INTRODUCTION
Glioma is the most common primary brain tumor, and is associated with a poor prognosis in part due to of its invasive nature. Early diagnosis and prompt treatment of glioma can be used to reduce mortality and improve prognosis. However, at present, the diagnosis of glioma mainly depends on imaging examination, which may misrepresent true intracranial tumor burden and phenotype. Although biopsy is used to confirm diagnosis of glioma, it is an invasive approach. Tumor biomarkers such as Alpha Fetoprotein, Carcino Embryonie Antigen, Cancer Antigen 125 have been reported to be useful to screen early malignancy and aid in cancer diagnosis without invasiveness. Furthermore, following a diagnosis of cancer, biomarkers may be used in order to determine prognosis and predict therapeutic response [1, 2]. Unfortunately, to date, there is no such biomarker for glioma patients. Therefore, it is desirable to identify biomarkers for use in the early diagnosis of glioma.
Cell-free DNA can be detected in body fluids such as serum/plasma, urine and sputum, and are released into the blood following apoptosis and necrosis of cancer cells in the tumor microenvironment. Therefore, there are greater amounts cell-free DNA in cancer patients compared to healthy controls. As is known, aberrant DNA methylation including global hypomethylation and regional hypermethylation, play an important role in the formation and progression of cancer. Over the past decades, many researchers have demonstrated the presence of aberrant methylation in the serum cell-free DNA of patients with various types of tumors [3–7]. However, few studies have investigated serum cell-free DNA in glioma. Furthermore, the published data on tumor suppressor genes MGMT, RASSF1A and P16 are inconsistent due to the different genes or different methods [8–15]. Moreover, almost all previous studies regarding aberrant methylation in glioma are focused on promoter hypermethylation, which is generally associated with gene silencing[16–20], with is limited information on global hypomethylation in serum cell-free DNA of glioma patients to date. Global hypomethylation mainly occurs in repetitive elements and leads to genomic instability and tumorigenesis [21, 22]. As such, global hypomethylation is considered a biomarker in many tumors [23]. Alu elements are the most abundant repetitive elements in the human genome, with the estimation of total methylation content of Alu elements reported to be useful in evaluating the global genomic methylation levels. Cadieux et al [24] reported that the degree of genomic hypomethylation was related with disease stage in glioma and thus may serve as a potential prognostic factor.
In the present study, we use the bisulfite sequencing (BSP), to detect the methylation status of Alu, MGMT, RASSF1A, and P16 in serum cell-free DNA of glioma patients compared to healthy controls. Identification of novel serum-based biomarkers may contribute significantly to the early diagnosis and thus improved prognosis of glioma patients.
RESULTS
Methylation levels of Alu, MGMT, RASSF1A, P16 in serum
The number of methylated CpG sites were defined by dividing the number of methylated CpG sites plus unmethylated CpG sites and expressed as the final methylation level (%) of each sample in the four genes. The median methylation level of Alu was 46.15% (IQR, 36.57%–54.00%) and 60.85% (IQR, 57.23%–65.68%) in glioma patients and healthy controls, respectively. Indeed, there was a significant difference (P<0.01, Figure 1A). The median methylation level of MGMT was significantly higher in glioma patients 64.65% (IQR, 54.87%–74.37%) compared to 38.30% (IQR, 34.13%–45.45%) in healthy controls (P<0.01, Figure 1B). However, the median methylation level of RASSF1A in patients and healthy controls did not reveal any significant differences (P=0.349), and were 15.50% (IQR, 5.85%–24.70%), 11.40% (IQR, 6.78%–20.60%), respectively (Figure 1C). In contrast to RASSF1A, the difference between patients and controls in P16 methylation levels was statistically significant (P=0.03), 31.35% (IQR, 22.58%–36.93%) and 26.65% (IQR, 17.15%–36.05%, Figure 1D).
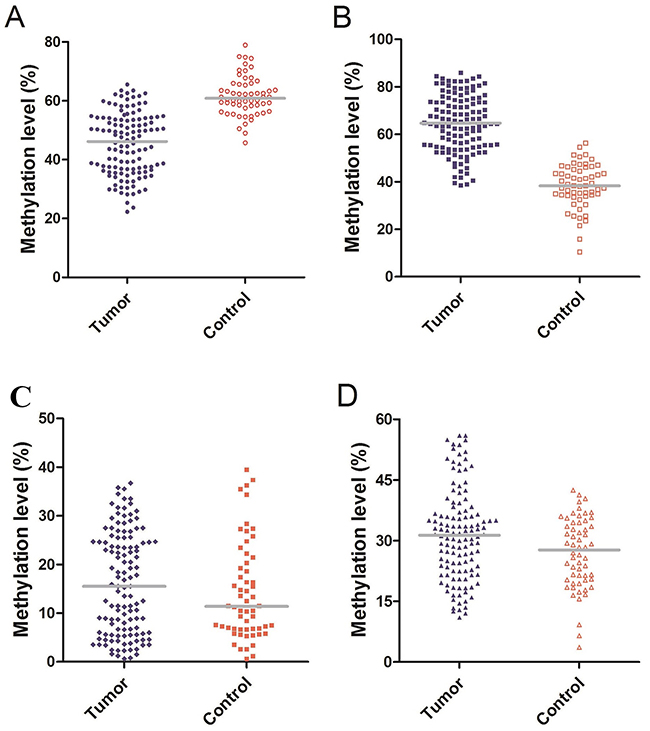
Figure 1: (A) Methylation level of Alu: The median methylation level of Alu was 46.15% (IQR, 36.57%–54.00%) and 60.85% (IQR, 57.23%–65.68%) in glioma patients and healthy controls. (B) Methylation level of MGMT: The median methylation level of MGMT was 64.65% (IQR, 54.87%–74.37%) and 38.30% (IQR, 34.13%–45.45%) in glioma patients and healthy controls. (C) Methylation level of RASSF1A: The median methylation level of RASSF1A was 15.50% (IQR, 5.85%–24.70%) and 11.40% (IQR, 6.78%–20.60%) in glioma patients and healthy controls. (D) Methylation level of P16: The median methylation level of P16 was 31.35% (IQR, 22.58%–36.93%) and 26.65% (IQR, 17.15%–36.05%) in glioma patients and healthy controls.
Diagnostic performance of serum cell-free DNA
ROC curves were constructed to assess the feasibility of serum cell-free DNA for the diagnosis of glioma patients. We analyzed the methylation levels of Alu, MGMT, P16 in serum between patients and healthy controls. The Area Under Curve was 0.904 (95% CI: 0.861–0.946, P<0.01, Figure 2A) in Alu, 0.962 (95% CI: 0.938–0.985, P<0.01, Figure 2B) in MGMT and 0.599 (95% CI: 0.514–0.683, P<0.01, Figure 2C) in P16. These results indicated that the methylation level of Alu and MGMT in serum may have good diagnostic value that is greater than P16. Further analysis revealed a 54.84% sensitivity and 98.28% specificity at the optimal cutoff value of 48.85% (Youden’s index) in Alu, compared to 76.61% sensitivity and 98.28% specificity at the optimal cutoff value of 48.85% (Youden’s index) in MGMT. Consequently, combination of Alu and MGMT may identify additional patients that may be missed by either diagnosis alone (Figure 3).
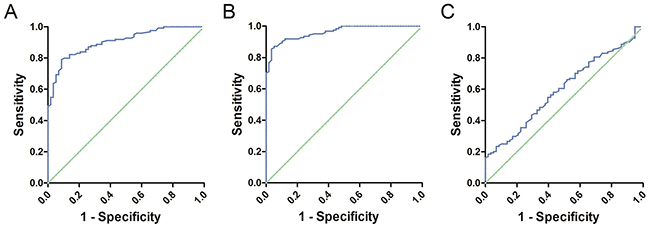
Figure 2: (A) ROC curve of Alu: The Area Under Curve was 0.904. (B) ROC curve of MGMT: The Area Under Curve was 0.962. (C) ROC curve of P16: The Area Under Curve was 0.599.
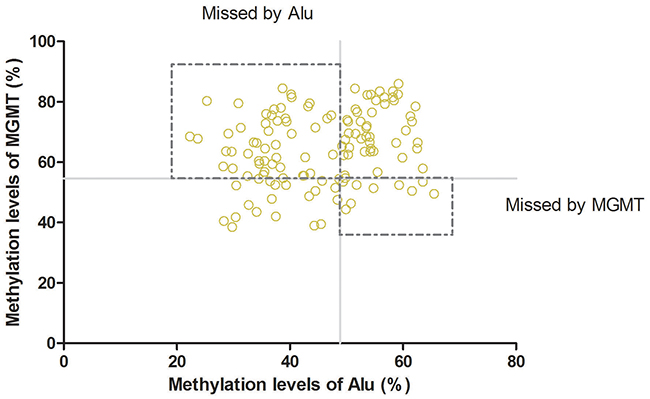
Figure 3: Combination of Alu and MGMT: Combination of Alu and MGMTcould improve the rate of diagnosis.
Methylation level and survival
In order to explore the relationship between serum cell-free DNA aberrant methylation and survival of glioma patients, we compared the methylation level of Alu, MGMT in different glioma grade groups and constructed the survival curves. The median methylation level of Alu in high-grade glioma was 37.90% (IQR, 34.15%–48.20%), while the low-grade was 52.20% (IQR, 43.53%–55.43%, P<0.01, Figure 4A). However, the median methylation level of MGMT in high-grade glioma was 71.75% (IQR, 63.75%–75.10%) compared to 56.55% of low-grade tumors (IQR, 52.33%–65.48%, P<0.01, Figure 4B).
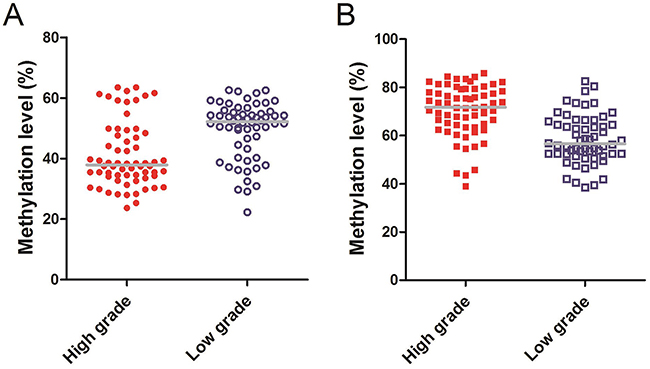
Figure 4: (A) Methylaiton level of Alu: The median methylation level of Alu in high-grade glioma was 37.90% (IQR, 34.15%–48.20%), while the low-grade was 52.20% (IQR, 43.53%–55.43%). (B) Methylaiton level of MGMT: The median methylation level of MGMT in high-grade glioma was 71.75% (IQR, 63.75%–75.10%), while the low-grade was 56.55% (IQR, 52.33%–65.48%).
In the present study, we defined > 46.15% as high methylation level and ≤ 46.15% as low methylation level for Alu, and > 64.65% as high methylation level and ≤ 64.65% as low methylation level for MGMT. In Alu group, the patients with high levels were associated with higher survival rates compared to those with low levels, with median OS time was 31 months in the high level group and 23 months in low level group (P<0.01, Figure 5A). Furthermore, similar results were obtained in the MGMT group, with median OS time 32 months in high level group compared to 24 months in the low level group (P<0.01, Figure 5B).
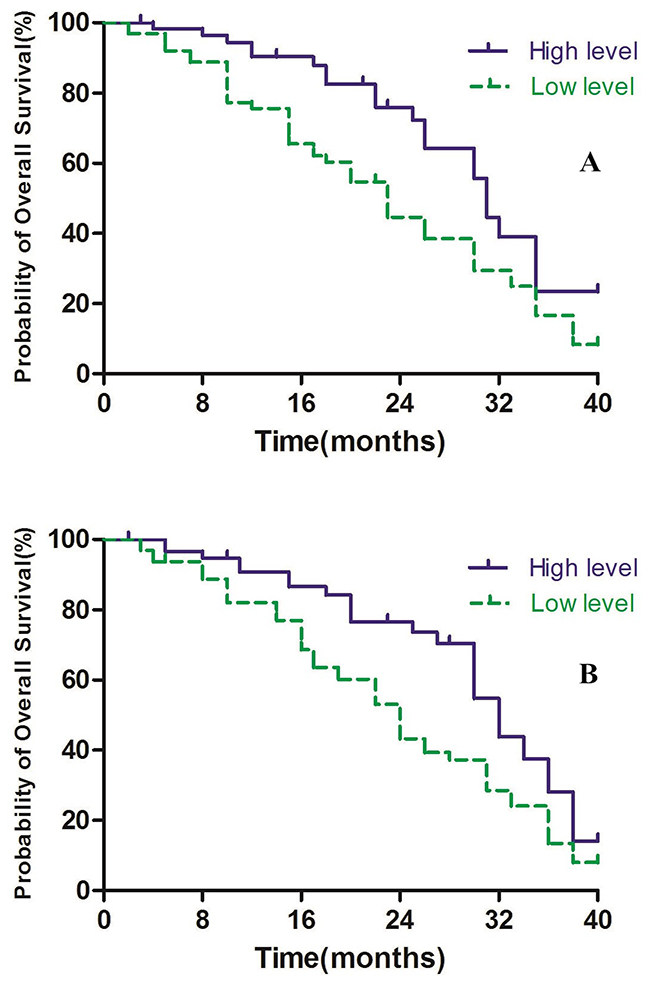
Figure 5: (A) Alu methylaiton level and overall survival. (B) MGMT methylaiton level and overall survival.
DISCUSSION
Early diagnosis and treatment plays a critical role in improving prognosis for glioma patients. However, currently there are no tumor-specific biomarkers for glioma to assist in early diagnosis. Previous studies[25–30] have reported that aberrant methylation of serum cell-free DNA offers the possibility to identify biomarkers for glioma early diagnosis and improve prognosis. Additionally, the detection of serum cell-free DNA as a noninvasive diagnostic method may provide an alternate diagnostic method to tumor tissue biopsy. Moreover, serum cell-free DNA is very stable and can survive harsh conditions for long time. In addition, serum cell-free DNA can be amplified by PCR or other technologies even if only small amounts are present. Moreover, aberrant methylation including global hypomethylation and regional hypermethylation occur in the early stages of tumorigenesis, and thus exist both in early and advanced cancers [31]. Furthermore, this study is the first to detect global hypomethylation and regional hypermethylation in serum at the same time.
In the present study, we used BSP which is the gold-standard technology to detect the methylation status of serum cell-free DNA, and it could sequence more CpG sites to get more accurate results than other technologies. We detected four genes (Alu, MGMT, RASSF1A, P16) in the serum of 124 glioma patients and 58 healthy controls. Results revealed the methylation levels of Alu, MGMT and P16 had significant differences between patients and controls. Cadieux et al[24] reported that Alu hypomethylation in glioblastoma multiforme, consistent with the results of our study, which demonstrated that Alu was hypomethylation in glioma patients and heavily methylated in healthy people. Similarly, we also found MGMT hypermethylation in patients while hypomethylation in controls consistent with previous reports [32–34]. P16 has been reported as hypermethylation in glioma [11], with the present study demonstrating a similar trend that was not significantly different. However, this study obtained different results to the literature in RASSF1A [19, 35, 36], as we did not identify RASSF1A hypermethylation in patients and hypomethylation in healthy controls. For further analysis of diagnostic value of serum cell-free DNA for glioma, we constructed the ROC curves, the AUC of Alu, MGMT, P16 was 0.904, 0.962, 0.599, respectively and identified that the methylation levels of Alu and MGMT in the serum had better diagnostic value than P16. Furthermore, combined analysis of Alu and MGMT revealed higher sensitivity for glioma diagnosis.
To evaluate the prognostic significance of Alu and MGMT, we analyzed the methylation level of Alu and MGMT in high-grade and low-grade groups. In Alu, the low-grade glioma patients had higher levels than high-grade group. The high-grade group had higher level than low-grade group in MGMT. Moreover, we performed the overall survival curves according to the follow-up data of patients. In the Alu group, results showed that patients in the high level group had higher survival rates compared to the low level group. MGMT analysis, also showed that the high level group had longer survival time compared to the low level group, perhaps due to epigenetically silenced MGMT treated with radiotherapy and alkylating chemotherapy may improve the survival of patients with glioblastoma without MGMT expression [37, 38].
In the present study, we detected increased global hypomethylation (Alu) and regional hypermethylation (MGMT, RASSF1A, P16) in serum cell-free DNA of glioma patients compared to healthy controls, and demonstrate that both Alu and MGMT may have good diagnostic value. However, our results suggest that there was much better diagnostic efficiency if a combination of Alu and MGMT was used. Therefore, both serum Alu and MGMT methylation level could be useful tools to predict the prognosis of glioma patients. But could patients that have hypermethylated MGMT with hypomethylated Alu have worst overall survival than patents with only hypermethylated MGMT, hypomethylated Alu only, and hypomethylated MGMT with hypermethylated Alu? We plan to increase the number of patients in the future research for getting more real and effective results.
MATERIALS AND METHODS
Patients
The study involved 124 glioma patients, treated at Department of Neurosurgery at the Changshu No.2 People’s Hospital/ The 5th Affiliated Hospital of Yangzhou University (Changshu, People’s Republic of China) and the Affiliated Hospital of Nantong University (Nantong, People’s Republic of China) between 2003 and 2012. The sample population contained 60 low-grade gliomas (32 grade I, 28 grade II), and 64 high-grade gliomas (34 grade III, 30 grade IV). All patients included in the study underwent surgical tumor resection, and diagnosis of glioma was confirmed by histological examination. Patients then underwent the accepted standard radiotherapy and chemotherapy if necessary. The following clinical data determined for patients are presented in Table 1 : age at the time of operation, sex, Karnofsky performance score, tumor diameter, tumor location (region, side) and the World Health Organization grade. The study protocol was approved by the Independent Ethics Committee of Affiliated Hospital of Nantong University, with all patients and healthy controls providing informed consent prior to obtaining the serum samples.
Table 1: Patients’ chracteristics
Characteristic |
No. of patients (%) |
|---|---|
Age at diagnosis, years |
|
≤50 |
46 (37.10) |
<50 |
78 (62.90) |
Gender |
|
Male |
55 (44.35) |
Female |
69 (55.65) |
Karnofsky performance score(KPS) |
|
<70 |
26 (20.97) |
≥70 |
98 (79.03) |
Tumor diameter |
|
≤3cm |
39 (31.45) |
<3cm |
85 (68.55) |
Tumor location (region) |
|
Frontal |
45 (36.29) |
Temporal |
37 (29.84) |
Parietal |
29 (23.39) |
Occipital |
10 (8.06) |
Other |
3 (2.42) |
Tumor location (side) |
|
Left |
38 (30.65) |
Right |
46 (37.10) |
Middle |
40 (32.26) |
WHO grade |
|
Low grade |
60 (48.39) |
High grade |
64 (51.61) |
Cell-free DNA extraction and sodium bisulfite modification
Peripheral venous blood (5–10 ml) was collected prior to surgery from 124 glioma patients and 58 blood samples from healthy people were also obtained as controls. Serum was isolated by centrifugation at 3500 rpm for 10 min at room temperature and stored at −80°C until use. Serum cell-free DNA was obtained by QIAamp MinElute Virus Spin Kit (Qiagen, Germany), according to the manufacturer’s instructions and stored at −20°C until use. Sodium bisulfite modification of cell-free DNA was performed with Epitect Bisulfite Kit (Qiagen) according to the manufacturer’s instructions. Briefly, 1 μg prepared DNA were bisulfite treated to convert unmethylated cytosine to uracil and the elutions were stored at −20°C until use.
PCR of Alu, MGMT, RASSF1A and P16
The primers of Alu, MGMT, RASSF1A and P16 for BSP were designed using Methyl Primer Express® Software v1.0. Amplification of the four genes used a 50 μl PCR mixture, which contained 5 μl Buffer, 4 μl dNTP, 0.5 μl HotStar Taq DNA polymerase (Invitrogen, USA), 0.8 μl (10 μM) forward and reverse primers, 10 μl bisulfite converted DNA and 28.9 μl ddH2O. The PCR of Alu was performed as follows: initial denaturing at 94°C for 3 min; 37 cycles of denaturing at 94°C for 20 s, annealing at 55°C for 30 s, extension for 30 s at 72°C, and a final extension at 72°C for 5 min. The PCR cycling conditions of RASSF1A were 94°C for 2 min, 37 cycles at 94°C for 20 s, 57°C for 30 s, and 72°C for 30 s, followed by 72°C for 10 min. The PCR products were analyzed on a 1.5% agarose gel stained with ethidium bromide and subjected to UV illumination. (MGMT, P16)
Cloning and sequencing
The separated PCR products by the gel were purified using QIAquick Gel Extraction kit (Qiagen) and then cloned using the TOPO TA-cloning kit (Invitrogen) according to the manufacturer’s instructions. Colony PCR was undertaken to screen the positive colonies, and the clones of the right size of PCR product were sequenced using the ABI 3730 DNA Analyzer. Additionally, 10 clones of each sample were subjected to standard sequencing analysis.
Statistical analysis
Analysis of all data was performed with GraphPad Prism software (version 5.01). Comparison between two groups was tested by using the t test or Mann–Whitney test. Receiver Operator Characteristic curve curve was constructed to assess the diagnostic value of serum cell-free DNA for glioma. Overall survival (OS) was calculated from the day of first surgery until death or end of follow-up, and analysis performed by Kaplan–Meier and the log rank test. The level of statistical significance was set at P<0.05.
Abbreviations
MGMT: O6-methylguanine-DNA-methyltransferase, RASSFIA: ras association domain family gene 1A, IQR: interquartile range, BSP: bisulfite sequencing, ROC: receiver operator characteristic, AUC: area under curve, OS: overall survival, KPS: Karnofsky performance score.
Author contributions
Mingjie Gong and Rongtao Chu conceived and designed the study. Mingjie Gong, Wei Shi, Jing Qi and Guoping Shao performed the experiments. Mingjie Gong, Zhenghua Shi and Junxiang Wang wrote the paper. Jian Chen and Rongtao Chu reviewed and edited the manuscript. All authors read and approved the manuscript.
ACKNOWLEDGMENTS
We would like to thank Dr. Jian Li, Dr. Weining Hui for the collection of clinical data and for providing technical advice regarding bisulfite sequencing.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by the National Natural Science Foundation of China (No.81201975), Medicine and Technology Project For Youth Changshu CSWSQ201501, Medicine and Technology Project of Changshu CSWS201621, All the authors report no conflict of interest disclosures relevant to the manuscript.
REFERENCEs
1. Duffy MJ. Role of tumor markers in patients with solid cancers: a critical review. Eur J Intern Med. 2007; 18:175-184.
2. Bosnyak E, Michelhaugh SK, Klinger NV, Kamson DO, Barger GR, Mittal S, Juhasz C. Prognostic molecular and imaging biomarkers in primary glioblastoma. Clin Nucl Med. 2017.
3. Kanekiyo S, Iizuka N, Tsunedomi R, Tokumitsu Y, Hashimoto N, Tokuhisa Y, Maeda Y, Iida M, Sakamoto K, Tamesa T, Fujita Y, Yoshino S, Hazama S, et al. Preoperative serum methylation signature as prognostic tool after curative hepatectomy in patients with hepatocellular carcinoma. Anticancer Res. 2015; 35:997-1007.
4. Lazarev I, Leibovitch L, Czeiger D, Sion-Vardi N, Geffen DB, Douvdevani A, Ariad S. Cell-free DNA blood levels in colorectal cancer patients do not correlate with mismatch repair-proficiency. In Vivo. 2014; 28:349-354.
5. Nie K, Jia Y, Zhang X. Cell-free circulating tumor DNA in plasma/serum of non-small cell lung cancer. Tumour Biol. 2015; 36:7-19.
6. Kato K, Iida S, Uetake H, Takagi Y, Yamashita T, Inokuchi M, Yamada H, Kojima K, Sugihara K. Methylated TMS1 and DAPK genes predict prognosis and response to chemotherapy in gastric cancer. Int J Cancer. 2008; 122:603-608.
7. Wallner M, Herbst A, Behrens A, Crispin A, Stieber P, Goke B, Lamerz R, Kolligs FT. Methylation of serum DNA is an independent prognostic marker in colorectal cancer. Clin Cancer Res. 2006; 12:7347-7352.
8. Balana C, Ramirez JL, Taron M, Roussos Y, Ariza A, Ballester R, Sarries C, Mendez P, Sanchez JJ, Rosell R. O6-methyl-guanine-DNA methyltransferase methylation in serum and tumor DNA predicts response to 1,3-bis(2-chloroethyl)-1-nitrosourea but not to temozolamide plus cisplatin in glioblastoma multiforme. Clin Cancer Res. 2003; 9:1461-1468.
9. Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neurooncol. 2010; 12:173-180.
10. Vendrell JA, Mau-Them FT, Beganton B, Godreuil S, Coopman P, Solassol J. Circulating cell free tumor DNA detection as a routine tool for lung cancer patient management. Int J Mol Sci. 2017; 18.
11. Wakabayashi T, Natsume A, Hatano H, Fujii M, Shimato S, Ito M, Ohno M, Ito S, Ogura M, Yoshida J. p16 promoter methylation in the serum as a basis for the molecular diagnosis of gliomas. Neurosurgery. 2009; 64:455-461; discussion 461-452.
12. Watanabe T, Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H. Promoter hypermethylation and homozygous deletion of the p14ARF and p16INK4a genes in oligodendrogliomas. Acta Neuropathol. 2001; 101:185-189.
13. Havik AB, Brandal P, Honne H, Dahlback HS, Scheie D, Hektoen M, Meling TR, Helseth E, Heim S, Lothe RA, Lind GE. MGMT promoter methylation in gliomas-assessment by pyrosequencing and quantitative methylation-specific PCR. J Transl Med. 2012; 10:36.
14. Balana C, Carrato C, Ramirez JL, Cardona AF, Berdiel M, Sanchez JJ, Taron M, Hostalot C, Musulen E, Ariza A, Rosell R. Tumour and serum MGMT promoter methylation and protein expression in glioblastoma patients. Clin Transl Oncol. 2011; 13:677-685.
15. Wang Z, Jiang W, Wang Y, Guo Y, Cong Z, Du F, Song B. MGMT promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. Biomed Rep. 2015; 3:543-548.
16. Biswas S, Rao CM. Epigenetics in cancer: fundamentals and Beyond. Pharmacol Ther. 2017.
17. Keil KP, Abler LL, Laporta J, Altmann HM, Yang B, Jarrard DF, Hernandez LL, Vezina CM. Androgen receptor DNA methylation regulates the timing and androgen sensitivity of mouse prostate ductal development. Dev Biol. 2014; 396:237-245.
18. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987-996.
19. Horiguchi K, Tomizawa Y, Tosaka M, Ishiuchi S, Kurihara H, Mori M, Saito N. Epigenetic inactivation of RASSF1A candidate tumor suppressor gene at 3p21.3 in brain tumors. Oncogene. 2003; 22:7862-7865.
20. Wittenberger T, Sleigh S, Reisel D, Zikan M, Wahl B, Alunni-Fabbroni M, Jones A, Evans I, Koch J, Paprotka T, Lempiainen H, Rujan T, Rack B, et al. DNA methylation markers for early detection of women’s cancer: promise and challenges. Epigenomics. 2014; 6:311-327.
21. Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998; 395:89-93.
22. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003; 300:455.
23. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004; 4:143-153.
24. Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006; 66:8469-8476.
25. Wu YC, Lv P, Han J, Yu JL, Zhu X, Hong LL, Zhu WY, Yu QM, Wang XB, Li P, Ling ZQ. Enhanced serum methylated p16 DNAs is associated with the progression of gastric cancer. Int J Clin Exp Pathol. 2014; 7:1553-1562.
26. Fiegl H, Millinger S, Mueller-Holzner E, Marth C, Ensinger C, Berger A, Klocker H, Goebel G, Widschwendter M. Circulating tumor-specific DNA: a marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer Res. 2005; 65:1141-1145.
27. Warton K, Lin V, Navin T, Armstrong NJ, Kaplan W, Ying K, Gloss B, Mangs H, Nair SS, Hacker NF, Sutherland RL, Clark SJ, Samimi G. Methylation-capture and next-generation sequencing of free circulating DNA from human plasma. BMC Genomics. 2014; 15:476.
28. Mori T, O’Day SJ, Umetani N, Martinez SR, Kitago M, Koyanagi K, Kuo C, Takeshima TL, Milford R, Wang HJ, Vu VD, Nguyen SL, Hoon DS. Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J Clin Oncol. 2005; 23:9351-9358.
29. An Q, Liu Y, Gao Y, Huang J, Fong X, Li L, Zhang D, Cheng S. Detection of p16 hypermethylation in circulating plasma DNA of non-small cell lung cancer patients. Cancer Lett. 2002; 188:109-114.
30. Dumache R, Puiu M, Motoc M, Vernic C, Dumitrascu V. Prostate cancer molecular detection in plasma samples by glutathione S-transferase P1 (GSTP1) methylation analysis. Clin Lab. 2014; 60:847-852.
31. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108-112.
32. Zhang Z, Chen Y, Tang J, Xie X. Frequent loss expression of dab2 and promotor hypermethylation in human cancers: a meta-analysis and systematic review. Pak J Med Sci. 2014; 30:432-437.
33. Vlassenbroeck I, Califice S, Diserens AC, Migliavacca E, Straub J, Di Stefano I, Moreau F, Hamou MF, Renard I, Delorenzi M, Flamion B, DiGuiseppi J, Bierau K, Hegi ME. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn. 2008; 10:332-337.
34. Kramar F, Minarik M, Benesova L, Halkova T, Netuka D, Bradac O, Benes V. IDH1/2 mutation and MGMT promoter methylation - the relevant survival predictors in Czech patients with brain gliomas. Folia Biol. 2016; 62:194-202.
35. Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005; 65:3497-3508.
36. Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res. 2001; 61:7034-7038.
37. Jaeckle KA, Eyre HJ, Townsend JJ, Schulman S, Knudson HM, Belanich M, Yarosh DB, Bearman SI, Giroux DJ, Schold SC. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol. 1998; 16:3310-3315.
38. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000; 343:1350-1354.