INTRODUCTION
Breast cancer is one of the most common cancers and the second leading cause of mortality for women worldwide [1]. Almost one of eight to ten women will suffer breast cancer during their lifetime [2]. Incidence rate of breast cancer has been on the increase for several years and the average onset age is dropping, which probably occasioned by the changes of lifestyle, environment and the development of screening methods [3–5]. Recent advances of the chemotherapy, radiotherapy, hormone therapy and immuno-biological therapy have dramatically improved the survival for patients with breast cancer. Nevertheless, great individual differences have been found in the outcomes of breast cancer treatments due to the tumor heterogeneity.
Chemotherapy is the chief means of HER2-negative breast cancer treatment, among which, taxane-anthracycline combination regimens have been advised as standard neoadjuvant and adjuvant strategies [6]. At present, chemo-resistance still poses a major obstacle to successful treatment of breast cancer, with a lot of patients being under- or over-treated. Though great efforts have been made on the development of effective prognostic indicators through molecular and cell biological studies, outcomes of patients with breast cancer are still predicted largely on the basis of conventional clinicopathological and molecular prognostic factors [7–9]. However, quite a few patients show distinct responses to chemotherapy even if they have same or similar clinicopathological characteristics. Thus, there is a critical need for innovative biomarkers to accurately predict the therapeutic sensitivity and prognosis of HER-2 negative breast cancer.
In this study, we first performed univariate survival analysis and identified 3145 distant relapse–free survival (DRFS) associated genes in 310 patients with HER-2 negative breast cancer from the GSE25055 dataset. After that, a 4-gene prognostic signature was developed by using robust likelihood-based survival model and unsupervised hierarchical clustering analysis. A risk score model was then built by multivariate survival analysis and the prognostic value was further validated in GSE25065 dataset. Our findings suggested that this 4-gene signature could serve as an effective biomarker to predict the chemosensitivity and prognosis for patients with HER-2 negative breast cancer receiving taxane/anthracycline-based therapy.
RESULTS
Identification of differentially expressed genes associated with prognosis in the training dataset
The overall flow diagram of present study was summarized in Figure 1. The 310 breast cancer samples with expression values of 22283 genes were acquired from the GSE25055 dataset. All the patients were diagnosed HER-2 negative and treated with taxane and anthracycline-based chemotherapy. 13510 differentially expressed probes were selected for further analysis according to the screening criteria described in the Materials and Methods part (Supplementary Table 1). A univariate survival analysis was conducted using Cox proportional hazard regression model based on the expression level of these genes. Finally, 3145 seed genes significantly associated with DRFS (p < 0.05) were identified (Supplementary Table 2). The top 20 genes with most remarkable changes are listed in Table 1.
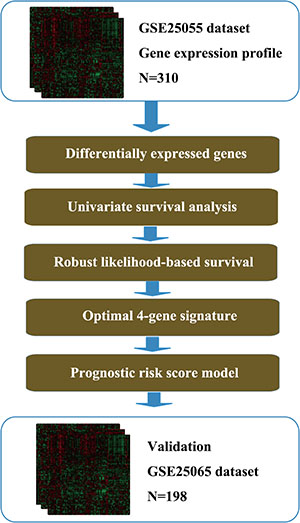
Figure 1: Flow diagram of methods for developing the prognostic 4-gene signature.
Table 1: The top 20 genes with most remarkable changes in training set
Probe ID |
Gene symbol |
p value |
|---|---|---|
211110_s_at |
AR |
1.39E–08 |
210476_s_at |
PRLR |
5.21E–08 |
200810_s_at |
CIRBP |
1.24E–07 |
219648_at |
MREG |
1.31E–07 |
202171_at |
VEZF1 |
1.38E–07 |
205862_at |
GREB1 |
1.54E–07 |
212811_x_at |
SLC1A4 |
1.87E–07 |
208935_s_at |
LGALS8 |
2.18E–07 |
221874_at |
KIAA1324 |
2.36E–07 |
205428_s_at |
CALB2 |
2.38E–07 |
203860_at |
PCCA |
3.16E–07 |
202200_s_at |
SRPK1 |
4.53E–07 |
214552_s_at |
RABEP1 |
5.15E–07 |
205597_at |
SLC44A4 |
5.24E–07 |
206401_s_at |
MAPT |
5.92E–07 |
201951_at |
ALCAM |
6.67E–07 |
209696_at |
FBP1 |
7.44E–07 |
212095_s_at |
MTUS1 |
8.02E–07 |
218692_at |
SYBU |
8.11E–07 |
208682_s_at |
MAGED2 |
9.01E–07 |
In order to investigate the main function of the aforesaid seed genes, we performed KEGG pathway enrichment analysis using clusterProfiler. The result showed that these genes were enriched in several key cancer-related signaling pathways such as cell cycle, cellular senescence, pathways in cancer (Supplementary Table 3). The top 10 pathways were shown in Figure 2.
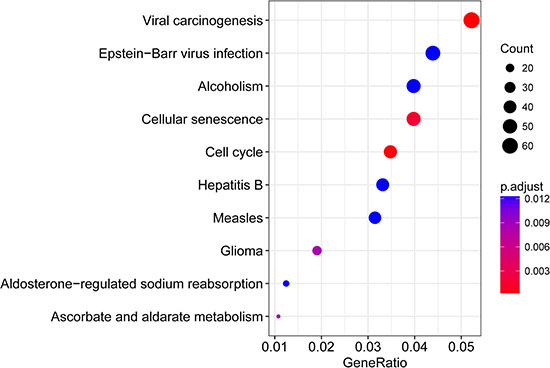
Figure 2: The top 10 enriched pathways for 3145 seed genes significantly associated with DRFS.
Development of the 4-gene signature for prognosis prediction in the training dataset
Given the difficulty in using such a large number of genes for clinical diagnosis, we next screened the optimal survival-associated signature genes by a robust likelihood-based survival model. Four genes (SRPK1, PCCA, PRLR and FBP1) were selected as signature genes that can optimally predict the DRFS of patients in the training dataset, as shown in Table 2. KEGG pathway functional annotation was then adopted to explore the function of these four signature genes. As shown in Table 3, these four genes involved in several signaling pathways related to the development and progression of breast cancer.
Table 2: Survival-associated gene signature screening using forward selection
Probe ID |
Gene symbol |
nloglik |
AIC |
|---|---|---|---|
202200_s_at |
SRPK1 |
342.16 |
686.33* |
203860_at |
PCCA |
335.30 |
674.60* |
210476_s_at |
PRLR |
329.94 |
665.88* |
209696_at |
FBP1 |
328.87 |
665.75* |
212956_at |
TBC1D9 |
328.87 |
667.73 |
206401_s_at |
MAPT |
327.93 |
667.86 |
214552_s_at |
RABEP1 |
327.58 |
669.16 |
208682_s_at |
MAGED2 |
326.85 |
669.71 |
212492_s_at |
KDM4B |
326.82 |
671.65 |
212811_x_at |
SLC1A4 |
323.86 |
667.71 |
200670_at |
XBP1 |
323.71 |
669.42 |
211110_s_at |
AR |
322.83 |
669.65 |
205597_at |
SLC44A4 |
322.46 |
670.93 |
221874_at |
KIAA1324 |
322.38 |
672.77 |
219197_s_at |
SCUBE2 |
322.00 |
673.99 |
200810_s_at |
CIRBP |
321.74 |
675.48 |
219648_at |
MREG |
321.03 |
676.06 |
205862_at |
GREB1 |
320.37 |
676.73 |
202171_at |
VEZF1 |
316.68 |
671.35 |
Table 3: Results of function annotation analysis for 4 signature genes
KEGG pathway |
Gene symbol |
|---|---|
Pentose phosphate pathway |
FBP1 |
Fructose and mannose metabolism |
FBP1 |
Glycolysis/Gluconeogenesis |
FBP1 |
Glucagon signaling pathway |
FBP1 |
AMPK signaling pathway |
FBP1 |
Insulin signaling pathway |
FBP1 |
Glyoxylate and dicarboxylate metabolism |
PCCA |
Propanoate metabolism |
PCCA |
Valine, leucine and isoleucine degradation |
PCCA |
Carbon metabolism |
PCCA/FBP1 |
Prolactin signaling pathway |
PRLR |
Jak-STAT signaling pathway |
PRLR |
Cytokine-cytokine receptor interaction |
PRLR |
Neuroactive ligand-recptor interaction |
PRLR |
PI3K-Akt signaling pathway |
PRLR |
Herpes simplex infection |
SRPK1 |
With the selected gene signature, unsupervised hierarchical clustering analysis was carried out, and the patient population was divided into three sub-classes (Cluster 1, Cluster 2 and Cluster 3), with 132, 69 and 109 samples respectively (Figure 3A). As depicted in Figure 3B, comparing with the other two sub-classes, patients in cluster 3 had much worse outcomes (p = 1.07e–10). A closer look at the clinical characteristics revealed that patients in cluster 3 were mostly basal-like subtype (86.2%, 94/109), and the number was only 39.4% in the whole cohort. Thus, basal-like breast cancer was distributed mainly in cluster 3, consisting with the well-known fact that the triple negative patients always had poor outcomes in clinical practice. Moreover, SRPK1 expression was significantly elevated in triple negative samples while PCCA, PRLR and FBP1 expressions were decreased dramatically (Figure 3C).
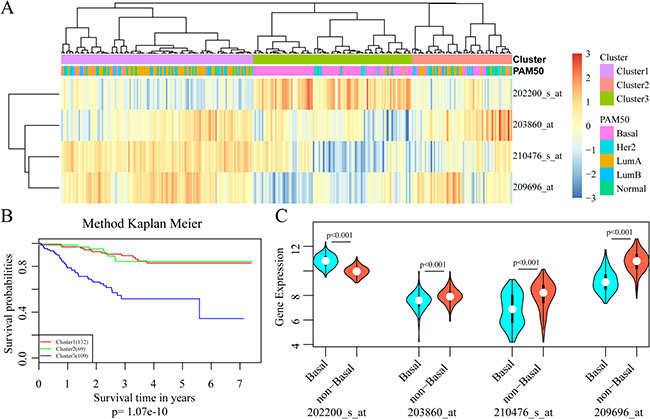
Figure 3: Development of the 4-gene signature for prognosis prediction. (A) Results of unsupervised hierarchical clustering analysis based on the expression levels of the four signature genes. (B) Kaplan–Meier curves for patients in different clusters. (C) The mRNA expression of four signature genes in Basal-like and non-basal-like patients.
All these results suggested that this 4-gene signature may have important application in predicting the prognosis for patients with HER-2 negative breast cancer receiving taxane and anthracycline combination regimens.
Construction and assessment of prognostic risk score model based on 4-gene signature
The regression coefficients of the four signature genes were generated by multivariate survival analysis. A risk score model was then built as follow: Risk Score = 0.38*exp (SRPK1)-0.56*exp (PCCA)-0.3*exp (PRLR)-0.22*exp (FBP1). With the risk score of each sample, the prognostic differences was evaluated (Figure 4A). As illustrated in Figure 4B, higher risk score indicated greater mortality risk for patient with HER-2 negative breast cancer. We also observed that along with the increase of risk score, the expression level of SRPK1 was up-regulated while the other three were declined (Figure 4C). Receiver operating characteristic (ROC) curve analysis was performed to evaluate the prediction power of the risk model. The area under ROC curve (AUC) was 0.883, indicating good performance of this model for prognosis prediction (Figure 5A). The 310 patients were divided into high and low-risk groups using the optimal cut-off score. As showed in Figure 5B, the DRFS of patients in the high-risk group was significantly shorter than that of the low-risk group (p = 8.24e–11).
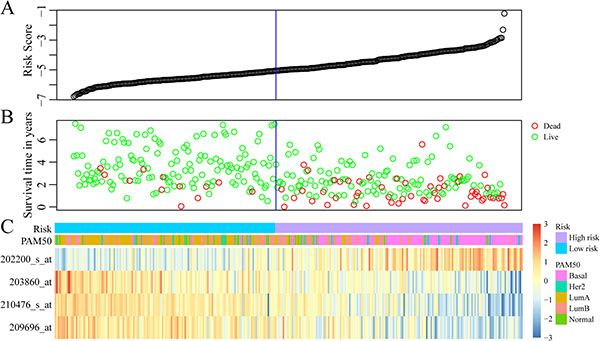
Figure 4: Construction of prognostic risk score model based on 4-gene signature. (A) The risk score of each sample. (B) Different survival status according to the risk score. (C) The mRNA expression of four signature genes in each sample with different risk score.
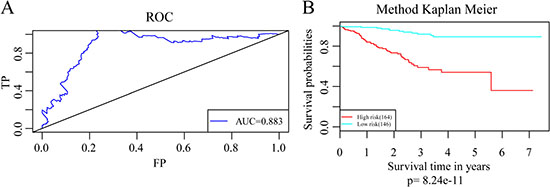
Figure 5: Assessment of the the prediction power of the risk model. (A) The ROC curve for survival predictions with an AUC of 0.883. (B) Kaplan–Meier curves for patients in high and low-risk groups divided with the optimal cut-off score.
To investigate the impact on prognosis of each single signature gene, patients of the training dataset were classified into different groups using the median expression level as the threshold. The results of Kaplan-Meier analysis showed that each of the genes had predictive ability on DRFS, especially PRLR and FBP1. However, the prognostic power was found much stronger when these four genes used in combination (Figure 6). We further compared the prediction capacity of this risk score model with other clinical classification systems including tumor size (T), lymph node invasion (N), TNM stage and PAM50 signature. As shown in Figure 7, the 4-gene signature was proved to have the most robust prognostic power among these clinical signatures.
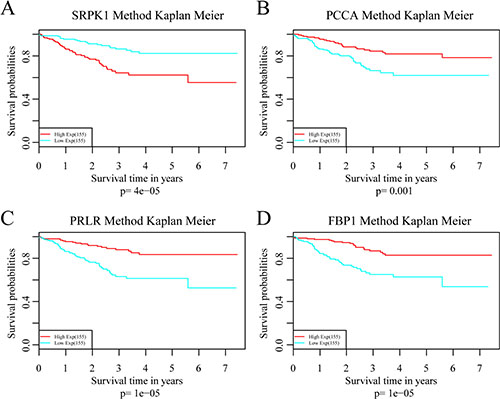
Figure 6: Analysis of the prognostic impact of each single signature gene. (A) Kaplan–Meier curves for patients in SRPK1 high and low-expression groups. (B) Kaplan–Meier curves for patients in PCAA high and low-expression groups. (C) Kaplan–Meier curves for patients in PRLR high and low-expression groups. (D) Kaplan–Meier curves for patients in FBP1 high and low-expression groups.
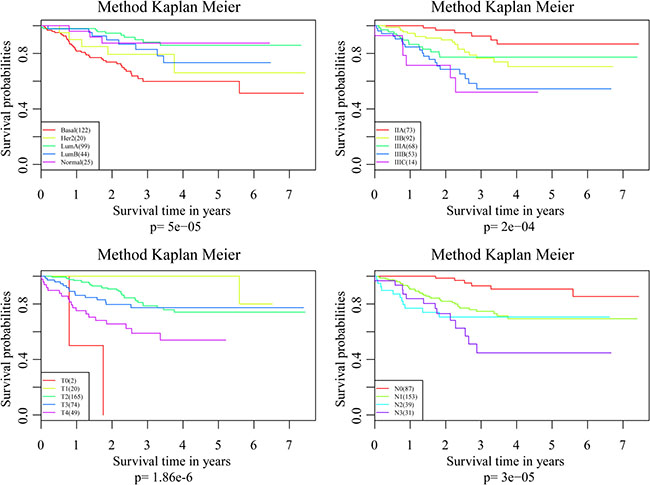
Figure 7: Comparisons of the prediction capacity between 4-gene signature and other clinical classification systems.
External validation of 4-gene signature
To validate the accuracy and repeatability of the prognostic 4-gene signature, the risk score model was applied to the GSE25065 dataset (n = 198). Like that of GSE25055, all the samples of GSE25065 were HER-2 negative following taxane and anthracycline chemotherapy. The prognostic risk score of each patient was then calculated according to the formula. As in the training dataset, higher risk score indicated greater mortality risk for patient with HER-2 negative breast cancer (Figure 8A). Besides, both the expression levels of four signature genes and the proportion of basal-like breast cancer in the GSE25065 dataset were in line with that in the training dataset. The samples were further divided into high-risk group and low-risk group based on the optimal cut-off risk scores. The Kaplan-Meier univariate analysis indicated a statistical significance on DRFS between the two groups (p = 2.0e–6, Figure 8B). Furthermore, increased expression of SRPK1 and reduced expressions of the other three genes in basal-like breast cancer were also observed in the validation dataset. Therefore, this 4-gene signature was an effective marker in predicting prognosis for patients with HER-2 negative breast cancer following taxane and anthracycline-based chemotherapy.
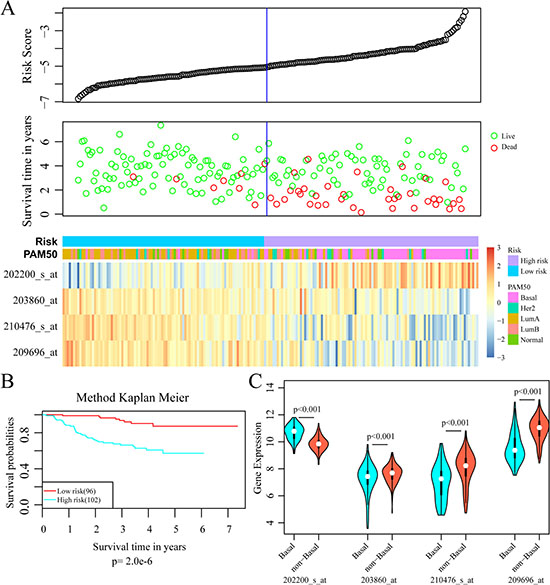
Figure 8: External validation of 4-gene signature. (A) Top: The risk score of each sample in validation set; Middle: Corresponding survival status of each sample; Bottom: The mRNA expression of four signature genes in each sample with different risk score. (B) Kaplan–Meier curves for patients in high and low-risk groups. (C) The mRNA expression of four signature genes in Basal-like and non-basal-like patients.
DISCUSSION
Breast cancer is a heterogeneous group of diseases with diverse clinicopathological features and gene dysregulations [10, 11]. Despite the rapid development of therapeutic approaches, there are still many patients suffer from tumor recurrence and metastasis, which is mainly caused by chemo-resistance. Conventional clinicopathological and molecular prognostic factors, such as TNM stage, histological grade, expression of the oestrogen and progesterone receptors, can not effectively estimate the benefits of chemotherapy in HER-2 negative breast cancer. Additionally, tests designed for molecular classification or prognosis without chemotherapy were found lacking of clinical usefulness in the prediction of survival outcomes in chemosensitive patients [12–14]. Therefore, reliable prognostic factors are urgently needed for HER-2 negative breast cancer patients treated with chemotherapy.
Various genetic changes have been found to play important roles in breast cancer initiation and progression [15, 16]. For example, women with pathogenic variants in breast cancer 1 (BRCA1) and BRCA2 were reported to have a cumulative lifetime risk of developing breast cancer between 41% and 90% [17, 18]. Besides, mutations in tumor protein p53 (TP53) [19], phosphatase and tensin homolog (PTEN) [20], serine/threonine kinase 11 (STK11), cadherin 1 (CDH1) [21], partner and localizer of BRCA2 (PALB2) [22] and checkpoint kinase 2 (CHEK2) [23] were also associated with an increased risk of breast cancer. However, there are currently few systemic evaluations on clinical application of these genes as most studies only focused on one or a few genes. In recent decades, high-throughput genomic technologies, such as DNA microarrays and next-generation sequencing, have been widely applied in the studies of cancer heterogeneity. Several risk models have been constructed to predict tumor metastasis, recurrence, treatment response and prognosis by using miRNA [24, 25] and lncRNA expression profiling [26–28].
In the current study, we developed a robust 4-gene signature (SRPK1, PCCA, PRLR and FBP1) to predict DRFS for patients with HER2-negative breast cancer receiving chemotherapy by analyzing the publicly available gene expression profiles from the GEO database.
The GSE25055 dataset was used as training set and total 3145 genes were identified to be significantly associated with DRFS. Results of KEGG analysis revealed that these genes were enriched in several key cancer-related signaling pathways such as cell cycle, cellular senescence, pathways in cancer. We eventually selected four genes (SRPK1, PCCA, PRLR and FBP1) as signature genes for prognosis prediction by using robust likelihood-based survival model. Among which, PCCA was predicted to be associated with energy metabolism. However, there has been no reported study in breast cancer so far. SRPK1, a protein kinase that specifically phosphorylates serine/arginine-rich (SR) splicing factors, has been reported to be involved in a number of biological and pathological processes [29]. Studies have found that SRPK1 expression was up-regulated in breast cancer, which correlated with poor outcome and preferential metastasis to the lungs and brain [30, 31]. Targeted inhibition of SRPK1 may exert some of its antitumor effects in breast cancer through altering the splice pattern and sensitivity to apoptotic signals [32, 33]. PRLR is a type 1 cytokine receptor that has been implicated in the pathology of breast cancer. Emerging evidence suggests that targeting the PRLR signaling pathway may represent a novel antihormonal approach for the treatment of breast cancer [34, 35]. FBP1, the rate-limiting enzyme in gluconeogenesis, is a critical modulator in breast cancer progression by altering glucose metabolism [36, 37]. Recent studies have demonstrated that low or absent expression levels of FBP1 was a critical oncogenic event in epithelial-mesenchymal transition and might be associated with reduced disease-free survival in basal-like breast cancer [38, 39]. Therefore, all the four signature genes may play key roles in the development and progression of breast cancer and are worth further investigation.
The 4-gene signature was first assessed in the training set. Results of ROC analysis showed robust prognostic power with an AUC of 0.883. Patients in the training set were successfully divided into high- and low-risk groups with significant differences in DRFS between the two groups. The predictive value was further validated in another GEO dataset and similar results were observed. Moreover, the 4-gene signature was proved to have superior prognostic power compared with several clinical signatures such as tumor size, lymph node invasion, TNM stage and PAM50 signature. These results indicated that this 4-gene signature was an effective prognostic predictor for patients with HER-2 negative breast cancer following taxane and anthracycline-based chemotherapy.
High-throughput genomic studies have provided new insights into the molecular mechanisms of breast cancer. However, the clinical applicability of this technology was restricted by the cost and information overflow. The 4-gene prognostic signature obtained from our study can overcome this hurdle to some extent. Considering the limited sample size of our study, large-scale cohort studies will be performed in the future to evaluate the prognostic value of this 4-gene signature. In addition, the biological functions of these four signature genes in breast cancer metastasis have not been fully revealed. Thus, further experimental studies should be conducted to uncover the detailed effects of these four genes on the biological behavior and pathogenesis of breast cancer.
In conclusion, our findings demonstrated that the 4-gene signature was a promising prognostic indicator with a good prospect of clinical application for HER-2 negative breast cancer patients receiving taxane-anthracycline combination therapy.
MATERIALS AND METHODS
Microarray data acquisition and processing
Two independent breast cancer gene expression profile datasets on the Affymetrix Human Genome U133A platform with corresponding clinical information were downloaded from the publicly available GEO database. All the patients were diagnosed HER-2 negative and treated with taxane and anthracycline-based chemotherapy. The GSE25055 dataset containing 310 samples was used as training set to construct risk model and the GSE25065 dataset containing 198 samples was used as validation set to confirm the prognostic power of the model. The MAS5.0 signal intensity for each probe was log2 transformed and quantile normalized to obtain equal distributions.
Four-gene signature identification
Differentially expressed probes were preliminary picked out with the criteria as follows: (i) the median expression level of A gene in each sample is 20% higher than that of the whole genome; (ii) the expression level variance of A gene in each sample is 20% higher than that of the whole genome. A univariate Cox proportional hazard regression survival analysis was conducted by using the R package “survival” to obtain the prognosis-related seed genes with p < 0.05. KEGG Pathway enrichment analysis was performed to investigate the functions of these seed genes by using the clusterProfiler package in R [40]. A robust likelihood-based survival modeling approach [41–43] was used to select the optimal survival-associated gene signature by using the R package “rbsurv”. The detailed algorithmic procedure is as follows: (i) The samples were randomly split into the training set with N*(1 − p) samples and the validation set with N*p samples (p = 1/3). The parameter estimate for each gene was obtained after fitting to the training sample set. With the parameter estimate, log likelihood was computed in both sample sets; (ii) The above procedure was repeated 10 times and the best gene with the largest mean log likelihood was first picked out; (iii) By evaluating every two-gene model, the one with the largest mean log likelihood was selected as the next most appropriate gene. Such forward gene selection process was continued and eventually generated a set of different candidate models; (iv) Akaike information criterion (AIC) statistics was applied for all the candidate models generated in the previous steps. Finally, an optimal model with the smallest AIC was obtained. KEGG pathway functional annotation was further adopted to explore the function of these four signature genes.
Unsupervised hierarchical clustering and multivariate survival analysis
By unsupervised hierarchical clustering analysis, samples were divided into three sub-classes according to the expression levels of four signature genes [44]. Prognostic differences between these sub-classes were further analyzed with Kaplan-Meier survival analysis [45].
Prognostic risk score model construction and external data validation
The regression coefficients of the four signature genes were generated by multivariate survival analysis. A prognostic risk score model was constructed to evaluate the effects of this 4-gene signature on prognosis. The risk score of each patient was calculated according to the formula as follow: Risk Score = 0.38*exp (SRPK1)-0.56*exp (PCCA)-0.3*exp (PRLR)-0.22*exp (FBP1). Receiver operating characteristic (ROC) curve analysis was performed to evaluate the prediction power of the risk model by R package “survivalROC” [46] and the area under the curve (AUC) was calculated. The optimal threshold for risk classification based on ROC curve was obtained. The risk score model was applied to the GSE25065 dataset to validate the accuracy and repeatability of the prognostic 4-gene signature.
ACKNOWLEDGMENTS AND FUNDING
We gratefully acknowledge financial support by the National Natural Science Foundation of China (81702803), the Natural Science Foundation of Zhejiang Province (LY18H160001).
CONFLICTS OF INTEREST
The authors have declared that no competing interests exist.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67:7–30. https://doi.org/10.3322/caac.21387.
2. Harbeck N, Gnant M. Breast cancer. Lancet. 2017; 389:1134–50. https://doi.org/10.1016/s0140-6736(16)31891-8.
3. Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016; 66:271–89. https://doi.org/10.3322/caac.21349.
4. Isik A, Firat D. Bilateral intra-areolar polythelia. Breast J. 2017. https://doi.org/10.1111/tbj.12838.
5. Isik A, Karavas E, Peker K, Soyturk M, Yilmaz I. Male Mondor’s Disease is a Rare Entity. Breast J. 2016; 22:700–1. https://doi.org/10.1111/tbj.12657.
6. Kuijer A, Straver M, den Dekker B, van Bommel AC, Elias SG, Smorenburg CH, Wesseling J, Linn SC, Rutgers EJ, Siesling S, van Dalen T. Impact of 70-Gene Signature Use on Adjuvant Chemotherapy Decisions in Patients With Estrogen Receptor-Positive Early Breast Cancer: Results of a Prospective Cohort Study. J Clin Oncol. 2017; 35:2814–9. https://doi.org/10.1200/jco.2016.70.3959.
7. Hess KR, Anderson K, Symmans WF, Valero V, Ibrahim N, Mejia JA, Booser D, Theriault RL, Buzdar AU, Dempsey PJ, Rouzier R, Sneige N, Ross JS, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006; 24:4236–44. https://doi.org/10.1200/jco.2006.05.6861.
8. Lee JK, Coutant C, Kim YC, Qi Y, Theodorescu D, Symmans WF, Baggerly K, Rouzier R, Pusztai L. Prospective comparison of clinical and genomic multivariate predictors of response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2010; 16:711–8. https://doi.org/10.1158/1078-0432.ccr-09-2247.
9. Popovici V, Chen W, Gallas BG, Hatzis C, Shi W, Samuelson FW, Nikolsky Y, Tsyganova M, Ishkin A, Nikolskaya T, Hess KR, Valero V, Booser D, et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 2010; 12: R5. https://doi.org/10.1186/bcr2468.
10. Genomic Analysis Detects Recurrent Promoter Mutations in Breast Cancer. Cancer Discov. 2017. https://doi.org/10.1158/2159-8290.cd-rw2017-130.
11. Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, Lawrence MS, Taylor-Weiner A, Rodriguez-Cuevas S, Rosenberg M, Hess J, Stewart C, Maruvka YE, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017; 547:55–60. https://doi.org/10.1038/nature22992.
12. Albain KS, Barlow WE, Shak S, Hortobagyi GN, Livingston RB, Yeh IT, Ravdin P, Bugarini R, Baehner FL, Davidson NE, Sledge GW, Winer EP, Hudis C, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol. 2010; 11:55–65. https://doi.org/10.1016/s1470-2045(09)70314-6.
13. Straver ME, Glas AM, Hannemann J, Wesseling J, van de Vijver MJ, Rutgers EJ, Vrancken Peeters MJ, van Tinteren H, Van’t Veer LJ, Rodenhuis S. The 70-gene signature as a response predictor for neoadjuvant chemotherapy in breast cancer. Breast Cancer Res Treat. 2010; 119:551–8. https://doi.org/10.1007/s10549-009-0333-1.
14. Liedtke C, Hatzis C, Symmans WF, Desmedt C, Haibe-Kains B, Valero V, Kuerer H, Hortobagyi GN, Piccart-Gebhart M, Sotiriou C, Pusztai L. Genomic grade index is associated with response to chemotherapy in patients with breast cancer. J Clin Oncol. 2009; 27:3185–91. https://doi.org/10.1200/jco.2008.18.5934.
15. Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, Sharma L, Saam J, Lancaster J, Daly MB. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. 2017; 123:1721–30. https://doi.org/10.1002/cncr.30498.
16. Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, Massard C, Levy C, Arnedos M, Lacroix-Triki M, Garrabey J, Boursin Y, Deloger M, et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016; 13:e1002201. https://doi.org/10.1371/journal.pmed.1002201.
17. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007; 25:1329–33. https://doi.org/10.1200/jco.2006.09.1066.
18. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003; 72:1117–30. https://doi.org/10.1086/375033.
19. Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, Platzer P, Eng C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med. 2007; 357:2543–51. https://doi.org/10.1056/NEJMoa071825.
20. Li S, Shen Y, Wang M, Yang J, Lv M, Li P, Chen Z, Yang J. Loss of PTEN expression in breast cancer: association with clinicopathological characteristics and prognosis. Oncotarget. 2017; 8:32043–54. https://doi.org/10.18632/oncotarget.16761.
21. Han MR, Zheng W, Cai Q, Gao YT, Zheng Y, Bolla MK, Michailidou K, Dennis J, Wang Q, Dunning AM, Brennan P, Chen ST, Choi JY, et al. Evaluating genetic variants associated with breast cancer risk in high and moderate-penetrance genes in Asians. Carcinogenesis. 2017; 38: 511–8. https://doi.org/10.1093/carcin/bgx010.
22. Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, Berman SH, Blecua P, Riaz N, Huo Y, Ding YC, Neuhausen SL, Weigelt B, et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene. 2017; 36:4161–70. https://doi.org/10.1038/onc.2017.46.
23. Schoolmeester JK, Moyer AM, Goodenberger ML, Keeney GL, Carter JM, Bakkum-Gamez JN. Pathologic Findings in Breast, Fallopian Tube and Ovary Specimens in non-BRCA Hereditary Breast and/or Ovarian Cancer Syndromes: A Study of 18 Patients with Deleterious Germline Mutations in RAD51C, BARD1, BRIP1, PALB2, MUTYH or CHEK2. Hum Pathol. 2017. https://doi.org/10.1016/j.humpath.2017.06.018.
24. Du F, Yuan P, Zhao ZT, Yang Z, Wang T, Zhao JD, Luo Y, Ma F, Wang JY, Fan Y, Cai RG, Zhang P, Li Q, et al. A miRNA-based signature predicts development of disease recurrence in HER2 positive breast cancer after adjuvant trastuzumab-based treatment. Sci Rep. 2016; 6: 33825. https://doi.org/10.1038/srep33825.
25. Bing Z, Tian J, Zhang J, Li X, Wang X, Yang K. An Integrative Model of miRNA and mRNA Expression Signature for Patients of Breast Invasive Carcinoma with Radiotherapy Prognosis. Cancer Biother Radiopharm. 2016; 31:253–60. https://doi.org/10.1089/cbr.2016.2059.
26. Zhong L, Lou G, Zhou X, Qin Y, Liu L, Jiang W. A six-long non-coding RNAs signature as a potential prognostic marker for survival prediction of ER-positive breast cancer patients. Oncotarget. 2017; 8:67861–67870. https://doi.org/10.18632/oncotarget.18919.
27. Guo W, Wang Q, Zhan Y, Chen X, Yu Q, Zhang J, Wang Y, Xu XJ, Zhu L. Transcriptome sequencing uncovers a three-long noncoding RNA signature in predicting breast cancer survival. Sci Rep. 2016; 6:27931. https://doi.org/10.1038/srep27931.
28. Sun J, Chen X, Wang Z, Guo M, Shi H, Wang X, Cheng L, Zhou M. A potential prognostic long non-coding RNA signature to predict metastasis-free survival of breast cancer patients. Sci Rep. 2015; 5:16553. https://doi.org/10.1038/srep16553.
29. Bullock N, Oltean S. The many faces of SRPK1. J Pathol. 2017; 241:437–40. https://doi.org/10.1002/path.4846.
30. van Roosmalen W, Le Devedec SE, Golani O, Smid M, Pulyakhina I, Timmermans AM, Look MP, Zi D, Pont C, de Graauw M, Naffar-Abu-Amara S, Kirsanova C, Rustici G, et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J Clin Invest. 2015; 125:1648–64. https://doi.org/10.1172/jci74440.
31. Li XH, Song JW, Liu JL, Wu S, Wang LS, Gong LY, Lin X. Serine-arginine protein kinase 1 is associated with breast cancer progression and poor patient survival. Med Oncol. 2014; 31: 83. https://doi.org/10.1007/s12032-014-0083-8.
32. Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007; 67:2072–80. https://doi.org/10.1158/0008-5472.can-06-2969.
33. Lin JC, Lin CY, Tarn WY, Li FY. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. Rna. 2014; 20:1621–31. https://doi.org/10.1261/rna.045583.114.
34. Damiano JS, Wasserman E. Molecular pathways: blockade of the PRLR signaling pathway as a novel antihormonal approach for the treatment of breast and prostate cancer. Clin Cancer Res. 2013; 19:1644–50. https://doi.org/10.1158/1078-0432.ccr-12-0138.
35. Kelly MP, Hickey C, Makonnen S, Coetzee S, Jalal S, Wang Y, Delfino F, Shan J, Potocky TB, Chatterjee I, Andreev J, Kunz A, D’Souza C, et al. Preclinical Activity of the Novel Anti-Prolactin Receptor (PRLR) Antibody-Drug Conjugate REGN2878-DM1 in PRLR-Positive Breast Cancers. Mol Cancer Ther. 2017; 16:1299–311. https://doi.org/10.1158/1535-7163.mct-16-0839.
36. Shi L, He C, Li Z, Wang Z, Zhang Q. FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1alpha. Neoplasma. 2017; 64:535–42. https://doi.org/10.4149/neo_2017_407.
37. Li K, Ying M, Feng D, Du J, Chen S, Dan B, Wang C, Wang Y. Fructose-1,6-bisphosphatase is a novel regulator of Wnt/beta-Catenin pathway in breast cancer. Biomed Pharmacother. 2016; 84:1144–9. https://doi.org/10.1016/j.biopha.2016.10.050.
38. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, Lorkiewicz P, St Clair D, Hung MC, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013; 23:316–31. https://doi.org/10.1016/j.ccr.2013.01.022.
39. Shi L, Zhao C, Pu H, Zhang Q. FBP1 expression is associated with basal-like breast carcinoma. Oncol Lett. 2017; 13:3046–56. https://doi.org/10.3892/ol.2017.5860.
40. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012; 16:284–7. https://doi.org/10.1089/omi.2011.0118.
41. Wang JY, Tai JJ. Robust quantitative trait association tests in the parent-offspring triad design: conditional likelihood-based approaches. Ann Hum Genet. 2009; 73:231–44. https://doi.org/10.1111/j.1469-1809.2008.00502.x.
42. Renaud G, Stenzel U, Maricic T, Wiebe V, Kelso J. deML: robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics. 2015; 31:770–2. https://doi.org/10.1093/bioinformatics/btu719.
43. Kendall WL, Pollock KH, Brownie C. A likelihood-based approach to capture-recapture estimation of demographic parameters under the robust design. Biometrics. 1995; 51:293–308.
44. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998; 95:14863–8.
45. Stel VS, Dekker FW, Tripepi G, Zoccali C, Jager KJ. Survival analysis I: the Kaplan-Meier method. Nephron Clin Pract. 2011; 119:c83–8. https://doi.org/10.1159/000324758.
46. Zou KH, O’Malley AJ, Mauri L. Receiver-operating characteristic analysis for evaluating diagnostic tests and predictive models. Circulation. 2007; 115:654–7. https://doi.org/10.1161/circulationaha.105.594929.