INTRODUCTION
During spermatogenesis, Sertoli cells offer nutritional and physical support to the developing germ cells, and creates an immuno-privileged microenvironment by blood-testis barrier (BTB) spatially sequestering germ cells from autoimmunity and ectogenesis stimulus and thereby, fostering the completion of meiosis [1, 2]. BTB which is composed of the tight junction (TJ) together with basal ectoplasmic specialization (basal ES) and gap junction (GJ) by the adjacent Sertoli cells undergoes periodically reconstruction to facilitate the transition of preleptotene spermatocytes from basal to apical compartment during seminiferous epithelial cycle promoted by cytokines and chemicals [3, 4]. BTB catastrophe has been recognized as an important contributor for the environmental endocrine-disrupting compounds (EDCs) and other chemicals-induced dysfunction of spermatogenesis [5–7]. The damaging effects of these toxicants to testicular function are mediated by mitogen-activated protein kinases downstream, which in turn perturbs the actin bundling and accelerates the actin-branching activity, result in impaired spermatogenesis [8, 9]. In addition, oxidative stress-caused by inflammatory cytokines and ectogenesis stimulus induced TJ barrier disruption [10–13].
Olaquindox (OLA, 2-(N-2-hydroxyethyl-carba monyl)-3-methyl-quinoxaline-N1, N4-dioxide), a common medicinal feed additive in livestock husbandry for growth promoting and antibacterial purpose to prevent dysentery and bacterial enteritis, has been banned for using by the European Commission of the European Community and Canada since 1998 due to its potential risks such as genotoxic, mutagenic and photoallergenic effects [14–16]. Notably, it is still being widely used in China due to its beneficial effects, which potentially exposes our human, if consume those animal products with residual OLA due to its illogical usage, into the risks of its biological hazards. Recent studies have demonstrated that a relatively low concentration of olaquindox significantly induced DNA mutation, and the frequency of which was increased by up to 12-fold [17], and the cumulative toxicity of olaquindox has also been reported [18–20]. More importantly, OLA could incite DNA damage and apoptosis [21–23]. Despite OLA could be categorized as mutagenic and carcinogenic with developmental and reproductive toxicities [24] whether OLA has deleterious effects on mammalian spermatogenesis, or more specifically, the BTB dynamic and if so, what the underlying mechanisms might be involved in still remain to be explored.
In this study, we in depth determine that OLA perturbs the TJ permeability function by attenuating the cell-cell interface distribution of the tight junction proteins and the architecture of F-actin. Moreover, OLA potentiates DNA damage and apoptosis due to aggravation of ROS production, and disrupts the epigenetic status in Sertoli cells. Overall, our study pinpoint that olaquindox targets Sertoli cells to affect BTB function, which will help us to develop a better appreciation of the toxicological feature of OLA in male reproductive biology.
RESULTS
OLA compromises Sertoli cells survival
To investigate the effect of OLA on Sertoli cells viability, CCK-8 assay was performed after Sertoli cells treated with a concentration gradient of OLA for 24 h and 48 h. The cell viability was significantly decreased along with the increase of OLA concentration after 24 h and 48 h treatment, respectively, indicating that OLA effectively inhibited cell viability in a dose-dependent manner (Figure 1A). The abundance of ATP, which is the product of cellular energy metabolism in metabolically active cells, also experienced a conspicuous decline after 24 h treatment with 400- or 800 μg/ml OLA (Figure 1B). Moreover, exposure with different concentrations of OLA for 24 h and 48 h significantly led to a reduction in the number of living cells and morphology changes with shrinking, rounding, cell vacuolization and detachment morphologies in Sertoli cells (Figure 1C), further confirming that OLA exposure compromises Sertoli cells survival.
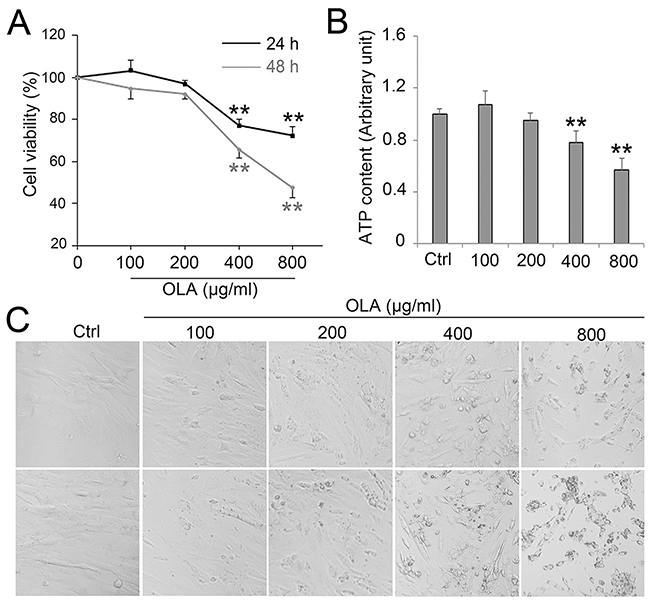
Figure 1: OLA decreased Sertoli cell viability. (A) Cell viability was determined by CCK-8 assay in Sertoli cells treated with the indicated concentrations of OLA for 24 h and 48 h. Cells treated with vehicle (0.2% DMSO) were used as negative control (0). Date are presented as mean ± S.E.M. (three independent replicates per groups). * p < 0.05; ** p < 0.01. (B) Content of ATP was measured by luciferase assay followed the indicated concentrations of OLA for 24 h. Cells treated with vehicle (0.1% DMSO) were used as negative control. Date are presented as mean ± S.E.M. (three independent replicates per groups). * p < 0.05; ** p < 0.01. (C) Cell morphology were observed using an inverted microscope (Nikon, Japan).
OLA disrupts the TJ permeability barrier
Sertoli cells cultured in vitro in ~2-3 d are known to establish a functional permeability barrier that mimic the BTB in vivo, and this modle has been widely used by investigators in this field to study Sertoli cell TJ barrier function [3, 25]. To uncover whether OLA could influence BTB integrity, Sertoli cells were cultured for 3 d and treated with a concentration gradient of OLA for 24 h followed by determination of trans-epithelial electrical resistance (TER). Of note, OLA was found to induce a disruption of the TJ barrier function evidenced by the precipitous decrease in TER starting from the concentration of 400 μg/ml (Figure 2A). The BTB-associated proteins were then examined to confirm the dysfunction of BTB integrity. Significantly, the TJ proteins [e.g., zonula occludens 1 (ZO-1), occludin], basal ES protein (e.g., N-cadherin) and GJ protein (p-Connexin 43) were down-regulated. Additionally, the expression of the TJ regulator FAK (focal adhesion kinase) remains unchanged while its phosphorylated form (p-FAKY397) was declined following OLA treatment. It was notable that p-p38 MAPK was also activated in a dose-dependent manner (Figure 2B). After OLA treatment (400 μg/ml), the weakened expression and mis-localization of TJ protein ZO-1 and basal ES protein N-cadherin at the Sertoli cell-cell interface were also noted wherein these proteins no longer tightly associated with the cell cortical zone, redistributing from the cell-cell interface to the cell cytosol (Figure 2C, white arrow). Taken together, we conclude that OLA could disrupt BTB integrity by interfering with the dynamics of the TJ and basal ES proteins at the BTB.
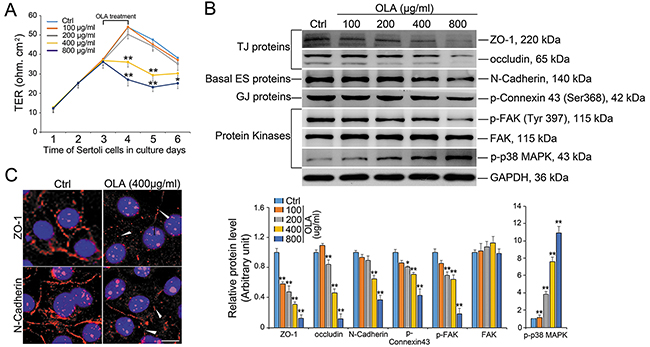
Figure 2: OLA perturbs Sertoli cell BTB function. Sertoli cells cultured on Matrigel-coated bicameral units, dishes or coverslips were treated on day 3 with 100-, 200-, 400- or 800 μg/ml of OLA for 24 h. Thereafter, cells were washed twice with PBS to remove residual OLA and terminated for immunoblot (IB) or IF on day 4. Cells treated with vehicle (0.2% DMSO) were used as negative control. (A) Graph showing the integrity of TJ permeability barrier after OLA vs. vehicle control (0.2% DMSO) treatment. Date are presented as mean ± S.E.M. (three independent replicates per groups). * p < 0.05; ** p < 0.01. (B) Immunoblot analysis to assess the effects of OLA on the expression of TJ proteins: ZO-1 and occludin; basal ES proteins: N-cadherin; protein kinases: FAK, p-FAK and p-p38MAPK. GAPDH served as protein loading control. Semiquantitative analysis of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01. (C) Immunofluorescence analysis to assess the effects of OLA at 400 μg/ml vs. control on the distribution of TJ proteins: ZO-1 and basal ES proteins: N-cadherin. OLA caused internalization of TJ and basal ES proteins in Sertoli cells in which these proteins no longer tightly localized at the Sertoli cell-cell interface. ZO-1 and N-cadherin at the cell-cell interface were diminished following OLA treatment (white arrowheads). Nuclei were visualized by DAPI (blue). Scale bar, 20μm.
OLA perturbs Sertoli cell cytoskeleton
The regulation of actin dynamics in unique testicular junctions is crucial to spermatogenesis [26–28]. To pinpoint whether destruction of TJ barrier function in OLA-treated Sertoli cells could be ascribe to disrupted F-actin architecture, F-actin organization was therefore analyzed. As expected, F-actin architecture in Sertoli cells was significantly affected by 400 μg/ml OLA exposure with actin microfilaments highly aggregated at the cell periphery (white arrow) and few stretches fibers running across the cells (Figure 3A), implicating that the dis-organization of F-actin is likely to be the causative of the disrupted TJ function. To deeply uncover the underlying mechanism by which OLA dictates F-actin dynamics, the expression of several actin regulating proteins were then examined. As shown in Figure 3B, the abundance of N-WASP, the regulator of actin polymerization and activator of Arp2/3 complex, was increased while the branched actin polymerization protein Arp3 remains unchanged. The small GTPases of Rho family including Rac1 and CDC42, which act as the upstream activators of N-WASP, are the major regulators of actin cytoskeleton [29, 30]. Noteworthly, the expression of both Rac1 and CDC42 were abruptly elevated following OLA treatment starting from the dosage of 100 μg/ml. Palladin, a cytoskeletal scaffolding molecule that promotes actin bundling, associates with the actin filaments in Sertoli cells and thereby maintains the TJ function and spermatid transport [31]. Intriguingly, the abundance of palladin was decreased by a relative high concentration (400 μg/ml) of OLA exposure (Figure 3B). Overall, our results indicated that OLA disrupts F-actin architecture which consequently triggers the loss of cell polarity in Sertoli cells.
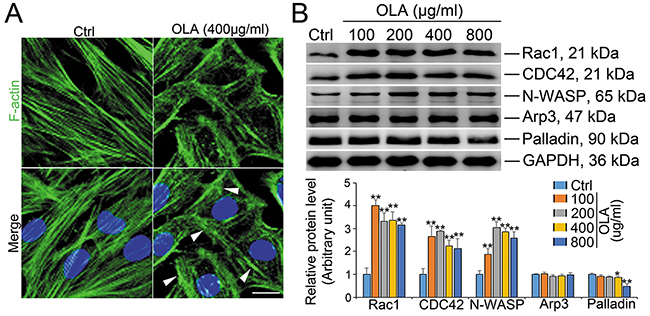
Figure 3: OLA disrupts actin microfilaments organization in Sertoli cells. (A) Immunofluorescence analysis to assess the effects of OLA on the organization of actin microfilaments in Sertoli cells. Sertoli cells cultured for 2 d were treated with 400 μg/ml OLA or control (0.1% DMSO) for 24 h. Cells were fixed and processed for FITC-phalloidin (F-actin, green) and DAPI (nuclei, blue) staining. Significantly, OLA was prominently disorganized actin microfilaments with dense clusters (annotated by white asterisk). Scale bar, 20 μm. (B) Immunoblot analysis to assess the effects of OLA on the expression of actin regulatory proteins: Rac1, CDC42, N-WASP, Arp3 and Palladin. GAPDH served as protein loading control. Semiquantitative analysis of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01.
OLA attenuates mTORC2 complex activity
The mammalian target of rapamycin (mTOR) is a well known non-receptor protein Ser/Thr kinase that orhestrate a spectrum of cellular biological events including cytoskeleton remodeling to assist BTB reconstruction during the epithelial cycle of spermatogenesis [32–36]. As such, we next examined the mTOR and rictor, which together with other binding partners form the mammalian target of rapamycin (mTOR) complex 2 (mTORC2) to regulate blood-testis barrier dynamics via effecting gap junction communications and actin cytoskeleton [34, 37]. As shown in Figure 4, rictor as well as the phosphorylated form of mTOR (p-mTORS2481) were significantly decreased by OLA exposure in a dose dependent manner, implying that the level of functional mTORC2 was reduced following OLA treatment (Figure 4). mTORC2 has been implicated in regulation of BTB dynamics via PKCα and/or AKT pathway [37]. For this, we further determined the proteins in PKCα and AKT pathways by immunoblot analysis. As anticipated, the abundance of p-PKCα and p-AKTSer 473 were indeed reduced regardless of the total levels of PKC-α and AKT remained unaltered. mTOR plays a critical role in governing cell proliferation by interfering with several translational effectors including p70S6 kinase [38] and the level of p-p70S6 kinase was obviously reduced in a OLA-dose dependent manner (Figure 4), confirming that the cell proliferation activity was dampened.
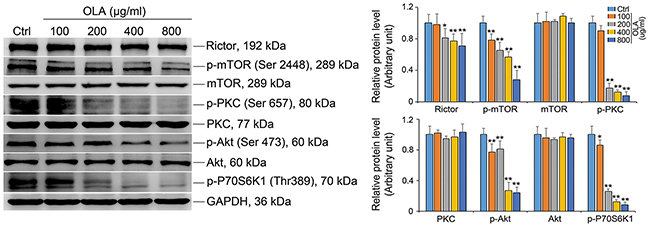
Figure 4: OLA attenuates mTORC2 complex activity. Sertoli cells cultured on dishes were treated on day 3 with 100-, 200-, 400- or 800 μg/ml OLA for 24 h. Cells treated with vehicle (0.2% DMSO) were used as negative control. Thereafter, cells were washed twice with PBS to remove residual OLA and terminated for immunoblot (IB). Immunoblot analysis to assess the effects of OLA on the expression of mTORC2 complex: Ritor and p-mTOR, downstream proteins: PKC, Akt, p-PKC and p-AktSer473. GAPDH served as protein loading control. Semiquantitative analysis of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01.
OLA induces ROS production
Oxidative stress represents as another common trigger of multiple chemicals-induced barrier disruption [10–12]. To reveal the effect of OLA on ROS production, Sertoli cells were treated with various concentrations of OLA for 24 h followed by determination of ROS generation with ROS-GloTM H2O2 Assay Kit. OLA strikingly aggravated the cellular reactive oxygen species (ROS) content in a dose-dependent manner (Figure 5A). Pretreatment with N-acetylcysteine (NAC), a thiol antioxidant, effectively ameliorated the OLA exposure induced ROS production in Sertoli cells (Figure 5B). More importantly, pretreatment with NAC could also effectively alleviate the dysfunction of protein expression regarding BTB integrity (ZO-1, N-Cadherin and p-FAK) and actin regulation (Rac1 and N-WASP) (Figure 5C). Overall, the results indicated that ROS stress might be an important contributor to the compromised permeability function by interfering with the expression of BTB integrity proteins following OLA exposure.
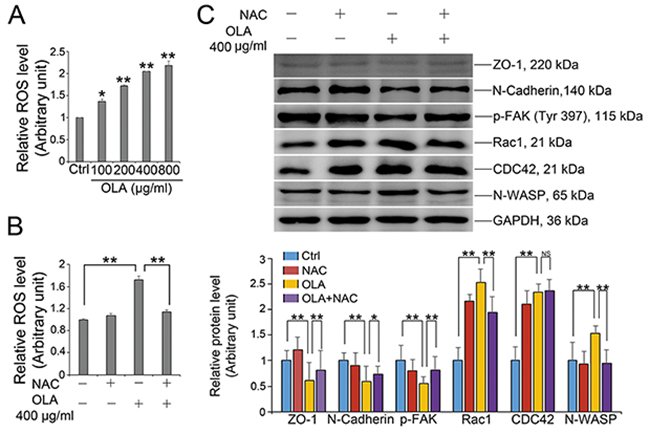
Figure 5: OLA induces ROS production. (A) The level of ROS was measured by ROS-GloTM H2O2 Assay Kit, after Sertoli cells were treated with the indicated concentrations of OLA for 24 h. Cells treated with vehicle (0.2% DMSO) were used as negative control. Date are presented as mean ± S.E.M. (three independent replicates per groups). * p < 0.05; ** p < 0.01. (B) Sertoli cells were pre-treated with NAC (10 mM) for 1 h, followed by OLA (400 μg/ml) treatment for 24 h prior to ROS assay. Date are presented as mean ± S.E.M. (three independent replicates per groups). * p < 0.05; ** p < 0.01. (C) Sertoli cells were pretreated with NAC(10 mM) for 1 h, followed by 400 μg/ml OLA treatment for 24 h prior to immunblot analysis for the expression of TJ protein (ZO-1), basal ES protein (N-Cadherin), p-FAK and actin regulator proteins (Rac1, CDC42, and N-WASP). GAPDH served as protein loading control. Semiquantitative analysis of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01, NS: not significant.
OLA causes DNA damage
Even though intracellular ROS is required for many cellular functions and mild oxidative stress facilitates cell survival and growth adaptation, severe oxidative stress could cause DNA damage, cell senescence and even cell death [39]. Indeed, the potentiated DNA damage, which was revealed by the increased expression of γH2A.X, was obviously observed in OLA-exposed Sertoli cells (Figure 6A), which was further confirmed by the conspicuous increased in the γH2A.X foci (Figure 6B). Pretreatment with NAC, significantly, reduced the expression of γH2A.X to a lesser extent in OLA-exposed Sertoli cells implies that, in OLA-exposed Sertoli cells, the DNA damage might be largely caused by ROS stress (Figure 6C). In response to DNA damage, cells must timely and properly activate the related signaling cascades to modulate chromatin structure and organization within the DNA lesion region and thereby, creating a permissive environment for recruiting DNA damage repair (DDR) factors [40]. To examine whether the potentiated DNA damage in OLA-exposed Sertoli cells could be attributed to the dysfunction of DNA damage repairing event, the homologous recombination (HR) DNA repairing factor RAD51C was analyzed. Significantly, the expression RAD51C was gradually decreased following OLA treatment (Figure 6A). These results indicated that the DNA damage caused by ROS stress in OLA-exposed Sertoli cells was ascribed to the attenuation of DNA repairing.
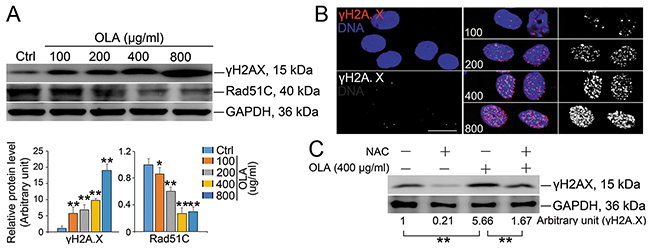
Figure 6: OLA induces DNA damage. (A) Sertoli cells were treated with the indicated concentrations of OLA for 24 h, and treated with vehicle (0.2% DMSO) were used as negative control followed by immunblot analysis for the expression of γH2A.X and Rad51C. Semiquantitative analyses of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01. (B) Representative images showing the immunofluorescence staining of γH2A.X (red) and DAPI (nuclei, blue) in Sertoli cells after treatment with the indicated concentrations of OLA for 24 h. Scale bar, 20μm. (C) Sertoli cells were pretreated with NAC (10 mM) for 1 h, followed by 400 μg/ml OLA treatment for 24 h prior to immunblot analysis for the expression of γH2A.X. GAPDH served as protein loading control. Semiquantitative analysis of protein expression in following number (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01.
OLA inhibits autophagic flux and induces apoptosis in Sertoli cells
Autophagy and apoptosis are two main interlinked mechanisms for cells to eliminate the unwanted cellular materials and to determine the cell fate upon intrinsic and extrinsic stress insult [41, 42]. Autophagy in Sertoli cells was further assessed by examining autophagic flux, which is the dynamic process of autophagy. Intriguingly, Beclin-1 which is a critical factor that initiates autophagy was somehow significantly decreased in OLA-exposed Sertoli cells in a dose dependent manner (Figure 7A). Autophagic flux can be measured by the level of SQSTM1/p62 and LC3-II. The accumulation of SQSTM1/p62 and the strikingly reduction in LC3II/I ratio indicating that a weakened autophagic flux occurred in OLA treated Sertoli cells (Figure 7A). Pretreatment with NAC markedly inhibited the OLA exposure induced accumulation of SQSTM1/p62 (Figure 7D). When DNA damage cannot be efficiently repaired, cells will ultimately undergo necrosis and programmed cell death or namely apoptosis [42]. As shown in Figure 7B, the apoptosis, which was revealed by the caspase-3/7 activity, was steeply induced by OLA exposure in a dose-dependent manner. The increased expression of cleaved-PARP, cleaved-caspase 3 and Bax, which are well-recognized apoptosis markers, further confirmed the occurrence of apoptosis in OLA treated Sertoli cells (Figure 7C). Notably, the OLA induced apoptosis in Sertoli cells could be efficiently rescued by NAC pretreatment evidenced from the decline in the expression of cleaved-caspase 3 and cleaved-PARP (Figure 7D). Taken together, these results potently indicated that OLA induces apoptosis while inhibit autophagy in Sertoli cells.
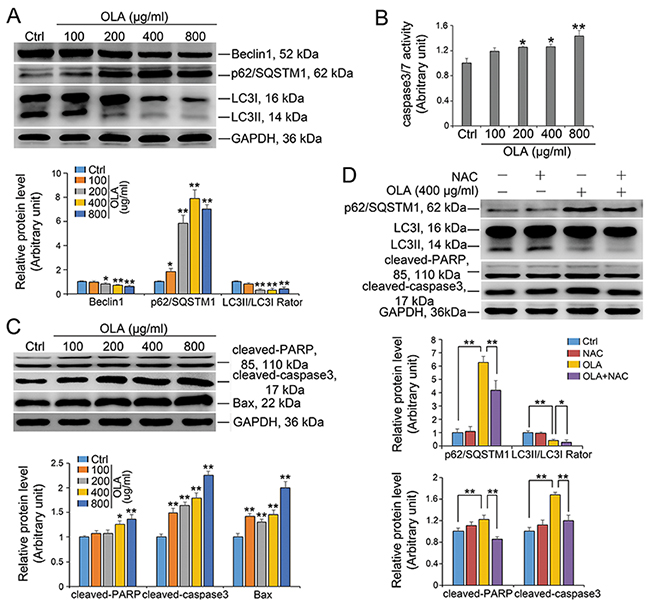
Figure 7: OLA inhibits autophagic flux and induces apoptosis. Sertoli cells cultured on dishes were treated on day 3 with 100-, 200-, 400- or 800 μg/ml OLA for 24 h. Cells treated with vehicle (0.2% DMSO) were used as negative control. (A) Immunoblot analysis to assess the expression of autophagic flux. GAPDH served as protein loading control. Semiquantitative analyses of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05, ** p < 0.01. (B) Caspase-3/7 activity was examined using the Caspase-3/7 assay Kit (Promega) according to the manufacturer’s protocol. Date are presented as mean ± S.E.M. (three independent replicates per groups). * p < 0.05; ** p < 0.01; NS: not significant. (C) Immunoblot analysis to assess the expression of cleaved-PARP, cleaved-caspase3 and Bax. Semiquantitative analyses of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01. (D) Sertoli cells were pretreated with NAC (10 mM) for 1 h, followed by 400 μg/ml OLA treatment for 24 h prior to immunblot analysis for the expression of each protein. Semiquantitative analysis of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01.
OLA causes histone modification change
Histone modifications, such as methylation and acetylation, are critical for DNA damage repair and genome integrity [43–45]. Tri-methylation of histone H3 (H3K9me3) is locally and transiently up-regulated while the acetylation of H3 (H3K9ac) is down-regulated in the vicinity of DNA lesions [46, 47]. Of note, in OLA-treated (400 μg/ml) Sertoli cells, the methylation of H3K9 and H3K27, which mainly involves in regulating transcriptional activity and heterochromatic genome integrity was significantly augmented while the acetylation of H3K9 and H3K27, which are transcriptionally active chromatin marks [48], was concomitantly reduced (Figure 8). Notably, after NAC pretreatment, the hypoacetylation of H3K9 and H3K27 induced by OLA exposure could be efficiently rescued while the hypermethylation of H3K9 and H3K27 induced by OLA exposure was intriguingly somehow boosted (Supplementary Figure 1).
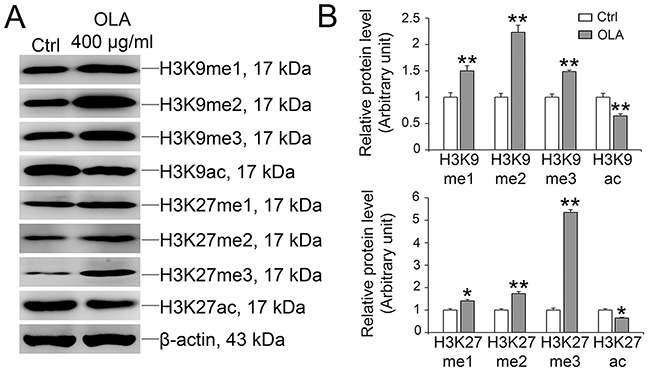
Figure 8: OLA increases the methylation and decreases acetylation of H3K9 and H3K27. Sertoli cells cultured on dishes were treated on day 3 with 400 μg/ml OLA for 24 h. Cells treated with vehicle (0.1% DMSO) were used as negative control. Thereafter, cells were washed twice with PBS to remove residual OLA and terminated for immunoblot (IB). (A) Immunoblot analysis to assess the effects of OLA on the expression of H3K9 and H3K27 modification. β-actin served as protein loading control. (B) Semiquantitative analysis of protein expression in following histogram (mean ± S.E.M., three independent replicates per groups). * p < 0.05; ** p < 0.01.
DISCUSSION
Spermatogenesis, which is largely dictated by environmental, endocrine, paracrine and metabolic cues, produces the male germ cells or termed sperms and thereby determines the reproductive potential of males. Sertoli cells play a crucial role during spermatogenesis by offering nutritional and physical support and creating an immune-privileged environment to protect germ cells from endogenous and exogenous insults [1, 2]. Unfortunately, Sertoli cells usually are the target of multiple reproductive toxicants which compromise the spermatogenesis and the consequent fertility in male [49]. We herein for the first time deciphered that OLA has deleterious effects on male reproduction by interfering with the BTB integrity, cytoskeleton architecture, DNA damage and even histone epigenetic status in Sertoli cells. More importantly, our study could broaden our knowledge of the biological hazards of OLA to human health, which might help to develop a better appreciation of the regulation of OLA usage and environmental contamination.
Primary Sertoli cells after 2-3 days of culturing are known to form a functional and structurally ultra-structures that mimics the BTB in vivo [3, 4]. In this context, we use in vitro model to determine the underlying mechanisms by which OLA induces reproductive toxicity. We found that OLA possess cytotoxicity to Sertoli cell by reducing cell viability and ATP production. Moreover, OLA increases the tight junction permeability accompanied by a decline in trans-epithelial electrical resistance in Sertoli cells. TJ proteins (e.g., ZO-1 and occludin), basal ES protein (e.g., N-Cadherin) and GJ protein (e.g., connexin 43) are important structural components in Sertoli cells, which are essential for the regulation of BTB function [3]. Significantly, the function of aforementioned proteins is usually affected by environmental toxicants which impair male fertility [8, 9]. Our results showed that OLA decreased the expression and localization of the junction proteins ZO-1, Occludin and N-cadherin at cell-cell interface. Mammalian target of rapamycin (mTOR) is a central regulator of cellular metabolic phenotype which integrates not only nutrient and energy-sensing pathways but also actin cytoskeleton organization [33, 34]. Herein, the steady-state protein level of rictor, a component of mTORC2 complex that is crucial for BTB dynamics [33, 37], was significantly decreased by OLA exposure, indicating that mTORC2 are potential mediators of OLA-induced BTB disruption. mTORC1/2 have been implicated in regulation of several testicular functions [36]. Depletion of mTOR in Sertoli cells impairs the organization of the seminiferous epithelium and the loss of cells polarity with a redistribution of BTB gap junction protein-1 (GJA1) [50]. In our study, the phosphorylation of mTORS2481, p70 S6KThr389 and AktSer-473 were reduced in OLA-exposed Sertoli cells, indicating that both mTORC1 and mTORC2 are molecular targets of OLA. Notably, mTORC1 and mTORC2 have been reported to have opposing effects on BTB dynamics as ribosomal protein S6 (rpS6, a pivotal downstream effector of mTORC1) compromises the permeability of BTB through down-regulating the expression of tight junction proteins whereas rictor, by orchestrating cytoskeleton architecture, promotes the BTB integrity [35, 37, 51]. Indeed, depletion of rictor was found to perturb the Sertoli cell TJ-barrier function and the BTB integrity due to the alteration of F-actin organization [37]. Herein, we also found that OLA could disrupt F-actin organization likely due to down-regulation of p-PKCα, which reminiscent of the finding that rictor knockdown leads to PKCα-mediated actin reorganization [52].
An overall reduction of F-actin bundles were found to across the entire cytosol with highly aggregated at the periphery of Sertoli cells. Actin regulator proteins such as small GTPases (e.g., Rac1 and CDC42) were strickly augmented. Moreover, CDC42 facilitates TGF-β3–mediated cascade of events that lead to the disruption of the TJ fibrils, which increases the endocytosis of occludin and ZO-1 at the BTB [53]. CDC42 accumulation promotes ZO-1 redistributed near the cell surface to cytosol [53]. In our study, the abundance of Arp2/3, which is a nucleation promoting factors and activated by ATP to induce branched actin nucleation [54], remains stable. However, the expression of N-WASP the activator of the Arp2/3 complex was drastically enhanced. Thus, we could not exclude the possibility that the Arp2/3 activity might be disrupted as the reduction in ATP content is induced in OLA-exposed Sertoli cells. Notably, palladin a actin-binding protein that promotes F-actin bundling also plays an important role in organizing actin filament arrays [55]. Both knockdown and overexpression of palladin result in the disorganization of actin cytoskeleton, and depletion of palladin leads to the loss of stress fibers and focal adhesions, and the cells become round up [56]. More importantly, palladin depletion also disrupts the permeability barrier integrity in Sertoli cell [31]. Collectively, these finding indicate that the F-actin disturbance induced by OLA exposure emerges as an important cue for the compromised TJ barrier permeability.
In mammalian cells, DNA damage was caused by intracellularly normal cellular metabolism, spontaneous mutations and external environment. Faithful maintenance and propagation of genetic and epigenetic information is essential for cell proliferation and survival [43]. Herein, we found that OLA-induced DNA damage which is presumably caused by ROS aggravation. The repair of DNA damage requires dynamic chromatin alterations comprising both transient decondensation and compaction of chromatin in the vicinity of DNA lesion sites [43, 57]. Double-strand break (DSB) repair initiates dynamic changes in histone modifications that are required to maintain genome stability [47, 58]. At DNA lesion sites, gene silencing is initiated by the recruitment of key proteins that involve in establishing and maintaining transcriptional repression, and conferring histone modifications including acetylation of H4K16 and methylation of H3K9me2/3 and H3K27me3 [59]. In higher eukaryotes, the methylation of H3K9 and H3K27 is linked to DNA replication and repair [47]. In our study, the global methylation of H3K9/H3K27 were significantly increased while the acetylation of H3K9/H3K27 were reduced after OLA-treatment (400 ug/ml), which sufficiently prove that methylation of H3K9 and H3K27 are linked to genotoxic stress in OLA-exposed Sertoli cells, implicating that the transient gene expression silencing is initiated until the completion of DNA repair is achieved.
Autophagy occurence in response to various stress stimulus, including oxidative stress, and DNA damage, which was considered to be a major protective mechanism against stress stimuli and plays an important role in many physiological processes [41, 60]. In this study, autophagic flux was inhibited while induced apoptosis in OLA-exposed Sertoli cells.Generally, autophagy block the induction of apoptosis, especially the stress is not lethal, while apoptosis-associated caspase activation shuts off the autophagic process when the intensity or duration of stress reaches the limit of the cell [61, 62]. Therefore, the aforementioned puzzle might be dispelled by the notion that the persistent DNA damage due to the failure in DNA repair revealed by the reduction in Rad51C expression predominately triggers apoptosis in the present OLA case in Sertoli cells. The accumulation of the autophagy receptor protein p62/SQSTM1 resulting from either inhibition or loss of autophagy can lead to impaired homologous recombination (HR)-mediated DNA damage repair [41, 63, 64]. Moreover, the up-regulation of SQSTM1/p62 also increases the ROS production [65]. In OLA-exposed Sertoli cells, the expression of SQSTM1/p62 is also increased, therefore, a possible mechanism could be inferred that the aggregation of ROS caused by SQSTM1/p62 up-regulation potentiates DNA damage. However, we could not exclude the possibility that a promiscuous interaction may exist among SQSTM1/p62, ROS and DNA damage as amelioration of ROS by NAC pretreatment could antagonize the accumulation of SQSTM1/p62 and DNA damage in OLA-exposed Sertoli cells. Autophagy is actived in Sertoli cells and plays an important role in maintain spermatogenesis [66, 67]. Sertoli cell-specific disruption of autophagy influences the fertility in male mouse caused by the resulting disorganized seminiferous tubules and abnormal spermatozoa with malformed heads due to disorganization of the cytoskeleton structures [67]. Thus, autophagy disruption might be another explanation for the disorganized cytoskeleton architecture induced by OLA exposure in Sertoli cells.
In conclusion, by deciphering that OLA perturbs TJ proteins cell-cell interface distribution and F-actin organization, which thereby compromises the BTB integrity and dynamics, we herein conceive that the Sertoli cells is sensitive to OLA-induced reproductive toxicity. Moreover, OLA could interferes with the epigenetic status of histone H3 presumably due to potentiated DNA damage, and the two main pathways apoptosis and autophagy that determine the cell fate in Sertoli cells. Our study provides the toxicological information of OLA in male fertility side, which might broaden our knowledge of the biological hazards of OLA to animal and our human health.
MATERIALS AND METHODS
Animals and ethnics statement
The present study was approved by the Ethical Committee of Hubei Research Center of Experimental Animals (Approval ID: SCXK (Hubei) 20080005). Wild-type Kunming (KM) mice were obtained from the local Central Animal Laboratory and housed in the experimental animal center of Huazhong Agricultural University under a 12 h light/12 h dark regimen at a temperature of 22°C with water and food ad libitum. All experimental procedures were performed in line with the guidelines of the Committee of Animal Research Institute, Huazhong Agricultural University, China.
Chemicals and regents
Olaquindox (OLA, 2-(N-2’(hydroxymethyl) carboamoyl)-3-methylquinoxaline N1, N4-dioxide), C12H13N3O4, MW 263.25, CAS no. 23696-28-8, 99%) was purchased from Sigma-Aldrich (St. Louis, MO, USA) and prepared in dimethyl sulfoxide (DMSO) as a 400 mg/ml stock and diluted to its desired concentration with DMEM/F12 medium. The same amount of DMSO was used in control. N-acetylcysteine (NAC) was from Sigma-Aldrich (St. Louis, MO, USA), 10 mM of NAC was added 1 h before OLA treatment.
Primary Sertoli cells isolation and treatment with OLA
Primary Sertoli cells were isolated from 3 weeks old male mice testes and cultured in serum-free DMEM/F12 medium supplemented with EGF (2.5 ng/ml), bovine insulin (10 μg/ml), human transferrin (5 μg/ml), bacitracin (5 μg/ml) and gentamicin (20 μg/ml) at 35°C with a humidified atmosphere of 5% CO2 in air as described [68]. Freshly isolated Sertoli cells were seeded on Matrigel-coated (Corning): (I) 6- or 12-well dishes at 0.5×106 cells per cm2 were subsequently used for lysate preparation, (II) coated coverslips at 0.04-0.08×106 cells per cm2 were processed to immunofluorescent analysis. The time of cell plating onto the dishes was defined as day 0. On day 2, cells were prior to hypotonic treatment using 20 mM Tris, pH 7.4, at 22°C for 2.5 minutes to lyse residual germ cells and then washed twice with DMEM/F12 medium. On day 3, OLA dissolved in DMSO was diluted in DMEM/F12 medium supplemented with various growth factors without bacitracin and gentamicin to obtain the desired final concentration. Sertoli cell were exposed to OLA for 24 h before termination. For the control, vehicle (i.e., DMEM/F12 medium containing DMSO at most 0.2% (V/V)) was used.
Cell viability assay and morphology observation
Cell viability was detected by Cell Counting Kit-8 (CCK8) assays (Dojindo molecular technologies, Inc., Japan) according to the manufacturer’s instructions. Briefly, Sertoli cells were seeded in 96-well culture plates at a density of 0.5×105 cells/well and treated with various concentrations of OLA (100-, 200-, 400-, 800 μg/ml) for different length of time (24-48 h). Cell treated with vehicle (0.2 % DMSO) were used as a reference group with cell viability set as 100%. After treatments, the plate was carefully washed once in PBS at the indicated time, and then, 100 μl of medium containing CCK-8 solution was added to each well (the ratio of medium and CCK-8 volume was 9 : 1). After the plates were incubated for 1h at 37°C, the absorption at 450 nm of each well was measured using EnSpire® Multimode Reader (PerkinElmer, Inc., Waltham, USA). Cell used in this study were constantly observed under an inverted microscope (Nikon, Japan). Photographs were taken after Sertoli cells were treated with different concentrations of OLA for 24- and 48 h a described in the text.
ATP assessment
ATP relative concentrations were evaluated by using the CellTiter-Glo® ATP Assay Kit (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. Briefly, Sertoli cells were seeded in 96-well black culture plates at a density of 0.5×105 cells/well and treated with various concentrations of OLA (100-, 200-, 400-, 800 μg/ml) for 24 h. After treatments, add a volume of CellTiter-Glo® Reagent equal to the volume of cell culture medium present in each well (add 100 μl of regent to 100 μl of medium containing cells for a 96-well plate) for 10 min at room temperature to stabilize luminescent signal. The plate was measured using EnSpire® Multimode Reader (PerkinElmer, Inc., Waltham, USA) to record luminescence. The data are shown as the average of five wells for each group.
Caspase-3/7 activity assay
Caspase-3/7 activity was evaluated by using the Caspase-Glo® 3/7 Assay Kit (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. Briefly, Sertoli cells were seeded in 96-well white culture plates at a density of 0.5×105 cells/well and treated with various concentrations of OLA (100-, 200-, 400-, 800 μg/ml) for 24 h. Afterwards, add a volume of Caspase-Glo® 3/7 Assay Reagent equal to the volume of cell culture medium present in each well for 1 h at room temperature to stabilize luminescent signal. The plate was measured using EnSpire® Multimode Reader (PerkinElmer, Inc., Waltham, USA) to record luminescence. The data are shown as the average of five wells for each group.
Functional assessment of the Sertoli cell TJ permeability barrier in vitro by trans-epithelial electrical resistance (TER) measurement
Primary Sertoli cell were plated on Millicell bicameral units (diameter, 12 mm; pore size, 0.45 μm, effective surface area, ~ 0.6 cm2; Millipore) at 1.2×106 cells per cm2 placed in 24-well dishes containing 0.5 ml F12/DMEM to monitored the Sertoli cell tight junction permeability barrier function by quantifying the trans-epithelial electrical resistance (TER) across the cell epithelium. On day 3, Sertoli cell were exposed to OLA (0, 100-, 200-, 400- and 800 μg/ml) for 24 h. Thereafter, cells were washed twice with F12/DMEM to remove chemicals reagents. The TER was assessed every 12 h until day 6 and each date was presented as mean ± S.E.M of n = 3 replicates as described. A total of four direction positions should be recorded for each bicameral culture unit. The true TER value was calculated as TERsample (Ωcm2) = (Resistancesample-Resistanceblank) (Ω) × Effective surface area (cm2) described by Mruk and Cheng [25].
ROS determination
H2O2 was evaluated using ROS-GloTM H2O2 Assay Kit (Promega Corp., Madison, WI, USA) according to the manufacturer’s protocol using a microplate reader. Sertoli cells plated at 1×105 cells cells/well to a 96-well white plate and exposed to various concentrations of OLA (100-, 200-, 400-, 800 μg/ml) for 18 h before termination, 10 mM of NAC was added 1 h before OLA treatment. Less than 80μl of medium is desirable to accommodate addition of test compounds. Add 20 μl of H2O2 Substrate solution to cells and mix. The final well volume will be 100 μl, and the final H2O2 Substrate concentration will be 25 μM, incubate at 37°C in 5% CO2 for the final 6 h of treatment. Add 100 μl of ROS-GloTM Detection Solution to each well and incubate for 20 mins at room temperature, and the luminescence was determined with EnSpire ® Multimode Reader (PerkinElmer, Inc., Waltham, USA).
Immunofluorescence
For immunofluorescent analysis, Sertoli cells cultured at 0.04 × 106 cells per cm2 on Matrigel-coated coverslips were fixed with 4% paraformaldehyde (wt/vol) in PBS for 10 min, permeabilized in 0.1% Triton X-100/PBS (vol/vol) for 4 mins. Cells were then blocked with 5% BSA/PBS (wt/vol) for 1 h, followed by an overnight incubation of primary antibodies. After washing in PBS/0.1%Tween/0.01%Triton X-100, cells were processed to incubation with corresponding secondary antibodies at 4°C for 16 h and 37°C for 2 h, respectively. For F-actin staining, Sertoli cells were incubated with FITC-conjugated phalloidin (Sigma, MO). DNA was visualized with DAPI (10 μg/ml) for 15 mins at room temperature and then coverslips were mounted to slides with DABCO followed by examination with confocal laser scanning microscope (ZEISS LSM 510 META, Carl Zeiss Imaging, Germany) equipped with a Plan-Apochromat 40 ×/1.4 oil DIC objective. Confocal images were processed using Zeiss LSM Image Browser software and Adobe Photoshop (Adobe Systems Inc., San Jose, CA). For the negative control, non-immunized rabbit or goat IgG were used to replace the primary antibodies. Antibodies and dilutions were listed in Table 1.
Table 1: The information of antibodies used in IF and WB analyses
Product |
Catalog |
Application and dilution |
|
|---|---|---|---|
WB |
IF |
||
ZO-1 antibody |
61-7300, Invitrogen |
1:100 |
1:50 |
occludin antibody |
71-1500, Invitrogen |
1:100 |
|
N-cadherin antibody |
Ab76011, Abcam |
1:1000 |
|
14215, CST |
1:100 |
||
Phospho-Connexin 43 antibody |
3511, CST |
1:1000 |
|
FAK |
AF6397, Affinity |
1:500 |
|
Phospho-FAK |
AF3398, Affinity |
1:500 |
|
Phospho-P38 MAPK antibody |
4511, CST |
1:1000 |
|
Phalloidin-FITC |
P5282, Sigma |
1:100 |
|
Rac1antibody |
2465, CST |
1:1000 |
|
CDC42 antibody |
ab187643, Abcam |
1:1000 |
|
N-WASP antibody |
ab126626, Abcam |
1:1000 |
|
Arp3 antibody |
ab181164, Abcam |
1:2000 |
|
Palladin |
DF9731, Affinity |
1:500 |
|
RICTOR |
DF7530, Affinity |
1:500 |
|
Phospho-mTOR antibody |
DF3308, Affinity |
1:500 |
|
Akt antibody |
AF6261, Affinity |
1:500 |
|
Phospho-Akt antibody |
7160, CST |
1:1000 |
|
Phospho-p70 S6 Kinase antibody |
AF3228, Affinity |
1:500 |
|
PKCαβ antibody |
ab179522, Abcam |
1:1000 |
|
Phospho-PKC antibody |
AF8396, Affinity |
1:500 |
|
Cleaved-PARP antibody |
AF7023, Affinity |
1:500 |
|
Cleaved-Caspase 3 antibody |
AF7022, Affinity |
1:500 |
|
Bax antibody |
AF0083, Affinity |
1:500 |
|
Beclin1 antibody |
AF5128, Affinity |
1:500 |
|
P62/SQSTM1 antibody |
AF5384, Affinity |
1:1000 |
|
LC3A/B antibody |
1274, CST |
1:1000 |
|
Phospho-H2A.X antibody |
9718, CST |
1:1000 |
1:100 |
Rad51C antibody |
NB100-177, Novus |
1:1000 |
|
H3K9ac antibody |
Ab10182, Abcam |
1:1000 |
|
H3K27ac antibody |
39134, Active Motif |
1:1000 |
|
H3K9me1 antibody |
DF6936, Affinity |
1:500 |
|
H3K9me2 antibody |
DF6937, Affinity |
1:500 |
|
H3K9me3 antibody |
39285, Active Motif |
1:500 |
|
H3K27me1 antibody |
DF6939, Affinity |
1:500 |
|
H3K27me2 antibody |
DF6940, Affinity |
1:500 |
|
H3K27me3 antibody |
39155, Active Motif |
1:1000 |
|
β-Actin antibody |
BF0198, Affinity |
1:2000 |
|
HRP-GAPDH antibody |
AB2000, Abways |
1:5000 |
|
HRP-goat anti-rabbit antibody |
sc2004, Santa Cruz |
1:3000 |
|
HRP-goat anti-mouse antibody |
sc-2005, Santa Cruz |
1:3000 |
|
Cy3-goat anti-rabbit antibody |
111-166-045, Jackson |
1:100 |
|
Immunoblot assay
Sertoli cell washed with ice-cold PBS, and lysed in extraction buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, pH 7.4) supplemented with protease and phosphatase inhibitor mixtures and PMSF (Sigma, MO), sonicating, and centrifuging to obtain the clear supernatant. Lysates were stored at -80°C until used. For immunoblot analysis, lysates (30 μg of protein) were separated by SDS-polyacrylamide gel, blotted onto PVDF membrane (Immobilon-P; Millipore), and blocked in TBS/0.1% Tween-20/5% BSA (TBS, 25 mM Tris, 150 mM NaCl, pH 7.4) for 1 h, followed by primary antibodies incubation for overnight at 4°C. After washing in TBS/0.1% Tween-20, membranes were subjected to incubation with corresponding HRP-conjugated secondary antibodies. The immunoblot bands were visualized with ECL kit and read using chemiluminescence system (Thermo Scientific, Waltham, MA). Antibodies and dilutions were listed in Table 1.
Statistical analysis
Data from at least 3 replicates was presented as mean ± S.E.M. Comparisons of two datasets were performed using unpaired two-tailed Student’s t-test. All other comparisons of multiple data-sets were performed using one-way ANOVA followed by Tukey’s post hoc test with p < 0.05 was considered to be statistically significant.
Abbreviations
OLA, olaquindox; NAC, N-acetylcysteine; BTB, blood-testis barrier; TJ, tight junction; basal ES, basal ectoplasmic specializations; GJ, gap junction; ROS, reactive oxygen species; TER, trans-epithelial electrical resistance.
Author contributions
D.W. and L.J.H. conceived the experiments; D.W. and C.J.H. performed experiments and interpreted the data; X.F.J., Z.M.D., J.Y.Z., F.C., Y.S.W., and X.L. contributed reagents/ materials/ analysis tools; D.W. wrote the draft, and L.J.H. revised the paper. All the authors have approved the final version for submission.
ACKNOWLEDGMENTS
Sincerely thanks were owed to beneficial suggestions from colleagues whose names were not listed here.
CONFLICTS OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interests.
FUNDING
This study was supported by the Fundamental Research Funds for the Central Universities (Program NO. 2014PY045) and National Key Technology R&D Program of China (2013BAD03B01).
REFERENCES
1. Oliveire PF, Alves MG. Sertoli Cell Metabolism and Spermatogenesis. Springer: New York. 2015.
2. Stanton PG. Regualtion of the blood-testis barrier. Semin Cell Dev Biol. 2016; 59:166-173.
3. Cheng CY, Mruk DD. The blood-testis barrier and its implications for male contraception. Pharmacol Rev. 2012; 64:16-64.
4. Mruk DD, Cheng CY. The mammalian blood-testis barrier: its biology and regulation. Endocr Rev. 2015; 36:564-591.
5. Chung NP, Cheng CY. Is cadmium chloride-induced inter-sertoli tight junction permeability barrier disruption a suitable in vitro model to study the events of junction disassembly during spermatogenesis in the rat testis. Endocrinology. 2001; 142:1878-1888.
6. Li MW, Mruk DD, Lee WM, Cheng CY. Disruption of the blood-testis barrier integrity by bisphenol A in vitro: is this a suitable model for studying blood–testis barrier dynamics? Int J Biochem Cell Biol. 2009; 41:2302-2314.
7. Wan HT, Mruk DD, Wong CK, Cheng CY. Perfluorooctanesulfonate (PFOS) perturbs male rat Sertoli cell blood–testis barrier function by affecting F-actin organization via p-FAK-(Tyr 407): an in vitro study. Endocrinology. 2014; 155:249-262.
8. Lu Y, Luo B, Li J, Dai J. Perfluorooctanoic acid disrupts the blood-testis barrier and activates the TNFα/p38 MAPK signaling pathway in vivo and in vitro. Arch Toxicol. 2016; 90:971-983.
9. Qiu L, Qian Y, Liu Z, Wang C, Qu J, Wang X, Wang S. Perfluorooctane sulfonate (PFOS) disrupts blood-testis barrier by down-regulating junction proteins via p38 MAPK/ATF2/MMP9 signaling pathway. Toxicology. 2016; 373:1-12.
10. Rao RK, Basuroy S, Rao VU, Karnaky KJ Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002; 368:471-481.
11. Suzuki T, Hara H. Role of flavonoids in intestinal tight junction regulation. J Nutr Biochem. 2011; 22:401-408.
12. Liu Q, Xu C, Ji G, Liu H, Mo Y, Tollerud DJ, Gu A, Zhang Q. Sublethal effects of zinc oxide nanoparticles on male reproductive cells. Toxicol In Vitro. 2016; 35:131-138.
13. Cao XN, Shen LJ, Wu SD, Yan C, Zhou Y, Xiong G, Wang YC, Liu Y, Liu B, Tang XL, Guo M, Liu DY, Long CL, et al. Urban fine particulate matter exposure causes male reproductive injury through destroying blood-testis barrier (BTB) integrity. Toxicol Lett. 2017; 266:1-12.
14. WHO (World Health Organization). Toxicological evaluation of certain veterinary drug residues in food. WHO Food Additives Series, No. 33, nos 804-817 on INCHEM. 1994.
15. EU (European Union). Commission Regulation No. 2788/98, Off J Eur Commun. 1998; 347/32.
16. SCAN (Standing Committee for Animal Nutrition). Short report of the meeting of the standing committee for animal nutrition. 1998.
17. Hao L, Chen Q, Xiao X. Molecular mechanism of mutagenesis induced by a shuttle vector pSP189/mammalian cell system. Mutat Res. 2006; 599:21-25.
18. Wang X, Zhang W, Wang YL, Ihsan A, Dai MH, Huang LL, Chen DM, Tao YF, Peng DP, Liu ZL, Yuan ZH. Two generation reproduction and teratogenicity studies of feeding quinocetone fed to Wistar rats. Food Chem Toxicol. 2012; 50:1600-1609.
19. Fang GJ, He QH, Zhou S,Wang DJ, Zhang YL, Yuan ZH. Subchronic oral toxicity study with cyadox in Wistar rats. Food Chem Toxicol. 2006; 44:36-41.
20. Liu ZY, Huang LL, Zhou XN, Chen DM, Tao YF, Zhang HH, Yuan ZH. The metabolism of olaquindox in rats, chickens and pigs. Toxicol Lett. 2011; 200:24-33.
21. Zhao WX, Tang SS, Jin X, Zhang CM, Zhang T, Wang CC, Sun Y, Xiao XL. Olaquindox-induced apoptosis is suppressed through p38 MAPK and ROS-mediated JNK pathways in HepG2 cells. Cell Biol Toxicol. 2013; 29:229-238.
22. Yang Y, Jiang L, She Y, Chen M, Li Q, Yang G, Geng C, Tang L, Zhong L, Jiang L, Liu X. Olaquindox induced DNA damage via the lysosomal and mitochondrial pathway involving ROS production and p53 activation in HEK293 cells. Environ Toxicol Pharmacol. 2015; 40:729-799.
23. Li D, Dai C, Yang X, Li B, Xiao X, Tang S. GADD45a regulates olaquindox-induced DNA damage and S-phase arrest in human hepatoma G2 cells via JNK/p38 pathway. Molecules. 2017; 22.
24. Woodward KN. Assessment of user safety, exposure and risk to veterinary medicinal products in the European Union. Regul Toxicol Pharmacol. 2008; 50:114-128.
25. Mruk DD, Cheng CY. An in vitro system to study Sertoli cell blood-testis barrier dynamics. Methods Mol Biol. 2011; 763:237-252.
26. Li N, Mruk DD, Cheng CY. Actin binding proteins in blood-testis barrier function. Curr Opin Endocrinol Diabetes Obes. 2015; 22:238-247.
27. Li N, Tang EI, Cheng CY. Regulation of blood-testis barrier by actin binding proteins and protein kinases. Reproduction. 2016; 151:R29-R41.
28. Qian X, Mruk DD, Cheng YH, Tang EI, Han D, Lee WM, Wong EW, Cheng CY. Actin binding proteins, spermatid transport and spermiation. Semin Cell Dev Biol. 2014; 30:75-85.
29. Takashima S, Kanatsu-Shinohara M, Tanaka T, Takehashi M, Morimoto H, Shinohara T. Rac mediates mouse spermatogonial stem cell homing to germline niches by regulating transmigration through the blood-testis barrier. Cell Stem Cell. 2011; 9:463-475.
30. Xiao X, Mruk DD, Tang EI, Massarwa R, Mok KW, Li N, Wong CK, Lee WM, Snapper SB, Shilo BZ, Schejter ED, Cheng CY. N-wasp is required for structural integrity of the blood-testis barrier. PLoS Genet. 2014; 10:e1004447.
31. Qian X, Mruk DD, Wong EW, Lie PP, Cheng CY. Palladin is a regulator of actin filament bundles at the ectoplasmic specialization in adult rat testes. Endocrinology. 2013; 154:1907-1920.
32. Weichhart T. Mammalian target of rapamycin: a signaling kinase for every aspect of cellular life. Methods Mol Biol. 2012; 821:1-14.
33. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21-35.
34. Mok KW, Mruk DD, Lee WM, Cheng CY. Rictor/mTORC2 regulates blood-testis barrier dynamics via its effects on gap junction communications and actin filament network. FASEB J. 2013; 27:1137-1152.
35. Mok KW, Chen H, Lee WM, Cheng CY. rpS6 regulates blood-testis barrier dynamics through Arp3-mediated actin microfilament organization in rat sertoli cells. An in vitro study. Endocrinology. 2015; 156:1900-1913.
36. Oliveira PF, Cheng CY, Alves MG. Emerging role of mammalian target of rapamycin in male fertility. Trends Endocrinol Metab. 2017; 28:165-167.
37. Dong H, Chen Z, Wang C, Xiong Z, Zhao W, Jia C, Lin J, Lin Y, Yuan W, Zhao AZ, Bai X. Rictor regulates spermatogenesis by controlling Sertoli cell cytoskeletal organization and cell polarity in the mouse testis. Endocrinology. 2015; 156:4244-4256.
38. Weber JD, Gutmann DH. Deconvoluting TOR biology. Cell Cycle. 2012; 11:236-248.
39. Halliwell B. Biochemistry of oxidative stress. Biochem Soc Trans. 2007; 35:1147-1150.
40. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012; 47:497-510.
41. Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J. Autophagy in DNA response. Int J Mol Sci. 2015; 16:2641-2662.
42. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016; 16:20-33.
43. Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128:693-705.
44. Chen Y, Zhu WG. Biological function and regulation of histone and non-histone lysine methylation in response to DNA damage. Acta Biochim Biophys Sin (Shanghai). 2016; 48:603-616.
45. Roos WP, Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016; 44:10017-10030.
46. Bártová E, Šustáèková G, Stixová L, Kozubek S, Legartová S, Foltánková V. Recruitment of Oct4 protein to UV-damaged chromatin in embryonic stem cells. PLoS One. 2011; 6:e27281.
47. Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double- strand breaks promote methylation of histone H3 on lysine 9 and transient formationof repressive chromatin. Proc Natl Acad Sci U S A. 2014; 111:9169-9174.
48. Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293:1074-1080.
49. Li N, Mruk DD, Lee WM, Wong CK, Cheng CY. Is toxicant-induced Sertoli cell injury in vitro a useful modle to study molecular mechanisms in spermatogenesis? Semin Cell Dev Biol. 2016; 59:141-156.
50. Boyer A, Girard M, Thimmanahalli DS, Levasseur A, Céleste C, Paquet M, Duggavathi R, Boerboom D. mTOR regulates gap junction alpha-1 protein trafficking in Sertoli cells and is required for the maintenance of spermatogenesis in mice. Biol Reprod. 2016; 95:13.
51. Mok KW, Mruk DD, Cheng CY. Regulation of blood-testis barrier (BTB) dynamics during spermatogenesis via “Yin” and “Yang” effects of mammalian target of repamycin complex 1 (mTORC1) and mTORC2. Int Rev Cell Mol Biol. 2013; 301:291-358.
52. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004; 14:1296-1302.
53. Wong EW, Mruk DD, Lee WM, Cheng CY. Regulation of blood–testis barrier dynamics by TGF-β3 is a Cdc42-dependent protein trafficking event. Proc Natl Acad Sci U S A. 2010; 107:11399-11404.
54. Rodnick-Smith M, Liu SL, Balzer CJ, Luan Q, Nolen BJ. Identification of an ATP-controlled allosteric switch that controls actin filament nucleation by Arp2/3 complex. Nat Commun. 2016; 7:12226.
55. Vattepu R, Beck MR. Actin-induced dimerization of palladin promotes actin-bundling. Protein Sci. 2015; 24:70-80.
56. Parast MM, Otey CA. Characterization of palladin, a novel protein localized to stress fibers and cell adhesions. J Cell Biol. 2000; 150:643-656.
57. Feng W, Hale CJ, Over RS, Cokus SJ, Jacobsen SE, Michaels SD. Large-scale heterochromatin remodeling linked to overreplication-associated DNA damage. Proc Natl Acad Sci U S A. 2017; 114:406-411.
58. Agarwal P, Miller KM. The nucleosome: orchestrating DNA damage signaling and repair whitin chromatin. Biochem Cell Biol. 2016; 94:381-395.
59. O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1 dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008; 4:e1000155.
60. Liang C. Negtive regulation of autophagy. Cell Death Differ. 2010; 17:1807-1815.
61. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S, Vandenabeele P. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptocit fators from mitochondria. Cell Death Dis. 2010; 1:e18.
62. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014; 15:81-94.
63. Bae H, Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res. 2011; 9:1232-1241.
64. Hewitt G, Korolchuk VI. Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol. 2017; 27:340-351.
65. Scheffler IE. Mitochondria, 2nd ed. Wiley: Hoboken, NJ, USA. 2008.
66. Yefimova MG, Messaddeq N, Harnois T, Meunier AC, Clarhaut J, Noblanc A, Weickert JL, Cantereau A, Philippe M, Bourmeyster N, Benzakour O. A chimerical phagocytosis model reveals the recruitment by Sertoli cells of autophagy for the degradation of ingested illegitimate substrates. Autophagy. 2013; 9:653-666.
67. Liu C, Wang H, Shang Y, Liu W, Song Z, Zhao H, Wang L, Jia P, Gao F, Xu Z, Yang L, Gao F, Li W. Autophagy is required for ectoplasmic specialization assemebly in sertoli cells. Autophagy. 2016; 12:814-832.
68. Wu D, Huang CJ, Khan FA, Jiao XF, Liu XM, Pandupuspitasari NS, Brohi RD, Huo LJ. SENP3 grants tight junction integrity and cytoskeleton architecture in mouse Sertoli cells. Oncotarget. 2017; 8:58430-58442. https://doi.org/10.18632/oncotarget.16915.