INTRODUCTION
MicroRNAs (miRNAs) are non-coding RNAs which are composed of approximately 22 nucleotides [1]. The miRNAs can influence some important biological process, such as cell development, proliferation, and apoptosis [2]. As an important research result, the miRNAs have been shown to be responsible for the regulation of approximately 60% of the coding genes in mammalian [3]. Furthermore, their regulatory functions have also been shown to be related to some special gene expressions in the post transcription stage [4]. For example, they can inhibit translation or cause miRNA degradation of target genes to repress gene expression [5]. The first miRNA was found about twenty years ago, after that a large amount of miRNAs have been discovered from a wide variety of species including human [6, 7]. Furthermore, increasing numbers of miRNAs have been experimentally shown to be associated with the development processes of various human diseases [8, 9]. For example, the study of Benjamin et al. has identified differentially expressed miRNAs between malignant pleural mesothelioma (MPM) and various carcinomas using microarrays. Among them, hsa-miR-193-3p was over-expressed in MPM, while hsa-miR-200c and hsa-miR-192 were over-expressed in peripheral lung adenocarcinoma and carcinomas that frequently metastasize to lung pleura [10]. Additionally, in the study of William et al. in 2010, they demonstrated the critical role of miR-155 in regulation of cell survival and chemosensitivity through down-regulation of FOXO3a in breast cancer [11]. Moreover, Wang et al. confirmed that the concentrations of plasma miR-17-5p/20a were significantly associated with the differentiation status and TNM stages of gastric cancer, where the TNM is a notation system that describes the stage of a cancer which originates from a solid tumour with alphanumeric codes [12]. Though so many associations between miRNAs and diseases have been discovered, the known associations are only the tip of the iceberg. As far as we know, a large amount of experiments has been implemented for discovering their potential associations because the identification of miRNA-disease association has great significance for the diagnosis and treatment of human complex diseases. However, experimental methods are expensive and time-consuming. Therefore, increasing studies have focused on the computational algorithms to predict the probabilities of the potential miRNA-disease associations [13–26]. By choosing the most promising associated miRNAs, experiment methods could be more effective [27, 28].
Based on the assumption that functionally similar miRNAs are more likely to have relevance to diseases with similar phenotypic traits, Jiang et al. [29] proposed a hypergeometric distribution-based model which could provide the prediction of miRNA-disease associations through the disease phenotype similarity network, miRNA functional similarity network, and known human disease-miRNA association network. However, this computational model strongly relies on predicted miRNA-target interactions which have high rates of false-positive and false-negative prediction results. Shi et al. [30] introduced a modified random walk algorithm by taking advantage of the miRNA-target interactions, disease-gene associations and protein-protein interactions (PPIs) to acquire potential associations between the miRNAs and diseases. Mork et al. [31] presented a miRNA-Protein-Disease based miRNA-disease association prediction (miRPD) method through combining the protein-disease associations with the protein-miRNA interactions based on the shared proteins between miRNAs and the diseases to provide miRNA-disease association prediction scores. Xu et al. [32] constructed a miRNA prioritization method by integrating known disease-gene associations and miRNA-target interactions. Instead of using the known miRNA-disease associations, this method analyzed the similarity between the targets of miRNAs and disease genes. All these aforementioned methods could not generate sufficient accurate prediction results, because they strongly rely on the predicted miRNA-target interactions, which have high rates of false positive and false negative. Another reason is that current disease-gene association network is incomplete.
Several computational methods were proposed to overcome limitations of the previous methods. Xuan et al. [33] constructed an effective miRNA-disease association prediction model named HDMP based on weighted k most similar neighbors by assigning higher weights to similarity scores between members in the same miRNA cluster or family when calculating miRNA similarity. However, the HDMP was not suitable for detecting the potential association with respect to new diseases or new miRNAs. Additionally, HDMP could not perform better than most of the previous models that were calculated based on the global network similarity measures. Chen et al. [34] were the first to propose the global-network-similarity-based computational model called Random Walk with Restart for MiRNA-Disease Association prediction (RWRMDA) based on the global information of human miRNA functional similarity network and known human miRNA-disease association network. RWRMDA performed well in the cross validations and case studies on several human complex diseases. However, its unavailability for new diseases without any known related miRNAs was still a certain limitation. Chen et al. [20] presented another model of Within and Between Score for MiRNA-Disease Association prediction (WBSMDA) to predict potential miRNA-disease associations based on miRNA-disease associations, integrated miRNA similarity and integrated disease similarity. WBSMDA could predict the potential related miRNAs for new diseases without any known related miRNAs and new miRNAs without any known associated diseases, which could overcome the limitation of RWRMDA and HDMP. Chen et al. [35] also developed a Heterogeneous Graph Inference model for MiRNA-Disease Association prediction (HGIMDA) by using an iterative process based on global network similarity information to find the optimal solutions, which had better performances in comparison with the previous models. Li et al. [36] developed a matrix completion for miRNA-disease association prediction model (MCMDA) based on the known miRNA-disease associations in the Human MicroRNA Disease Database (HMDD). MCMDA used the singular value thresholding (SVT) algorithm to accomplish the matrix completion procedure. It obtained excellent performances although it merely depended on known miRNA-disease associations.
More and more studies have taken focus on the using of machine learning methods to predict associations between miRNAs and diseases [37–40]. Among those studies, Xu et al. [41] proposed a miRNA-target dysregulated network (MTDN) through combining miRNA-target interactions and expression pattern of miRNAs and mRNAs. They constructed the support vector machine (SVM) classifier to distinguish positive miRNA-disease associations from negative samples based on the network topologic information feature. However, it was hard to obtain the true negative miRNA-disease associations, which influenced the accuracy of the supervised classifier. Chen et al. [42] used semi-supervised learning to construct the Regularized Least Squares for MiRNA-Disease Association prediction (RLSMDA) model with disease semantic similarity, miRNA functional similarity and known miRNA-disease associations. RLSMDA could calculate the associated probabilities of related miRNAs for diseases without any known associated miRNAs. Meanwhile, it could avoid the problem of using negative associations between miRNAs and diseases. However, the choice of parameters for RLSMDA and the ways of combining the classifiers in different spaces together could influence the accuracy of prediction results. Furthermore, Chen et al. [14] developed the Restricted Boltzmann Machine for Multiple types of MiRNA-Disease Association prediction (RBMMMDA) method based on miRNA-disease associations. This model presented restricted Boltzmann machine (RBM) with a two-layer (visible and hidden) undirected graph RBMMMDA could obtain both new miRNA-disease associations and their corresponding association types.
In this paper, we proposed Spy and Super Cluster strategy for MiRNA-Disease Association prediction (SSCMDA) model based on the Regularized Least Square (RLS) classifier to predict the potential miRNA-disease associations. To fully integrate the known information, we used the known miRNA-disease associations, integrated miRNA similarity and integrated disease similarity as our input data. Firstly, we took spy samples from positive samples to determine potential negative samples with a high degree of confidence. Furthermore, the super cluster strategy has been used to increase as many positive samples as possible to improve the prediction accuracy. According to the results of cross validation, the areas under the ROC curves (AUCs) of global and local leave-one-out cross validation (LOOCV) were 0.9007 and 0.8747, respectively. The 5-fold cross validation showed an average AUC of 0.8806+/-0.0025. For further validation, we carried out the case studies based on known miRNA-disease associations in the latest version and previous version of HMDD database to test whether the predicted associations had been verified by reports in other databases, respectively. We also implemented case studies for new diseases that did not have any known related miRNAs. As a result, there were high proportions of the predicted miRNAs confirmed by recent experimental reports. According to the performances of all the evaluation methods implemented above, SSCMDA has shown anticipated prediction accuracy.
RESULTS
Performance evaluation
As is known, the receiver operating characteristic curve (ROC) is popular to indicate the evaluation performance of the prediction accuracy based on corresponding cross validations. Therefore, we drew ROC curve to present the visible accuracy description [43]. Through plotting the true positive rate (TPR, sensitivity) versus the false positive rate (FPR, 1-specificity) at different thresholds, we obtained the curve that is closer to the upper left corner to mean the better prediction effectiveness. Specifically, sensitivity refers to the percentage of the true positive samples whose rankings are higher than the given threshold in the whole positive samples. Meanwhile, specificity denotes the percentage of negative samples with rankings lower than the threshold in the whole negative samples. Furthermore, AUC was calculated to demonstrate the prediction ability of SSCMDA. If the AUC equals to 1, it indicates that the model has perfect prediction performance. If the AUC equals to 0.5, it indicates that the model only has a random prediction performance. According to the validation index mentioned above, we firstly used global LOOCV based on the known miRNA-disease associations in latest version of HMDD database to evaluate the accuracy of SSCMDA. In each round, we changed one of the known miRNA-disease associations to candidate one which was deemed as unknown miRNA-disease pair. Then we used the SSCMDA to predict all the probability scores of the candidate pairs, and compared the score of the test sample which was changed above with the scores of other candidate pairs to observe whether it ranked above the given threshold. Different from the global LOOCV, local LOOCV just considered miRNAs that were associated with the investigated disease. Specifically, we compared the score of the test sample with the scores of candidate miRNA-disease pairs that were just related to the investigated disease. As a result, SSCMDA obtained the AUC of 0.9007 in the global LOOCV and AUC of 0.8747 in the local LOOCV as shown in Figure 1. In comparison with some previous models, the SSCMDA has really shown an improvement of the prediction accuracy in some extent. For example, the AUCs obtained from MCMDA, HGIMDA, RLSMDA, HDMP, WBSMDA were 0.8749, 0.8781, 0.8426, 0.8366, 0.8030 in global LOOCV and 0.7718, 0.8077, 0.6953, 0.7702, 0.8031 in local LOOCV, respectively. However, RWRMDA only had AUC of local LOOCV (0.7891) which was a defect because it could not uncover the missing associations for all the diseases simultaneously. In conclusion, the proposed model SSCMDA performed well in the global LOOCV and local LOOCV (See Figure 1). To show the comparison with a more clear form, we also concluded the global and local Growth Rate (GR) of SSCMDA compared with the previous models and show them in Table 1 together with their AUCs of global and local LOOCV (See Table 1).
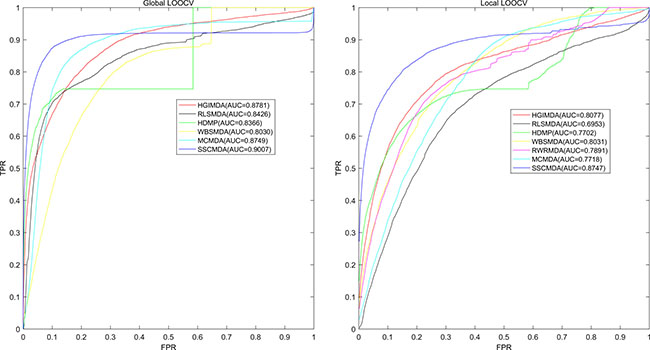
Figure 1: AUC of global LOOCV (left) compared with MCMDA, HGIMDA, RLSMDA, HDMP and WBSMDA; AUC of local LOOCV (right) compared with MCMDA, HGIMDA, RLSMDA, HDMP, WBSMDA and RWRMDA. As a result, SSCMDA achieved AUCs of 0. 9007 and 0.8747 in the global and local LOOCV, which exceed all the previous classical models.
Table 1: Comparison of the proposed method with all the previous methods according to the AUCs of global and local LOOCV and their Growth Rate (GR) of SSCMDA compared with the other models
Model |
Global LOOCV |
Global GR of SSCMDA |
Local LOOCV |
Local GR of SSCMDA |
|---|---|---|---|---|
SSCMDA |
0.9007 |
—— |
0.8747 |
—— |
HGIMDA |
0.8781 |
2.57% |
0.8077 |
8.30% |
RLSMDA |
0.8426 |
6.90% |
0.6953 |
25.80% |
HDMP |
0.8366 |
7.66% |
0.7702 |
13.57% |
WBSMDA |
0.8030 |
12.17% |
0.8031 |
8.92% |
RWRMDA |
—— |
—— |
0.7891 |
10.85% |
MCMDA |
0.8749 |
2.95% |
0.7718 |
13.33% |
We also carried out 5-fold cross validation for a further evaluation for the accuracy of the SSCMDA model. Firstly, we randomly divided the whole known associations equally into five sections and treated each section as test samples in turn by removing the associations of these test samples simultaneously. The miRNA-disease pairs without known association evidences were regarded as candidate pairs. Afterwards, all the test samples would be scored and compared with the scores of candidate pairs. We repeated the procedure of 5-fold cross validation 100 times randomly to obtain the average AUC value. In comparison with MCMDA, RLSMDA, HDMP and WBSMDA, whose average AUCs were 0.8767+/-0.0011, 0.8569+/-0.0020, 0.8342+/-0.0010 and 0.8185+/-0.0009, we could further confirm the effectiveness of SSCMDA for potential miRNA-disease association prediction with the average AUC of 5-fold cross validation 0.8806+/-0.0025.
Case Studies
We studied three different typical cases that were case studies for known diseases, case study for new disease, and case study based on old data. We used a total of five major human diseases including breast neoplasm, esophageal neoplasm, lymphoma, hepatocellular carcinoma and glioblastoma to test the prediction effect of the proposed model in all the types of case studies. To present the results of case studies, we further counted the number of the miRNAs verified by at least one database in the top 10, top 20 and even top 50 prediction results of the investigated disease-related miRNAs. For the first case, the numbers of verified miRNAs related with breast neoplasm, esophageal neoplasm and lymphoma in top 50 were 42, 40 and 39, respectively. For the second case, the number of verified miRNAs related with hepatocellular carcinoma in top 50 was 41. For the third case, the number of verified miRNAs related with Glioblastoma in top 50 was 29.
Case studies for known diseases
We have studied three known diseases based on the confirmation from the other two miRNA-disease association databases dbDEMC [44] and miR2Disease [45] to examine the effectiveness of predicting miRNAs by SSCMDA.
Breast neoplasm is the most common malignancy for women’s health, causing large amounts of death each year. The breast neoplasms consist of multiple types of breast neoplasm cells, but only a minority of breast neoplasm cells had the ability to form new tumors [46]. In the western world, over eighty percent of breast neoplasms are hormone-receptor positive [47]. Nowadays, lots of scientists have paid attention to the original etiology of breast cancers in the perspective of miRNAs. Increasing numbers of evidences showed that some miRNAs were highly correlated with breast neoplasm and played important roles in the development of breast neoplasm. For example, among the differentially expressed miRNAs, miR-10b, miR-125b, miR145, miR-21, and miR-155 emerged to be the most consistently deregulated in breast neoplasm. Three of them, miR-10b, miR-125b, and miR-145, were down-regulated and the remaining two, miR-21 and miR-155, were up-regulated, suggesting that they may potentially act as tumor suppressor genes or oncogenes, respectively [48]. Through implementing SSCMDA, we obtained the total ranking of the unknown miRNA-disease pairs. As the result shown, among the top 10, 20 and 50 potential associations between breast neoplasm and miRNAs, there were 9, 19 and 42 miRNA-disease associations confirmed by experiments, respectively (See Table 2). Taking the hsa-mir-215 as an example, which was ranked the first in our prediction results, recent study had shown that its difference in expression was observed between serum samples from healthy volunteers and serum samples from untreated patients with metastatic breast cancer [49].
Table 2: Prediction of the top 50 predicted miRNAs associated with breast neoplasms based on known associations in HMDD database
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-215 |
dbdemc |
hsa-mir-18a |
dbdemc;miR2Disease |
hsa-mir-15b |
dbdemc |
hsa-mir-590 |
dbdemc |
hsa-mir-16 |
dbdemc |
hsa-mir-143 |
dbdemc;miR2Disease |
hsa-mir-29b |
dbdemc;miR2Disease |
hsa-mir-675 |
unconfirmed |
hsa-mir-221 |
dbdemc;miR2Disease |
hsa-mir-326 |
dbdemc |
hsa-mir-15a |
dbdemc |
hsa-mir-194 |
dbdemc |
hsa-mir-345 |
dbdemc |
hsa-mir-218 |
dbdemc |
hsa-mir-99b |
dbdemc |
hsa-mir-183 |
dbdemc |
hsa-mir-483 |
dbdemc |
hsa-mir-1302 |
unconfirmed |
hsa-let-7g |
dbdemc |
hsa-mir-922 |
unconfirmed |
hsa-mir-150 |
dbdemc |
hsa-mir-202 |
dbdemc;miR2Disease |
hsa-let-7c |
dbdemc |
hsa-mir-195 |
dbdemc;miR2Disease |
hsa-mir-498 |
dbdemc |
hsa-mir-181 |
unconfirmed |
hsa-let-7b |
dbdemc |
hsa-mir-765 |
dbdemc |
hsa-let-7f |
dbdemc;miR2Disease |
hsa-mir-100 |
dbdemc |
hsa-mir-145 |
dbdemc;miR2Disease |
hsa-mir-30d |
dbdemc |
hsa-mir-92 |
dbdemc |
hsa-mir-1323 |
unconfirmed |
hsa-mir-1247 |
unconfirmed |
hsa-mir-200b |
dbdemc;miR2Disease |
hsa-let-7d |
dbdemc;miR2Disease |
hsa-let-7a |
dbdemc;miR2Disease |
hsa-let-7e |
dbdemc |
hsa-mir-103a |
unconfirmed |
hsa-mir-223 |
dbdemc |
hsa-mir-10b |
dbdemc;miR2Disease |
hsa-mir-200a |
dbdemc;miR2Disease |
hsa-mir-9 |
dbdemc;miR2Disease |
hsa-mir-29a |
dbdemc |
hsa-mir-17 |
miR2Disease |
hsa-mir-198 |
dbdemc |
hsa-mir-2355 |
unconfirmed |
hsa-mir-25 |
dbdemc |
hsa-mir-141 |
dbdemc;miR2Disease |
The first column records top 1-25 related miRNAs. The second column records the top 26-50 related miRNAs.
Esophageal neoplasm is a deadly cancer but rarely studied worldwide. Additionally, the esophageal neoplasm is age-specific which means that the incidence and mortality rates increased with age [50]. Through the efforts of researchers, great development has been achieved in the epidemiologic patterns associated with this disease during the past three decades. Recent advances in the diagnosis, staging, treatment and prognosis of esophageal neoplasm have led to small but significant improvements in survival [51]. Furthermore, recent researches have shown that the expression of miRNAs has tight associations with the development of esophageal neoplasm. For example, low expression of let-7b and let-7c in biopsies from 74 untreated patients of the training set significantly correlated with poor response to chemotherapy of both clinic and histopathology [52]. Based on the aforementioned facts, SSCMDA was implemented to identify potential related miRNAs for esophageal neoplasm based on known associations in the HMDD database. As a result, 9 out of the top 10 and 40 out of the top 50 predicted associations between esophageal neoplasm and miRNAs were confirmed by experimental reports from dbDEMC database and miR2Disease database (See Table 3). For example, hsa-mir-150 was ranked third among the predicted potential associations and it has been verified to regulate the EMT-inducer ZEB1 in esophageal squamous cell carcinoma [53].
Table 3: Prediction of the top 50 predicted miRNAs associated with esophageal neoplasm based on known associations in HMDD database
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-215 |
dbdemc |
hsa-mir-106b |
dbdemc |
hsa-mir-15b |
dbdemc |
hsa-mir-191 |
dbdemc |
hsa-mir-150 |
dbdemc |
hsa-mir-222 |
dbdemc |
hsa-mir-675 |
unconfirmed |
hsa-mir-1915 |
unconfirmed |
hsa-mir-15a |
dbdemc |
hsa-mir-18b |
dbdemc |
hsa-mir-141 |
dbdemc |
hsa-mir-22 |
dbdemc |
hsa-mir-146a |
dbdemc |
hsa-mir-10b |
dbdemc |
hsa-let-7a |
dbdemc |
hsa-mir-29b |
dbdemc |
hsa-mir-143 |
dbdemc |
hsa-mir-155 |
dbdemc |
hsa-mir-200b |
dbdemc |
hsa-mir-26b |
dbdemc |
hsa-mir-29a |
dbdemc |
hsa-mir-590 |
dbdemc |
hsa-mir-17 |
dbdemc |
hsa-mir-301b |
unconfirmed |
hsa-mir-198 |
dbdemc |
hsa-mir-26a |
dbdemc |
hsa-mir-125a |
dbdemc |
hsa-mir-92a |
unconfirmed |
hsa-mir-2355 |
unconfirmed |
hsa-mir-28 |
dbdemc |
hsa-mir-1247 |
unconfirmed |
hsa-mir-34b |
dbdemc |
hsa-mir-100 |
dbdemc |
hsa-mir-130b |
dbdemc |
hsa-mir-219 |
unconfirmed |
hsa-mir-520b |
dbdemc |
hsa-mir-145 |
dbdemc |
hsa-mir-192 |
dbdemc;miR2Disease |
hsa-mir-450b |
unconfirmed |
hsa-let-7c |
dbdemc |
hsa-mir-20a |
dbdemc |
hsa-mir-335 |
dbdemc |
hsa-mir-765 |
dbdemc |
hsa-mir-181b |
dbdemc |
hsa-mir-345 |
dbdemc |
hsa-mir-181 |
unconfirmed |
hsa-mir-328 |
dbdemc |
hsa-mir-527 |
dbdemc |
hsa-mir-1302 |
unconfirmed |
hsa-mir-373 |
dbdemc;miR2Disease |
The first column records top 1-25 related miRNAs. The second column records the top 26-50 related miRNAs.
Lymphoma is a form of cancer which encompasses a variety of cancers specific to the lymphatic system [54]. Some of the cells in the lymphatic system grow abnormally and are out of control when lymphoma occurs [54]. Eventually, they may form a tumor whose cells will continue to grow as the cancerous cells and will also continue to reproduce [54]. If all these cancerous cells are the same, they are called malignant or cancerous, because they will continue to grow and eventually harm the body’s systems [55]. Because there is lymph tissue throughout the body, the cancer cells may spread to other organs, or even into the bone marrow [55]. Recent experimental studies showed that profound mRNA expression changes of potential target genes involving cell cycle control, apoptosis and B-cell differentiation was concerned with the down-regulation of miR-16, miR-26a, miR-101, miR-29c and miR138 in the t(14;18)-negative follicular lymphoma subset [56]. Therefore, it is important to take lymphomas as a case to study. Through implementing SSCMDA for potential miRNA-disease association prediction, 6 out of top 10 potential lymphoma-associated miRNAs in the prediction result list have been verified by the recent studies. Furthermore, for the top 50 lymphoma-associated miRNAs predicted by SSCMDA, 39 of them have experimental literature evidences (See Table 4). For example, the hsa-mir-221 was ranked first in the prediction and it has been confirmed to have a significant difference in expression of lymphoid tissues between the lymphoma patient and healthy groups [57].
Table 4: Prediction of the top 50 predicted miRNAs associated with lymphoma based on known associations in HMDD database
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-221 |
dbdemc;miR2Disease |
hsa-mir-422a |
dbdemc |
hsa-mir-2355 |
unconfirmed |
hsa-mir-17 |
dbdemc;miR2Disease |
hsa-mir-1247 |
unconfirmed |
hsa-mir-29c |
dbdemc |
hsa-mir-215 |
dbdemc |
hsa-mir-222 |
dbdemc |
hsa-mir-922 |
unconfirmed |
hsa-mir-181c |
dbdemc |
hsa-mir-9 |
dbdemc |
hsa-mir-26a |
dbdemc |
hsa-mir-202 |
unconfirmed |
hsa-mir-183 |
dbdemc |
hsa-mir-29b |
dbdemc |
hsa-mir-200b |
dbdemc |
hsa-mir-326 |
dbdemc |
hsa-mir-92a |
dbdemc |
hsa-mir-1915 |
unconfirmed |
hsa-mir-103a |
unconfirmed |
hsa-mir-10b |
dbdemc |
hsa-mir-204 |
dbdemc |
hsa-let-7d |
dbdemc |
hsa-mir-139 |
dbdemc;miR2Disease |
hsa-mir-1302 |
unconfirmed |
hsa-mir-181a |
dbdemc |
hsa-mir-18a |
dbdemc |
hsa-mir-92 |
dbdemc;miR2Disease |
hsa-mir-15b |
dbdemc |
hsa-mir-182 |
dbdemc |
hsa-mir-99b |
dbdemc |
hsa-let-7a |
dbdemc |
hsa-let-7e |
dbdemc;miR2Disease |
hsa-mir-195 |
dbdemc |
hsa-let-7f |
dbdemc |
hsa-mir-21 |
dbdemc;miR2Disease |
hsa-mir-498 |
unconfirmed |
hsa-mir-200a |
dbdemc |
hsa-let-7g |
dbdemc |
hsa-mir-33a |
dbdemc |
hsa-mir-518a |
unconfirmed |
hsa-mir-27a |
dbdemc |
hsa-let-7c |
dbdemc |
hsa-mir-181b |
dbdemc |
hsa-mir-10a |
dbdemc;miR2Disease |
hsa-mir-20a |
dbdemc;miR2Disease |
hsa-let-7b |
dbdemc |
hsa-mir-320e |
unconfirmed |
hsa-mir-483 |
unconfirmed |
hsa-mir-31 |
dbdemc |
The first column records top 1-25 related miRNAs. The second column records the top 26-50 related miRNAs.
For demonstrating all the prediction results, we showed the prediction scores of all the candidate miRNA-disease pairs in Supplementary Table 1. This table contains the potential miRNAs associated with all the human diseases investigated in HMDD database (See Supplementary Table 1).
Case study for new disease in the simulation experiments
To validate the prediction effectiveness for new diseases, we also conducted another case study by hiding the information of miRNAs related with the investigated disease. For each investigated disease, we removed the known associations between this disease and all its related miRNAs. Through implementing the SSCMDA based on the changed input association matrix, we obtained the ranking of all the candidate miRNA-disease pairs by comparing their prediction scores. We could find that 9, 18 and 41 out of the top 10, 20 and 50 miRNAs of hepatocellular carcinoma had been confirmed by at least one of the three databases, namely HMDD, dbDEMC and miR2Disease (See Table 5). Specially, the has-miR-16, whose ranking is the first of the prediction result, has been studied by researchers and they found that the cyclooxygenase-2 whose expression has been detected in human hepatocellular carcinoma is a target of has-miR-16 [58].
Table 5: Prediction of the top 50 miRNAs associated with Hepatocellular Carcinoma in HMDD database
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-16 |
dbdemc;miR2Disease;HMDD |
hsa-mir-126 |
dbdemc;miR2Disease;HMDD |
hsa-mir-182 |
miR2Disease;HMDD |
hsa-mir-139 |
miR2Disease;HMDD |
hsa-mir-143 |
dbdemc;miR2Disease |
hsa-mir-1302 |
unconfirmed |
hsa-mir-146a |
dbdemc;miR2Disease;HMDD |
hsa-mir-125a |
dbdemc;miR2Disease;HMDD |
hsa-mir-15a |
dbdemc;miR2Disease;HMDD |
hsa-mir-17 |
miR2Disease;HMDD |
hsa-mir-382 |
unconfirmed |
hsa-mir-26b |
dbdemc;miR2Disease |
hsa-mir-200c |
HMDD |
hsa-let-7i |
dbdemc;HMDD |
hsa-mir-107 |
dbdemc;miR2Disease;HMDD |
hsa-mir-223 |
miR2Disease;HMDD |
hsa-mir-345 |
HMDD |
hsa-mir-922 |
unconfirmed |
hsa-mir-103a |
HMDD |
hsa-let-7c |
dbdemc;miR2Disease;HMDD |
hsa-mir-200a |
dbdemc;miR2Disease;HMDD |
hsa-mir-1247 |
unconfirmed |
hsa-mir-655 |
unconfirmed |
hsa-mir-205 |
miR2Disease;HMDD |
hsa-mir-141 |
miR2Disease;HMDD |
hsa-mir-203 |
miR2Disease;HMDD |
hsa-mir-152 |
miR2Disease;HMDD |
hsa-mir-215 |
miR2Disease |
hsa-let-7d |
miR2Disease;HMDD |
hsa-mir-29b |
dbdemc;HMDD |
hsa-mir-10b |
HMDD |
hsa-mir-148b |
dbdemc;miR2Disease;HMDD |
hsa-mir-92a |
miR2Disease;HMDD |
hsa-mir-1972 |
unconfirmed |
hsa-let-7a |
dbdemc;miR2Disease;HMDD |
hsa-mir-191 |
dbdemc;HMDD |
hsa-mir-181b |
dbdemc;miR2Disease;HMDD |
hsa-mir-106a |
dbdemc;miR2Disease;HMDD |
hsa-mir-20a |
dbdemc;miR2Disease;HMDD |
hsa-mir-100 |
dbdemc;HMDD |
hsa-mir-132 |
miR2Disease |
hsa-mir-299 |
unconfirmed |
hsa-mir-194 |
dbdemc;miR2Disease |
hsa-mir-195 |
dbdemc;miR2Disease;HMDD |
hsa-mir-204 |
unconfirmed |
hsa-mir-27b |
dbdemc |
hsa-mir-9 |
miR2Disease |
hsa-mir-27a |
miR2Disease;HMDD |
hsa-mir-200b |
miR2Disease;HMDD |
hsa-mir-675 |
unconfirmed |
The hepatocellular carcinoma was treated as new disease which does not have any related miRNAs. The first column records top 1-25 related miRNAs. The second column records the top 26-50 related miRNAs.
Case study on previous version dataset
Finally, we tested our model based on the old version of the database HMDD to see whether the SSCMDA still performed well on it. Through the experiment, there were 7, 13 and 29 respectively out of top 10, 20 and 50 miRNAs related with the Glioblastoma confirmed by at least one of three databases mentioned above (See Table 6). Taking the hsa-mir-338 as an example, which ranked the first among all the miRNAs verified by dbDEMC or miR2Disease, recent research showed that the mir-338-3p inhibited malignant biological behaviors of Glioblastoma cells by targeting MACC1 gene [59].
Table 6: Prediction of the top 50 predicted miRNAs associated with Glioblastoma based on known associations in previous version HMDD database
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-561 |
unconfirmed |
hsa-mir-135b |
dbdemc |
hsa-mir-338 |
dbdemc |
hsa-mir-194 |
dbdemc |
hsa-mir-301a |
unconfirmed |
hsa-mir-423 |
unconfirmed |
hsa-mir-342 |
dbdemc;HMDD |
hsa-let-7b |
unconfirmed |
hsa-mir-25 |
dbdemc;miR2Disease;HMDD |
hsa-mir-185 |
dbdemc |
hsa-mir-499 |
unconfirmed |
hsa-mir-525 |
unconfirmed |
hsa-mir-370 |
dbdemc |
hsa-let-7e |
dbdemc |
hsa-mir-134 |
HMDD |
hsa-mir-532 |
unconfirmed |
hsa-mir-23b |
miR2Disease;HMDD |
hsa-mir-218 |
dbdemc;HMDD |
hsa-mir-421 |
dbdemc |
hsa-mir-181c |
dbdemc;miR2Disease;HMDD |
hsa-mir-136 |
dbdemc |
hsa-mir-514 |
unconfirmed |
hsa-mir-34a |
dbdemc;miR2Disease;HMDD |
hsa-mir-518c |
unconfirmed |
hsa-mir-330 |
unconfirmed |
hsa-mir-638 |
unconfirmed |
hsa-mir-199b |
dbdemc |
hsa-mir-19a |
dbdemc;HMDD |
hsa-mir-583 |
unconfirmed |
hsa-mir-335 |
unconfirmed |
hsa-mir-30b |
unconfirmed |
hsa-mir-325 |
unconfirmed |
hsa-mir-451 |
miR2Disease |
hsa-mir-126 |
unconfirmed |
hsa-mir-520f |
unconfirmed |
hsa-mir-383 |
dbdemc |
hsa-mir-520c |
dbdemc |
hsa-mir-539 |
HMDD |
hsa-mir-210 |
dbdemc;HMDD |
hsa-mir-135a |
unconfirmed |
hsa-mir-29a |
HMDD |
hsa-mir-365 |
unconfirmed |
hsa-mir-596 |
unconfirmed |
hsa-mir-153 |
dbdemc;miR2Disease;HMDD |
hsa-mir-300 |
dbdemc |
hsa-mir-379 |
dbdemc |
hsa-mir-206 |
dbdemc;HMDD |
hsa-mir-382 |
dbdemc |
hsa-mir-128a |
miR2Disease |
hsa-mir-130b |
unconfirmed |
The first column records top 1-25 related miRNAs. The second column records the top 26-50 related miRNAs.
DISCUSSION
In this paper, we proposed the Spy and Super Cluster strategy for MiRNA-Disease Association prediction (SSCMDA) based on known miRNA-disease associations, integrated miRNA similarity and integrated disease similarity. The Regularized Least Square (RLS) was used as the baseline classifier. The spy strategy was implemented to identify the negative samples with high degree of confidence from the mixed sample set which contains potential associations and real negative associations. Because the shortage of positive samples would lead to lower accuracy of prediction, the super cluster strategy was conducted to increase as many positive samples as possible. To examine the accuracy of the SSCMDA, three types of cross validations including global LOOCV, local LOOCV and 5-fold cross validation had been implemented. Furthermore, we also implemented three types of case studies based on different miRNA-disease association databases. As a result, SSCMDA performed well both in the cross validations and the case studies.
The excellent performances of SSCMDA could be attributed to the following several important factors. Firstly, the increasing disease-miRNA association data have been discovered and confirmed with the development of the biological experiments. This advantage was beneficial to the SSCMDA because the SSCMDA was dependent on the known associations to improve the prediction accuracy. Secondly, because SSCMDA was constructed based on the integrated disease similarity and integrated miRNA similarity, it could make full use of various similarity information to recover the potential miRNA-disease associations. Thirdly, to improve the prediction accuracy, SSCMDA could adopt spy strategy to identify reliable negative samples from all the unknown miRNA-disease pairs, which contained mixed training samples including both potential associations and real negative samples. Meanwhile, the super cluster strategy could add as many positive samples as possible with the help of the similarity information. Last but not least, the baseline method of RLS performed well in previous computational biology research, which guaranteed the basic accuracy of our proposed model. In view of reasons mentioned above, SSCMDA could improve the prediction accuracy in comparison with the previous proposed methods.
However, there are still some limitations in this model. First of all, the proposed model relied heavily on the known association data, which will lead to an unstable prediction effect when the training samples were changed. Secondly, the RLS is so sensitive to the changes of parameters that it is hard to obtain the optimal combining parameters to combine all the proposed strategies to improve prediction accuracy. Therefore, the method of obtaining optimal combining parameters is expected to be theoretically solved in the future. Furthermore, the literature of Wang et al. has pointed out the more important fact that it is unwise to use a single disease-related miRNA to judge cancer risks for all the persons [60]. The recent study has also found that cancer signals are of not stable, which motivates us to construct various cancer hallmark networks to effectively evaluate cancer risks based on the miRNA profiles of each person. Furthermore, the re-sampling test should be developed in the future for estimating the robustness of the models [60, 61]. Additionally, there are still some important problems in the personalized medicine and treatment in the framework of miRNA-disease association prediction [60, 62]. According to the recent studies on the genome sequencing technology of cancer systems biology, we can anticipate that cancer subclones and other clone-based analysis would be helpful for predicting and treating cancer in the future [63, 64].
MATERIALS AND METHODS
Human miRNA-disease associations
We downloaded the human miRNA-disease associations data from the HMDD v2.0 database [65], which contained 5430 distinct experimentally confirmed human miRNA-disease associations between 383 diseases and 495 miRNAs. To conveniently deal with all these data in the following computations, we constructed an adjacency matrix A ε R nd×nm to formalize the human miRNA-disease associations, where nm and nd were denoted as the number of miRNAs and diseases in HMDD v2.0 database, respectively. If miRNA mj had been experimentally verified to be associated with disease di, then Aij equaled to 1, otherwise 0.
MiRNA functional similarity
Based on the assumption that functionally similar miRNAs are more likely to be associated with diseases with similar characters, Wang et al. [66] proposed a method to calculate the miRNA functional similarity. We downloaded the miRNA functional similarity data from http://www.cuilab.cn/files/images/cuilab/misim.zip. We denoted the matrix MS to represent the miRNA functional similarity. The element MSij represented the value of similarity between the miRNA mi and mj.
Disease semantic similarity model 1
We constructed a Directed Acyclic Graph (DAG) to describe the diseases according to the literature of Chen et al. [26] based on the MeSH descriptors downloaded from the National Library of Medicine (http://www.nlm.nih.gov/). According to the DAG, we denoted the contribution values of disease d in DAG(D) to the semantic value of disease D as follows:
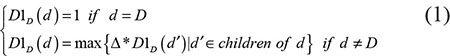
where Δ was the semantic contribution decay factor. The self semantic value of disease D was defined as follows:

where T(D) represented all ancestor nodes of D and D itself. According to the observation that two diseases with larger shared part of their DAGs had larger similarity score, the semantic similarity score between disease di and dj could be defined as follows:
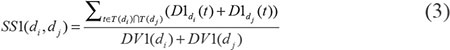
Disease semantic similarity model 2
As what we had considered, it was not reasonable to assign the same contribution value to the diseases in the same layer of DAG(D). As far as we have observed, a more specific disease which appears in less DAGs should contribute a higher value to the semantic similarity of disease D. Therefore, according to the model which was proposed by Xuan et al. [33], we defined the contribution of disease d in DAG(D) to the semantic value of disease D as follows:
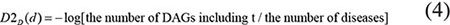
We defined the semantic similarity of disease di and dj as the ratio of the shared ancestor nodes’ contributions to all the ancestor nodes’ contributions. Therefore, disease semantic similarity model 2 was calculated as follows:
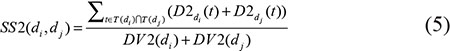
where

Gaussian interaction profile kernel similarity
Based on the Gaussian kernel function which is one of the Radial Basis function whose value depends only on the distance from the origin, Gaussian interaction profile kernel similarity were constructed as another algorithm of disease semantic similarity and miRNA functional similarity [67]. As the ith row and jth column of adjacent matrix A contains the information whether the disease or the miRNA associated with each of the miRNAs or the diseases, so we denoted vector IP(di) and IP(rj) to represent the ith row vector and jth column vector, respectively. Therefore, the similarity of diseases and miRNAs could be computed as follows:
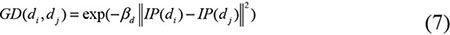
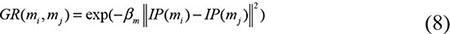
where adjustment coefficient βd and βm for the kernel bandwidth were denoted as follows:


where β′d and β′m were the original bandwidths. In the end, matrix GD and GR represented the Gaussian interaction profile kernel similarity of diseases and miRNAs, respectively.
Integrated similarity for miRNAs and diseases
Through combining the disease semantic similarity with disease Gaussian interaction profile kernel similarity, we obtained the integrated disease similarity. Specifically, if disease di and dj have semantic similarity, then the final integrated similarity is the average of SS1 and SS2, otherwise the integrated disease similarity equals to the value of Gaussian interaction profile kernel similarity. The formulations show as follows:
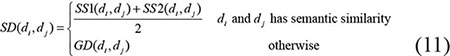
Furthermore, by combining the miRNA functional similarity with miRNA Gaussian interaction profile kernel similarity, we obtained the integrated miRNA similarity as follows:
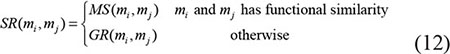
SSCMDA
The whole process of the proposed method was shown in Figure 2. Based on the materials prepared above, we adopted the RLS as our basic classifier, which was proposed to fit the known sample data and further predict the potential miRNA-disease associations [68]. According to the RLS and Lagrange multiplier method, we could construct our score function of association between disease di and miRNA mj as follows:
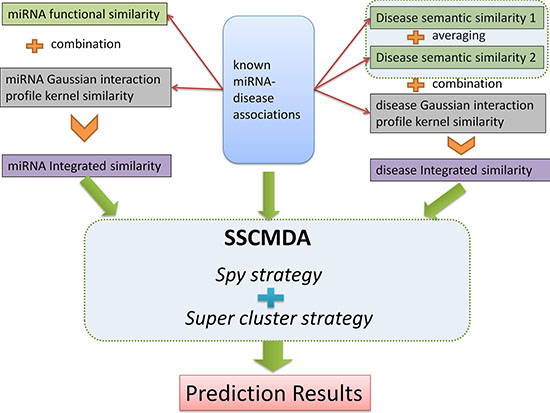
Figure 2: The whole process flowchart of the SSCMDA method. The upper half part shows the input data including the known miRNA-disease associations, miRNA integrated similarity, and disease integrated similarity. The middle part shows the main algorithm including the Spy strategy and the Super cluster strategy. The final part is the prediction results.

where Smj(di) was the predicted association score of disease di to miRNA mj, SDi was the ith row of the integrated disease similarity matrix, λd was the regularization parameter rooting in the lagrangian multiplier of RLS and experimentally set as 2 which was a common set in the experiments, I represented the identity matrix and ATj represents the nd×1 class label vector of training samples namely the association between all the diseases and the jth miRNA. On the other hand, the score function of miRNA to disease was as follows:

where Sdj(mi) was the predicted score of miRNA mi to disease dj, SRi was the ith row of the integrated miRNA similarity matrix, λr was also a regularization parameter rooting in the lagrangian multiplier of RLS and experimentally set as 2 which was also a common set in the experiments. Differently, Aj represented the 1×nm vector which was the jth column of the adjacent matrix A. Afterwards, we combined the two score functions as follows:

where FS(di, mj) was formed as the final basic score function based on the RLS.
Spy strategy
As far as we had realized, the unknown miRNA-disease pairs contained both the potential associations and the real negative samples. The fuzzy situation would lead to an inaccurate prediction result because the boundary of negative training samples was not exact. To solve this problem, we utilized the spy strategy which was a semi-supervised strategy to identify the reliable negative samples with high degree of confidence from all the unknown miRNA-disease pairs. Specifically, we randomly selected 10% from the positive associations as spy samples, and set these spy samples to unlabeled ones, namely changed them from 1 to 0. Based on the newly formed training samples, we used the RLS model to obtain the prediction score. Then, we took the minimum score of the spy samples as the threshold. If the prediction score of a candidate association was below the threshold, this association would be identified as a reliable negative sample which would be set as -1 in the miRNA-disease association adjacent matrix. Repeating the selection of spy samples for 200 times, we took the intersection of the reliable negative sets as the final reliable negative set to ensure its reliability. Finally, we obtained a new adjacent matrix AN with all the reliable negative samples. Figure 3 illustrates the procession of spy strategy and the prediction score by adopting spy strategy could be calculated as follows:
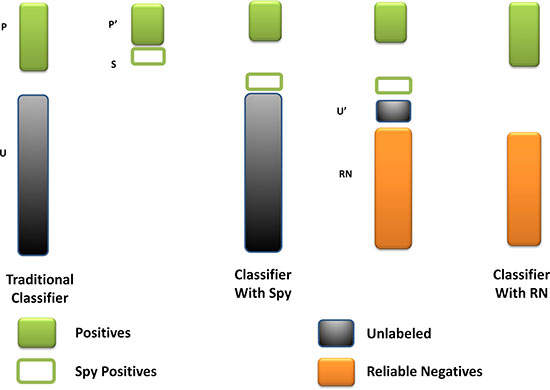
Figure 3: The flowchart of spy strategy. Spy instances (S) are randomly selected from positives (P). The spy-based classifier is built by remaining positives (P’) and new unknown miRNA-disease pairs (U’) combined S with U. And the reliable negatives (RN) are identified by comparing with the minimum score of S.
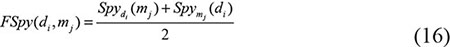
where

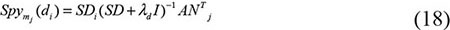
Super cluster strategy
In consideration that most of the diseases might associate with only one or a few miRNAs and vice versa. This imbalance would lead to a biased prediction which was likely to determine potential associations as negative associations. The shortage of known miRNA-disease associations would aggravate this bias. Therefore, we proposed the super cluster strategy to ameliorate this problem (See Figure 4). The main thought of super cluster strategy is to cluster as many as possible similar diseases or miRNAs as an integrated super-disease or super-miRNA. Furthermore, all the diseases related to at least one miRNA in the super-miRNA would be seemed as having association with the super-miRNA. On the other hand, the association between miRNA and super-disease would be constructed in the same way. Obviously, the integrated similarity of miRNA and disease would be used to construct the distances between two miRNAs or two diseases. According to these distances, we implemented the agglomerative hierarchical clustering to obtain super cluster. The agglomerative hierarchical clustering adopted a bottom-up strategy which deemed each entity as a cluster at the beginning of the process, then one cluster would merge the other clusters based on the linkage criterion of Ward’s minimum variance method [69]. After clustering, we cut the hierarchical clustering tree to obtain different super-clusters with a suitable threshold. However, there was a situation that diseases associated with miRNAs in super-miRNA are significantly different, which betrayed the assumption that similar diseases were likely to be associated with similar miRNAs. Therefore, we would remove the association between a disease associated with super-miRNA sr if there were not any of its k nearest neighbors simultaneously associated with sr. The probability score of disease di associated with super-miRNA srj could be calculated as follows:
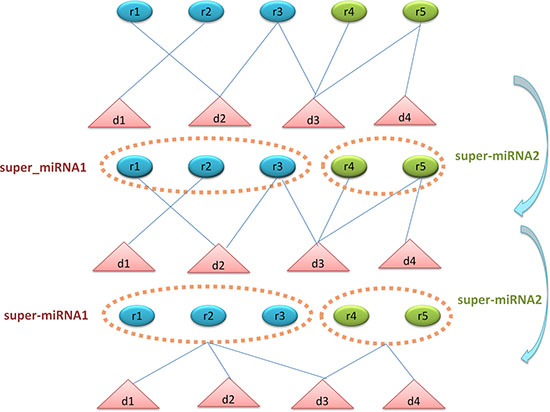
Figure 4: The flowchart of super-cluster strategy. The first section shows the original known associations between diseases and miRNAs. The second section shows the process of constructing super-miRNAs based on miRNA functional similarity and agglomerative hierarchical clustering. The third section shows the process of constructing the new associations between diseases and super-miRNAs. If the disease has association with the miRNAs in the super-miRNA, then this disease is associated with the super-miRNA.

where Asr was the new adjacent matrix formed according to the associations between diseases and super-miRNAs. Accordingly, the score of miRNA mi and super-disease sdj could be calculated as follows:

where Asd was the adjacent matrix of associations between miRNA and super-disease. The new associations are really not as accurate as the old associations. However, the main dilemma of us is not the accuracy of associations currently, but that the exact known association is rare, which prevented improving the accuracy of prediction. Thus, our aim is to excavate more useful information of the known associations based on which we can further separate the more likely miRNAs associated with disease from the mixed miRNAs. It is obvious that excessive sacrifices of known association accuracy will lead to predictive accuracy degradation. Therefore, it is a problem of clustering criterion to balance the relationship between the two situations. Fortunately, the experimental common clustering criterion used in other similar problems is still effective for our model. Furthermore, we also implemented the experiments to compare the accuracy of the model with and without the new associations, whose comparison results really reflect the effectiveness of this strategy to improve the prediction accuracy.
Two layer prediction model
We integrated the spy strategy and super-cluster strategy to form a two-layer prediction model. For disease di and miRNA mj, the prediction score obtained by adopting spy strategy and the prediction score obtained by adopting super-cluster strategy were combined as follows to compute the final miRNA-disease association prediction score:
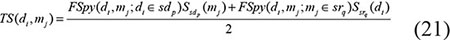
where 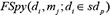 represented the prediction score of di and mj by adopting spy strategy which satisfied that
represented the prediction score of di and mj by adopting spy strategy which satisfied that 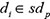 and
and 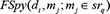 satisfied that
satisfied that 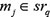 .
.
ACKNOWLEDGMENTS
XC was supported by National Natural Science Foundation of China under Grant Nos.61772531 and 11631014. QZ was supported by National Natural Science Foundation of China under Grant No.61772531, Innovation Team Project from the Education Department of Liaoning Province under Grant No.LT2015011 and the Doctor Startup Foundation from Liaoning Province under Grant No. 20170520217. HL was supported by National Natural Science Foundation of China under Grant No. 31570160 and Innovation Team Project from the Education Department of Liaoning Province under Grant No. LT2015011. FW was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the 2016 annual general university graduate research and innovation program of Jiangsu Province, China (Grant No. KYLX16_0526). GYY was supported by National Natural Science Foundation of China under Grant No. 11371355 and 11631014.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Ribeiro AO, Schoof CR, Izzotti A, Pereira LV, Vasques LR. MicroRNAs: modulators of cell identity, and their applications in tissue engineering. MicroRNA. 2014; 3:45–53.
2. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75:843–54.
3. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92–105.
4. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993; 75:855–62.
5. Dluzen DF, Noren Hooten N, Zhang Y, Kim Y, Glover FE, Tajuddin SM, Jacob KD, Zonderman AB, Evans MK. Racial differences in microRNA and gene expression in hypertensive women. Sci Rep. 2016; 6:35815.
6. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39:D152–57.
7. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005; 309:1577–81.
8. Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009; 84:55–71.
9. Meola N, Gennarino VA, Banfi S. microRNAs and genetic diseases. PathoGenetics. 2009; 2:7.
10. Benjamin H, Lebanony D, Rosenwald S, Cohen L, Gibori H, Barabash N, Ashkenazi K, Goren E, Meiri E, Morgenstern S, Perelman M, Barshack I, Goren Y, et al. A diagnostic assay based on microRNA expression accurately identifies malignant pleural mesothelioma. J Mol Diagn. 2010; 12:771–79.
11. Kong W, He L, Coppola M, Guo J, Esposito NN, Coppola D, Cheng JQ. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J Biol Chem. 2010; 285:17869–79.
12. Wang M, Gu H, Wang S, Qian H, Zhu W, Zhang L, Zhao C, Tao Y, Xu W. Circulating miR-17-5p and miR-20a: molecular markers for gastric cancer. Mol Med Rep. 2012; 5:1514–20.
13. Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci Rep. 2015; 5:13186.
14. Chen X, Yan CC, Zhang X, Li Z, Deng L, Zhang Y, Dai Q. RBMMMDA: predicting multiple types of disease-microRNA associations. Sci Rep. 2015; 5:13877.
15. Chen X, Yan CC, Luo C, Ji W, Zhang Y, Dai Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci Rep. 2015; 5:11338.
16. Chen X, Yan GY. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics. 2013; 29:2617–24.
17. Chen X, Yan CC, Zhang X, You ZH. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017; 18:558–76.
18. Chen X, Liu MX, Cui QH, Yan GY. Prediction of disease-related interactions between microRNAs and environmental factors based on a semi-supervised classifier. PLoS One. 2012; 7:e43425.
19. Chen X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci Rep. 2015; 5:16840.
20. Chen X, Yan CC, Zhang X, You ZH, Deng L, Liu Y, Zhang Y, Dai Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci Rep. 2016; 6:21106.
21. Chen X. miREFRWR: a novel disease-related microRNA-environmental factor interactions prediction method. Mol Biosyst. 2016; 12:624–33.
22. Huang YA, You ZH, Chen X, Chan K, Luo X. Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinformatics. 2016; 17:184.
23. Huang YA, Chen X, You ZH, Huang DS, Chan KC. ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget. 2016; 7:25902–14. https://doi.org/10.18632/oncotarget.8296.
24. Chen X, Huang YA, Wang XS, You ZH, Chan KC. FMLNCSIM: fuzzy measure-based lncRNA functional similarity calculation model. Oncotarget. 2016; 7:45948–58. https://doi.org/10.18632/oncotarget.10008.
25. Wong L, You ZH, Ming Z, Li J, Chen X, Huang YA. Detection of interactions between proteins through rotation forest and local phase quantization descriptors. Int J Mol Sci. 2015; 17:21.
26. Chen X, You ZH, Yan GY, Gong DW. IRWRLDA: improved random walk with restart for lncRNA-disease association prediction. Oncotarget. 2016; 7:57919–31. https://doi.org/10.18632/oncotarget.11141.
27. Pasquier C, Gardès J. Prediction of miRNA-disease associations with a vector space model. Sci Rep. 2016; 6:27036.
28. Bandyopadhyay S, Mitra R, Maulik U, Zhang MQ. Development of the human cancer microRNA network. Silence. 2010; 1:6.
29. Jiang Q, Hao Y, Wang G, Juan L, Zhang T, Teng M, Liu Y, Wang Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst Biol. 2010; 4:S2.
30. Shi H, Xu J, Zhang G, Xu L, Li C, Wang L, Zhao Z, Jiang W, Guo Z, Li X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst Biol. 2013; 7:101.
31. Mørk S, Pletscher-Frankild S, Palleja Caro A, Gorodkin J, Jensen LJ. Protein-driven inference of miRNA-disease associations. Bioinformatics. 2014; 30:392–97.
32. Xu C, Ping Y, Li X, Zhao H, Wang L, Fan H, Xiao Y, Li X. Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol Biosyst. 2014; 10:2800–09.
33. Xuan P, Han K, Guo M, Guo Y, Li J, Ding J, Liu Y, Dai Q, Li J, Teng Z, Huang Y. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS One. 2013; 8:e70204.
34. Chen X, Liu MX, Yan GY. RWRMDA: predicting novel human microRNA-disease associations. Mol Biosyst. 2012; 8:2792–98.
35. Chen X, Yan CC, Zhang X, You ZH, Huang YA, Yan GY. HGIMDA: heterogeneous graph inference for miRNA-disease association prediction. Oncotarget. 2016; 7:65257–69. https://doi.org/10.18632/oncotarget.11251.
36. Li JQ, Rong ZH, Chen X, Yan GY, You ZH. MCMDA: matrix completion for MiRNA-disease association prediction. Oncotarget. 2017; 8:21187–99. https://doi.org/10.18632/oncotarget.15061.
37. Chen X, Yan CC, Zhang X, Zhang X, Dai F, Yin J, Zhang Y. Drug-target interaction prediction: databases, web servers and computational models. Brief Bioinform. 2016; 17:696–712.
38. Chen X, Ren B, Chen M, Wang Q, Zhang L, Yan G. NLLSS: Predicting Synergistic Drug Combinations Based on Semi-supervised Learning. PLOS Comput Biol. 2016; 12:e1004975.
39. Hu H, Zhu C, Ai H, Zhang L, Zhao J, Zhao Q, Liu H. LPI-ETSLP: lncRNA-protein interaction prediction using eigenvalue transformation-based semi-supervised link prediction. Mol Biosyst. 2017; 13:1781–87.
40. Zhang L, Ai H, Zhao Q, Zhu J, Chen W, Wu X, Huang L, Yin Z, Zhao J, Liu H. Computational Prediction of Influenza Neuraminidase Inhibitors Using Machine Learning Algorithms and Recursive Feature Elimination Method. International Symposium on Bioinformatics Research and Applications. 2017: 344-49. https://doi.org/10.1007/978-3-319-59575-7_32.
41. Xu J, Li CX, Lv JY, Li YS, Xiao Y, Shao TT, Huo X, Li X, Zou Y, Han QL, Li X, Wang LH, Ren H. Prioritizing candidate disease miRNAs by topological features in the miRNA target-dysregulated network: case study of prostate cancer. Mol Cancer Ther. 2011; 10:1857–66.
42. Chen X, Yan GY. Semi-supervised learning for potential human microRNA-disease associations inference. Sci Rep. 2014; 4:5501.
43. Metz CE. Basic principles of ROC analysis. Semin Nucl Med. 1978; 8:283–98.
44. Yang Z, Ren F, Liu C, He S, Sun G, Gao Q, Yao L, Zhang Y, Miao R, Cao Y, Zhao Y, Zhong Y, Zhao H. dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genomics. 2010; 11:S5.
45. Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37:D98–104.
46. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003; 100:3983–88.
47. Van Asten K, Neven P, Lintermans A, Wildiers H, Paridaens R. Aromatase inhibitors in the breast cancer clinic: focus on exemestane. Endocr Relat Cancer. 2014; 21:R31–49.
48. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005; 65:7065–70.
49. van Schooneveld E, Wouters MC, Van der Auwera I, Peeters DJ, Wildiers H, Van Dam PA, Vergote I, Vermeulen PB, Dirix LY, Van Laere SJ. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. 2012; 14:R34.
50. Zeng H, Zheng R, Zhang S, Zuo T, Xia C, Zou X, Chen W. Esophageal cancer statistics in China, 2011: estimates based on 177 cancer registries. Thorac Cancer. 2016; 7:232–37.
51. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003; 349:2241–52.
52. Sugimura K, Miyata H, Tanaka K, Hamano R, Takahashi T, Kurokawa Y, Yamasaki M, Nakajima K, Takiguchi S, Mori M, Doki Y. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin Cancer Res. 2012; 18:5144–53.
53. Yokobori T, Suzuki S, Tanaka N, Inose T, Sohda M, Sano A, Sakai M, Nakajima M, Miyazaki T, Kato H. MiR-150 regulates the EMT-inducer ZEB1 in esophageal squamous cell carcinoma. Wound healing assays of premiR-150-treated esophageal squamous cell carcinoma TE-8 cells. Cancer Sci. 2013; 104.
54. England CG, Rui L, Cai W. Lymphoma: current status of clinical and preclinical imaging with radiolabeled antibodies. Eur J Nucl Med Mol Imaging. 2017; 44:517–32.
55. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403:503–11.
56. Leich E, Zamo A, Horn H, Haralambieva E, Puppe B, Gascoyne RD, Chan WC, Braziel RM, Rimsza LM, Weisenburger DD, Delabie J, Jaffe ES, Fitzgibbon J, et al. MicroRNA profiles of t(14;18)-negative follicular lymphoma support a late germinal center B-cell phenotype. Blood. 2011; 118:5550–58.
57. Guo HQ, Huang GL, Guo CC, Pu XX, Lin TY. Diagnostic and prognostic value of circulating miR-221 for extranodal natural killer/T-cell lymphoma. Dis Markers. 2010; 29:251–58.
58. Agra Andrieu N, Motiño O, Mayoral R, Llorente Izquierdo C, Fernández-Alvarez A, Boscá L, Casado M, Martín-Sanz P. Cyclooxygenase-2 is a target of microRNA-16 in human hepatoma cells. PLoS One. 2012; 7:e50935.
59. Shang C, Hong Y, Guo Y, Xue YX. Mir-338-3p Inhibits Malignant Biological Behaviors of Glioma Cells by Targeting MACC1 Gene. Med Sci Monit. 2016; 22:710–16.
60. Wang E, Zaman N, Mcgee S, Milanese JS, Masoudi-Nejad A, O’Connor-McCourt M. Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin Cancer Biol. 2015; 30:4–12.
61. Li J, Lenferink AE, Deng Y, Collins C, Cui Q, Purisima EO, O’Connor-McCourt MD, Wang E. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun. 2010; 1:34.
62. Wang E. Understanding genomic alterations in cancer genomes using an integrative network approach. Cancer Lett. 2013; 340:261–69.
63. Wang E, Zou J, Zaman N, Beitel LK, Trifiro M, Paliouras M. Cancer systems biology in the genome sequencing era: part 1, dissecting and modeling of tumor clones and their networks. Semin Cancer Biol. 2013; 23:279–85.
64. Wang E, Zou J, Zaman N, Beitel LK, Trifiro M, Paliouras M. Cancer systems biology in the genome sequencing era: part 2, evolutionary dynamics of tumor clonal networks and drug resistance. Semin Cancer Biol. 2013; 23:286–92.
65. Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014; 42:D1070–74.
66. Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010; 26:1644–50.
67. Chen X, Huang YA, You ZH, Yan GY, Wang XS. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics. 2017; 33:733–39.
68. Ancona N, Maglietta R, D’Addabbo A, Liuni S, Pesole G. Regularized Least Squares Cancer classifiers from DNA microarray data. BMC Bioinformatics. 2005; 6:S2.
69. Ward JH Jr. Hierarchical Grouping to Optimize an Objective Function. J Am Stat Assoc. 1963; 58:236–44.