In SPPR 2007 Proceedings of the Fourth Conference on IASTED International Conference. pp. 350–355. 2007.
11. Tesar L, Shimizu A, Smutek D, Kobatake H, Nawano S. Medical image analysis of 3D CT images based on extension of Haralick texture features. Comput Med Imaging Graph. 2008; 32:513–520.
12. Lee J, Narang S, Martinez J, Rao G, Rao A. Spatial habitat features derived from multiparametric magnetic resonance imaging data are associated with molecular subtype and 12-month survival status in glioblastoma multiforme. PLoS One. 2015; 10:e0136557.
13. Barajas RF Jr, Hodgson JG, Chang JS, Vandenberg SR, Yeh RF, Parsa AT, McDermott MW, Berger MS, Dillon WP, Cha S. Glioblastoma multiforme regional genetic and cellular expression patterns: influence on anatomic and physiologic MR imaging. Radiology. 2010; 254:564–576.
14. Levner I, Drabycz S, Roldan G, De Robles P, Cairncross JG, Mitchell R. “Predicting MGMT methylation status of glioblastomas from MRI texture.” In International Conference on Medical Image Computing and Computer-Assisted Intervention. Springer, Berlin, Heidelberg. pp. 522–530. 2009.
15. Rao A, Rao G, Gutman DA, Flanders AE, Hwang SN, Rubin DL, Colen RR, Zinn PO, Jain R, Wintermark M, Kirby JS, Jaffe CC, Freymann J, et al. A combinatorial radiographic phenotype may stratify patient survival and be associated with invasion and proliferation characteristics in glioblastoma. J Neurosurg. 2016; 124:1008–17.
16. Yang D, Rao G, Martinez J, Veeraraghavan A, Rao A. Evaluation of tumor-derived MRI-texture features for discrimination of molecular subtypes and prediction of 12-month survival status in glioblastoma. Med Phys. 2015; 42:6725.
17. Zhou M, Hall L, Goldgof D, Russo R, Balagurunathan Y, Gillies R, Gatenby R. Radiologically defined ecological dynamics and clinical outcomes in glioblastoma multiforme: preliminary results. Transl Oncol. 2014; 7:5–13.
18. Zinn PO, Mahajan B, Sathyan P, Singh SK, Majumder S, Jolesz FA, Colen RR. Radiogenomic mapping of edema/cellular invasion MRI-phenotypes in glioblastoma multiforme. PLoS One. 2011; 6:e25451.
19. Bauer S, Fejes T, Meier R, Reyes M, Slotboom J, Porz N, Pica A, Wiest R. BraTumIA - a software tool for automatic brain tumor image analysis. Technical Report. 2013.
20. Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010; 26:i237–i245.
21. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
22. Vlahopoulos SA, Cen O, Hengen N, Agan J, Moschovi M, Critselis E, Adamaki M, Bacopoulou F, Copland JA, Boldogh I, Karin M, Chrousos GP. Dynamic abberant NF-κβ spurs tumorigenesis: a new model encompassing the microenvironment. Cytokine Growth Factor Rev. 2015; 26:389–403.
23. Baylock RL. Immunoexcitatory mechanisms in glioma proliferation, invasion and occasional metastasis. Surg Neurol Int. 2013; 4:15.
24. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, Liebelt BD, Yaghi NK, Ezhilarasan R, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016; 1:e85841.
25. Rouzaire-Dubois B, Dubois JM. K+ channel block-induced mammalian neuroblastoma cell swelling: a possible mechanism to influence proliferation. J Physiology. 1998; 510:93–102.
26. Sforna L, Cenciarini M, Belia S, D’Adamo MC, Pessia M, Franciolini F, Catacuzzeno L. The role of ion channels in the hypoxia-induced aggressiveness of glioblastoma. Front Cell Neurosci. 2015; 8:457.
27. Hu LS, Ning S, Eschbacher JM, Gaw N, Dueck AC, Smith KA, Nakaji P, Plasencia J, Ranjbar S, Price SJ, Tran N, Loftus J, Jenkins R, et al. Multi-parametric MRI and texture analysis to visualize spatial histologic heterogeneity and tumor extent in glioblastoma. PLoS One. 2015; 10:e0141506.
28. Hu LS, Ning S, Eschbacher JM, Baxter LC, Gaw N, Ranjbar S, Plasencia J, Dueck AC, Peng S, Smith KA, Nakaji P, Karis JP, Quarles CC, et al. Radiogenomics to characterize regional genetic heterogeneity in glioblastoma. Neuro Oncol. 2016; 19:18–137.
29. Chowdhury R, Ganeshan B, Irshad S, Lawler K, Eisenblätter M, Milewicz H, Rodriguez-Justo M, Miles K, Ellis P, Groves A, Punwani S, Ng T. The use of molecular imaging combined with genomic techniques to understand the heterogeneity in cancer metastasis. Br J Radiol. 2014; 87:20140065.
30. Weiss GJ, Ganeshan B, Miles KA, Campbell DH, Cheung PY, Frank S, Korn RL. Noninvasive image texture analysis differentiates K-ras mutation from pan-wildtype NSCLC and is prognostic. PLoS One. 2014; 9:e100244.
31. Knogler T, Thomas K, El-Rabadi K, Weber M, Karanikas G, Mayerhoefer ME. Three-dimensional texture analysis of contrast enhanced CT images for treatment response assessment in Hodgkin lymphoma: comparison with F-18-FDG PET. Med Phys. 2014; 41:121904.
32. Ghosh P, Tamboli P, Vikram R, Rao A. Imaging-genomic pipeline for identifying gene mutations using three-dimensional intra-tumor heterogeneity features. J Med Imaging (Bellingham). 2015; 2:041009.
33. Maani R, Yang YH, Kalra S. Voxel-based texture analysis of the brain. PLoS One. 2015; 10:e0117759.
34. Chen W, Giger ML, Li H, Bick U, Newstead GM. Volumetric texture analysis of breast lesions on contrast-enhanced magnetic resonance images. Magn Reson Med. 2007; 58:562–571.
35. The Cancer Imaging Archive. https://public.cancerimagingarchive.net/ncia/. 2016.
36. cBioPortal for Cancer Genomics. http://www.cbioportal.org. 2016.
37. Porz N, Bauer S, Pica A, Schucht P, Beck J, Verma RK, Slotboom J, Reyes M, Wiest R. Multi-modal glioblastoma segmentation: man versus machine. PLoS One. 2014; 9:e96873.
38. Jain R, Poisson LM, Gutman D, Scarpace L, Hwang SN, Holder CA, Wintermark M, Rao A, Colen RR, Kirby J, Freymann J, Jaffe CC, Mikkelsen T, et al. Outcome prediction in patients with glioblastoma by using imaging, clinical, and genomic biomarkers: focus on the nonenhancing component of the tumor. Radiology. 2014; 272:484–493.
39. Ranjith G, Parvathy R, Vikas V, Chandrasekharan K, Nair S. Machine learning methods for the classification of gliomas: initial results using features extracted from MR spectroscopy. Neuroradiol J. 2015; 28:106–111.
40. Kim S, Park T, Kon M. Cancer survival classification using integrated data sets and intermediate information. Artif Intell Med. 2014; 62:23–31.
41. Lee J, Jain R, Khalil K, Griffith B, Bosca R, Rao G, Rao A. Texture feature ratios from relative CBV maps of perfusion MRI are associated with patient survival in glioblastoma. AJNR Am J Neuroradiol. 2016; 37:37–43.
42. Mauer M, Stupp R, Taphoorn MJ, Coens C, Osoba D, Marosi C, Wong R, de Witte O, Cairncross JG, Efficace F, Mirimanoff RO, Forsyth P, van den Bent MJ, et al. The prognostic value of health-related quality-of-life data in predicting survival in glioblastoma cancer patients: results from an international randomised phase III EORTC brain tumour and radiation oncology groups, and NCIC clinical trials group study. Br J Cancer. 2007; 97:302–307.