
INTRODUCTION
Endometrial Cancer (EC) is the fourth most common cancer affecting women. Over the last decade, the incidence of EC has been increasing globally. If current trends continue, in the United States the incidence of EC will double by 2030 [1]. Furthermore, the number of women dying from EC has also been increasing disproportionally to the rise in incidence, with rates exceeding those seen for most other solid tumors [2]. There are few therapeutic options for women diagnosed with recurrent or metastatic EC, and median overall survival (OS) is short. No new agents have been approved for the treatment of EC in the past two decades [3]. New therapeutic approaches are required to meet this significant unmet need. In many other cancers, a detailed understanding of underlying tumor biology has yielded remarkable advances in therapeutic interventions, most often when agents are administered in target-selected populations. Not only does this potentially enrich for benefit for patients and clarity in interpretation, but it would also allow differentiation between presence of mutation and functionally actionable events. Applying this approach to EC is critical if we are to improve outcomes for women diagnosed with this disease.
The US National Cancer Institute (NCI) Gynecologic Cancer Steering Committee (GCSC) identified the integration of molecular and/or histologic stratification into EC management as a top strategic priority in clinical trial planning. Based on this input the NCI convened a Uterine Cancer Clinical Trials Planning Meeting (UCTPM) in January 2016. The focus of the UCTPM was to review and apply emerging molecular knowledge of EC to yield clinical trial concepts for testing actionable events in molecularly defined recurrent EC patient populations. Prior to the January meeting, a group of experts were assembled to consider the published literature focusing particularly on reports related to human specimens, with the goal of identifying evidence to support therapeutic approaches for near-term clinical trial application. Reports were generated on a number of key areas including DNA repair, hormone-related pathways, ERBB2/HER2, PI3K/AKT/mTOR signaling, the ubiquitin-ligase complex, the WNT pathway, the immune system, and obesity-driven targets. We summarize the key findings from these individual reports below and indicate reasonable candidate approaches for near-term clinical trial planning.
MOLECULAR CLASSIFICATION OF ENDOMETRIAL CANCERS
Prognostic models for risk of metastatic disease or recurrence have been found to be of limited value in EC [4]. EC grade and histotype are major components of the models and are historically associated with poor reproducibility, with inter-observer disagreement in one third of high grade ECs [5, 6]. Thus, unlike other gynecologic cancers, it has become apparent that histologic subtype alone may not be the most effective approach to stratify and guide the treatment of EC. The traditional view of EC classification lacks the accuracy needed to sufficiently discriminate biologic variants to guide treatment.
The Cancer Genome Atlas (TCGA) project in 2013 examined nearly 500 samples of newly diagnosed endometrioid and serous ECs [7]. Detailed analysis allowed organization into four molecular subgroups based upon shared genomic features that correlated with clinical outcome. The four subgroups were: POLE-ultramutated (POLE), microsatellite instability-hypermutated (MSI), copy number low-microsatellite stable (CNL), and copy number high-serous-like (CNH). This classification is driving a paradigm shift in how EC is viewed, opening new possibilities for risk stratification, and identifying potential, actionable, subgroup-specific therapeutic targets.
The POLE subgroup is characterized by a very high mutation burden driven by POLE exonuclease-domain mutations. This subgroup has a very favorable clinical outcome despite including tumors of varied grade and histology. The MSI subgroup is characterized by MSI and has a high mutation burden with frequent MLH1 promoter methylation. The clinical significance of MSI in EC is uncertain, but it is frequently associated with an active immune cell infiltrate [8]. The CNL subgroup has low histological grade and frequent CTNNB1 mutations without MSI or TP53 mutations. PTEN mutations are very common in the MSI and CNL subgroups and infrequent in the CNH subgroup [7]. The CNH subgroup is defined by the presence of somatic TP53 mutations, and included nearly all uterine serous carcinomas and ~25% of high-grade endometrioid tumors. A number of groups are developing clinically applicable classifiers to identify these molecular subgroups, which are undergoing prospective validation [9, 10] (Figure 1).
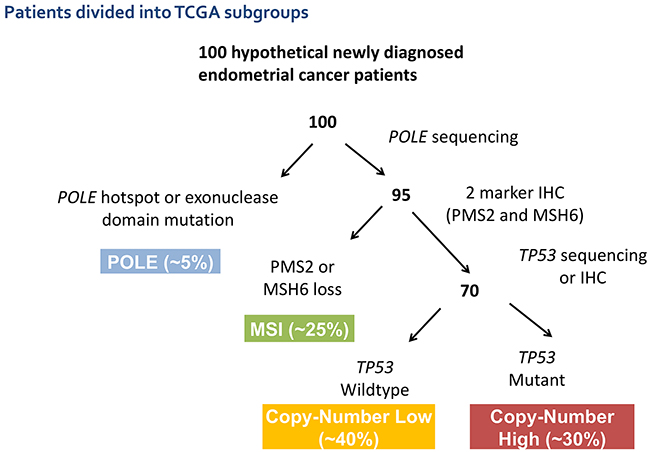
Figure 1: Suggested potential schema for molecular classification of endometrial cancer using sequencing and IHC results to segregate patients into the molecular subtypes previously defined by the TCGA.
Achieving the goal to minimize therapy where it is not needed, and to tailor treatment to the cancer and patient is most likely to be achieved by incorporating molecular subgroup stratification into current classification schemas. This will allow us to prospectively test the value of such molecular subgroup stratification on treatment selection and outcome. Consideration should be given to the impact of the molecular subgroups on outcome when analyzing and interpreting existing and upcoming data from clinical trials including mixed populations of EC patients.
Serous ECs are characterized by genomic instability, high rates of somatic mutations in the TP53, PPP2R1A, FBXW7, PIK3CA, PIK3R1, PTEN, SPOP, CHD4, and TAF1 genes [7, 11–16] frequent amplification and/or overexpression of the ERBB2/HER2 receptor tyrosine kinase [17, 18], and dysregulated expression of cyclin E, c-MYC, p16, E-cadherin, claudin-3, claudin-4, L1CAM and EpCAM [19]. Mutations in chromatin remodeling genes have also been reported [14, 16]. The TCGA classified 98% of serous ECs, 5% of low-grade endometrioid ECs (EECs), 19% of high grade EECs, and 75% of mixed histology ECs into a single molecular group referred to as “serous-like EC” because of their overall molecular resemblance to uterine serous carcinoma [7].
POTENTIAL TARGETS AND THERAPEUTIC OPPORTUNITIES
DNA repair
Classic cytotoxic agents cause DNA damage, and many newer agents lead to cell death through the inhibition of DNA repair. A major mechanism for augmentation of injury is to exploit DNA repair and cell cycle defects. Agents that prevent DNA repair or inhibit the cell cycle checkpoint cause rapid throughput in G1/S and G2/M. Such cell cycle progression results in cellular accumulation of DNA damage and subsequent apoptosis or mitotic catastrophic cell death.
The TCGA analysis identified genomic events that suggest EC, known to be susceptible to DNA damaging agents, may be affected by targeting DNA repair [7, 20]. These include: the high mutational profiles, TP53 mutation, PTEN loss, and ARID1A mutations. PTEN loss of function in EC, frequent in all TCGA subgroups except CNH, may confer a homologous recombination (HR) deficiency phenotype, similar to that seen in deleterious germline BRCA1 and BRCA2 mutations [21]. In vitro sensitivity to polyADP-ribose polymerase inhibitors (PARPi) has been demonstrated in PTEN-null cell lines [21]. This remains controversial, with others finding no association with PTEN loss and response to PARPi [22]. Cell line data from colorectal and endometrial cancers suggest MSI tumors may harbor mutations in other genes involved in HR repair of double strand DNA breaks, e.g., MRE11A and RAD50 [23–25]. ARID1A mutations are present in ~40% of MSI and CNL endometrioid tumors. ARID1A is recruited to DNA breakage sites through interaction with ATR and is required for normal G2/M checkpoint inhibition [26]. ARID1A functional loss impairs ATR activation by DNA double-strand breaks and is associated with sensitization to PARPi, and also may sensitize to platinum chemotherapy and radiation. The use of agents targeting DNA repair may also be of interest in the MSI subgroup of EC.
The number of classes of agents targeting inhibition of DNA repair continues to expand beyond the PARPi. Promising targets include ATM and ATR, and WEE1 and CHEK1 G2 checkpoint kinases. Preclinical data suggest that combining ATR inhibitors with platinum may provide an effective treatment of platinum resistant EC [27]. WEE1 and CHEK1 are involved in the normal G2/M transition. Data to date suggest that cancers with a dependence on G2/M DNA repair may be susceptible to inhibition with DNA repair inhibitors. Combining agents targeting DNA repair is an attractive potential therapeutic strategy [20, 28–30] (Figure 2). Combinations with other targeted agents, to create context-specific synthetic lethality are also promising.
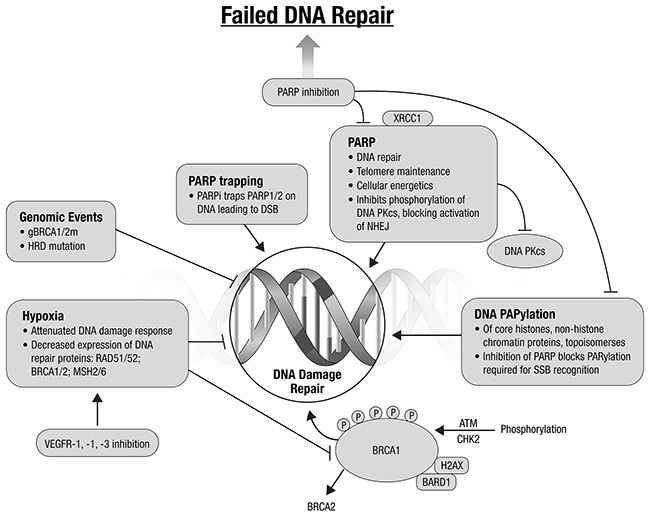
Figure 2: Augmenting DNA damage and repair: potential therapeutic directions (adapted from Ivy et al., 2016) [20].
Estrogen (ER) and progesterone (PR) receptor related pathways
Endocrine therapy has been investigated for the treatment of EC for over two decades. The 2010 Cochrane Review concluded that there was “no improvement in survival for women receiving endocrine therapy for advanced EC”. This conclusion, however, was limited by the lack of the large-scale randomized trials that would be necessary to show benefit [31]. There remains a strong rationale for endocrine therapy in EC, particularly in low grade cancers, which requires further investigation in light of our increased understanding of the EC molecular landscape.
Single agent progestins have yielded overall response rates of 20-25% [32], with some studies suggesting that ERα or PR expressing cancers are more likely to respond, although the overall data are inconsistent [32–34]. This serves to highlight the importance of standardizing tissue choice and handling, and analysis of receptor status when investigating endocrine therapy in EC. Many ECs have low levels or gradual loss of PR such that durable responses to therapy are rarely achieved. Agents targeting estrogen dependent pathways have also shown modest efficacy [35–38].
Explorations of strategies to increase hormone receptor expression are of interest. A 2004 GOG phase II study investigated tamoxifen alternating with megestrol acetate in order to increase PR expression in women with advanced and metastatic EC. A response rate of 27% was observed with a median OS of 14 months [39]. Recently in vitro studies suggest inducing changes in the epigenome could be a potential therapeutic strategy to increase hormone receptor expression. Differential methylation of multiple genes has been extensively reported in EC [40–43]. Aberrant DNA hypermethylation appears to be more frequent in lower grade endometrioid EC, with DNA hypomethylation a feature of serous-like EC [7]. Silencing of both ERα and PR by aberrant promoter methylation is reported in cell lines and patient samples with hypermethylation of the ERα-promoter C reported in over 90% and PR-promoter B in 70% of cases [44]. Treatment with a DNA methyltransferase inhibitor resulted in increased PR expression in several cell line studies. Histone modification also appears to influence PR expression. PR mRNA silencing has been shown to be reversed and functional PR expression restored with the use of epigenetic modulators in hormone-unresponsive EC cell lines [45]. HDAC inhibitors and demethylating agents are available for study and should be considered in combination with endocrine therapy.
Combining hormonal therapy with targeted therapies in rationally designed clinical trials is also an attractive therapeutic direction. Targeting the phosphoinosital-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway has been proposed as a mechanism for overcoming endocrine therapy resistance. Data predominantly in breast cancer suggest that cross regulation between the ER and PI3K/AKT/mTOR pathways makes targeting both pathways an attractive option [46]. Initial studies in EC with the rapalog class of mTOR inhibitors resulted in a clinical benefit rate (CBR) of 40% with an objective response rate of 32% for the combination of letrozole and everolimus [47]. The strongest indicator of lack of benefit from that treatment was serous histology. There was a statistically marginal benefit for the small subset of patients with catenin-beta 1 CTNNB1 mutations. However, given the intra-pathway feedback loops, inter-pathway crosstalk and the incomplete blockade of the mTOR complex by the rapalogs, the limited response and duration of this combination suggests there may be further scope to optimize this strategy.
Another direction to increase hormonal therapy efficacy would be to focus on its multiple escape pathways. Provisional data from a study combining metformin, letrozole and everolimus demonstrated a CBR of 60% [48]. However, the combination of temsirolimus with alternating megestrol acetate and daily tamoxifen did not add benefit relative to hormonal therapy alone, and was associated with high rates of venous thromboembolism [49]. Exploratory biomarker work in this study and the everolimus/letrozole trial suggests that tumors harboring CTNNB1 mutations may be more likely to respond, although responses were also seen in tumors with wild type CTNNB1 [50].
Extrapolating from the experience in breast cancer, other options include combinations of endocrine therapy with cyclin dependent kinase inhibitors [51]. Elevated CDK4 expression has been reported in 34-77% of endometrioid ECs [52, 53]. The combination of palbociclib and letrozole is currently under investigation (NCT 02730429). Correlative studies incorporated into clinical trials will be invaluable in optimizing endocrine therapy combination strategies and patient selection.
PI3K/AKT/mTOR pathway
The PIK3CA-PIK3R1-PTEN axis is somatically mutated in at least 40% of serous ECs and over 70% of endometrioid EC [7, 15, 54]. A large fraction of PIK3CA mutations are located in exons 1-8, in addition to exons 9 and 20 [11]. These mutations were found across all TCGA subgroups. Clinical trials of the rapalog class of mTOR inhibitors, temsirolimus, everolimus and ridaforolimus, have been completed in EC [49, 55–57]. Response rates have been modest with some patients experiencing prolonged stable disease. To date Correlative analyses of archival biospecimens have failed to identify a predictive biomarker [58, 59]. It is likely that a single or several biomarkers may be insufficient to predict clinical benefit due to complexity of this pathway and its many interactions in tumors, and the incomplete blockade of the pathway provided by these agents. There are multiple completed or ongoing single agent phase II clinical trials examining non-rapalog PI3K/mTOR agents in EC. A phase II trial (NCT01455493) of the PI3K/mTORC1/2 inhibitor, MK2206, in patients with advanced EC enrolled 56 patients, 3 patients had a confirmed clinical response and all 3 had one or more molecular abnormalities in PIK3CA, PTEN, or AKT [60]. A number of preclinical studies have shown that HER2-amplified serous cell lines were more sensitive to growth inhibition by mTORC1/2 inhibitors than HER2 non-amplified serous EC cell lines, a potential future direction. Targeting the PI3K/AKT/mTOR pathway alone or in combination remains an active area of research in EC. A new study has opened to examine the role of the PI3K inhibitor, copanlisib, in patients with PI3KCA hot spot mutations in their EC. (NRG GY008/NCT02728258).
Immune related pathways
Solid tumors frequently harbor an immune infiltrate that bi-directionally regulates cancer cell growth and metastatic potential, and in many cancers, has been demonstrated to have prognostic impact. Therapeutic immune modulation of this infiltrate could therefore be employed to optimize patient-tailored treatment and potentially outcome. The presence of tumor infiltrating lymphocytes (TiLs) and high ratios of CD8+TiLs to FoxP3+ T regulatory cells are associated in EC with a favorable prognosis [61], as are the presence of CD3+ T cells [61, 62] and CD45RO memory T cells [61]. On the other hand, the presence of CD163+ tumor-associated macrophages [63], CD4+CD25+ and CD4+FoxP3+ regulatory T cells, and high ratios of regulatory T cells to CD8+ cells were associated with a worse prognosis [64, 65]. This provides the impetus for using and understanding the role of agents that will enhance the presence of anti-tumor T-lymphocytes in the tumor tissue and microenvironment, and reduce the effect of local inhibitory factors, such as regulatory T cells.
The POLE subtype identified by TCGA has a good prognosis and displays enhanced cytotoxic T cell responses and high neoantigen load. Increased cytotoxic T cell responses are also seen in the TCGA identified MSI subgroup [8, 66]. MSI-associated EC are known to have a propensity for lower uterine segment involvement, intratumoral heterogeneity, and dense peritumoral lymphocytic infiltration [67]. Since both POLE and MSI subtypes demonstrate overexpression of PD1 and PDL1, these patients have been proposed to be excellent candidates for PD1-targeted therapies [68]. A recent meeting presentation reported 7 of 10 (70%) MSI EC responded to pembrolizumab within the KEYNOTE 158 study. There are multiple ongoing and planned trials of PD1-targeted therapy as single agents, with other immune modifying drugs, or with classic cytotoxic agents in microsatellite stable and unstable tumors. Further characterization of the immunological landscape of the copy-number low, copy-number high, and TP53 mutant high-risk ECs may result in additional patient-tailored immunological therapies.
Antigen-specific immunotherapy aims at activating the adaptive immune system towards a specific tumor-antigen. These vaccines may be the strategy of choice for patients with low anti-tumor immune responses, as these patients do not meet the criteria for checkpoint inhibition or adoptive T cell therapy. Important criteria for target tumor-associated antigens are no or low expression in healthy tissues and overexpression in EC. Examples of tumor-associated antigens that may be targeted using this strategy are survivin and Wilms’ tumor gene 1 (WT1). In an analysis of the immunogenicity of survivin, spontaneous T cell responses were seen in 10/39 EC patients [69]. Vaccination with autologous dendritic cells electroporated with WT1 mRNA generated a response in 3 out of 4 HLA-A2 patients, and a WT1-specific T cell response was seen in 2 of these patients [70]. It has been suggested that a combination of antigen-specific vaccines and chemotherapy may be synergistic as chemotherapy may lead to increased antigen uptake by antigen presenting cells and direct activation of dendritic cells.
Obesity-related pathways
Obesity, diabetes and insulin resistance are associated with increased risk of developing EC and worse prognosis for incident EC [71–75]. Epidemiological evidence suggests that use of metformin, as first line treatment for type 2 diabetes, lowers cancer risk and reduces cancer deaths among diabetic patients, including women with EC [75–79]. Although the hypotheses are controversial, metformin may exert anti-tumorigenic activity through indirect effects on the metabolic milieu via cation-selective transporters and direct effects on the tumor through inhibition of mitochondrial complex 1 and subsequent AMPK activation and mTOR pathway inhibition [80, 81].
Several pre-operative window studies of metformin in EC are reported [82–85]. Each reported a statistically significant decrease in expression of Ki-67, a marker of cell proliferation, and reduction in downstream markers of the MAPK and mTOR pathways. Metformin treatment also decreased circulating plasma factors, including insulin, glucose, insulin-like growth factor-1 (IGF-1), leptin and insulin-like growth factor binding protein-7 (IGFBP7). Discrepant results have been reported on the effect of metformin on phosphorylated-AMPK [82].
There are currently many studies of metformin for endometrial hyperplasia or cancer (Table 1). These include chemoprevention in the obese patient, treatment of endometrial hyperplasia, and combination therapy with other targeted agents. An ongoing randomized, placebo-controlled phase II/III trial is designed to assess efficacy of the addition of metformin to paclitaxel and carboplatin in women with advanced and recurrent EC (GOG-0286B; NCT02065687). Secondary endpoints are to estimate differences in obesity-related parameters, cation transport, and demographics.
Table 1: Table Obesity pathway vs. Table Metformin studies
Center |
Title |
Trial type |
Tumor types |
|---|---|---|---|
UNC Lineberger Comprehensive Cancer Center |
Metformin for the Treatment of Endometrial Hyperplasia |
Open label, safety/efficacy trial |
Simple or complex hyperplasia without atypia |
UNC Lineberger Comprehensive Cancer Center |
Metformin with the Levonorgestrel- Releasing Intrauterine Device for the Treatment of Complex Atypical Hyperplasia (CAH) and Endometrial Cancer (EC) in Non-Surgical Patients |
Open label, safety/efficacy trial |
Complex atypical hyperplasia and endometrial cancer |
M.D. Anderson Cancer Center |
An Endometrial Cancer Chemoprevention Study of Metformin Versus No Treatment in Women with a Body Mass Index (BMI) >/= 35 kg/m2 and Hyperinsulinemia |
Randomized, double blind, phase III efficacy trial |
Endometrial Cancer |
M.D. Anderson Cancer Center |
A Phase II, Single-Arm Study of RAD001 (Everolimus), Letrozole, and Metformin in Patients with Advanced or Recurrent Endometrial Carcinoma |
Open label, phase II, safety/efficacy trial |
Endometrial Cancer |
Gynecologic Oncology Group |
A Randomized Phase II/III Study of Paclitaxel/Carboplatin/Metformin (NSC#91485) Versus Paclitaxel/Carboplatin/Placebo as Initial Therapy for Measurable Stage III or IVA, Stage IVB, or Recurrent Endometrial Cancer |
Randomized, double blinded, placebo controlled, phase II/III trial |
Endometrial Cancer |
Queensland Centre for Gynaecological Cancer |
Improving the Treatment for Women with Early Stage Cancer of the Uterus |
Randomized, open label, efficacy, phase II, Mirena IUD versus, Mirena IUD + Metformin versus Mirena IUD + Weight Loss Intervention |
Endometrial hyperplasia with atypia, Endometrial Cancer |
Fudan University, China |
Megestrol Acetate Plus Metformin to Megestrol Acetate in Patients with Endometrial Atypical Hyperplasia or Early Stage Endometrial Adenocarcinoma |
Randomized, open label, efficacy, phase II |
Endometrial hyperplasia with atypia, Endometrial Cancer |
ERBB2/ HER2
ERBB2/HER2 is a receptor tyrosine kinase that mediates signaling via the PI3K and mitogen activated protein kinase (MAPK) pathways. It is amplified in 21%-47% of serous ECs, found in the TCGA CNH subgroup, and in 3%-21% of endometrioid ECs [86, 87]. Trastuzumab is a humanized monoclonal antibody that targets HER2. Although clinical responses to trastuzumab in HER2+ serous and endometrioid EC cancer patients have been noted in case reports [88, 89], a phase II trial (GOG181B) evaluating the activity of trastuzumab for HER2+ recurrent or advanced-stage EC observed no objective responses [89, 90]. Studies in tumor tissues and serum from EC patients raise the possibility that ECs may be intrinsically resistant to trastuzumab because they have relatively high levels of the p95 variant of HER2, which lacks the extracellular domain targeted by trastuzumab [91, 92], or because of the frequent somatic activation of the downstream PI3K pathway [93]. It has therefore been hypothesized that small molecule inhibitors that bind the intracellular domain of both HER2 and p95, such as lapatinib, might be more effective than trastuzumab in EC. A phase II trial of lapatinib in unselected patients with recurrent or persistent EC observed limited clinical activity [94]. Potential explanations for the limited activity included recruitment of unselected patients, and/or intrinsic lapatinib-resistance due to activation of the PI3K pathway. Afatinib, a pan-ERBB inhibitor, is currently under investigation in a phase II clinical trial in patients with persistent or recurrent HER2+ uterine serous carcinoma (NCT02491099).
TCGA has shown that amplification of HER2 and somatic mutations of PIK3CA, the p110 kinase subunit of PI3K, often co-occur in serous/serous-like EC [7]. Whether these events occur within the same or distinct subpopulations of a given tumor remains to be determined and may have clinical implications, as noted for breast cancer [95]. It would seem prudent that future clinical trial design of HER2 targeted therapies in EC should include a comprehensive molecular assessment of the PI3K pathway, including PIK3CA (all exons), PTEN, and PIK3R1, the gene encoding the p85α regulatory subunit of PI3K. Combination therapy of pan-ERBB inhibitors with PI3K inhibitors has been shown to be synergistic in preclinical models of serous cancer [96]. Importantly, dual inhibition initiated after tumor progression with single agent treatment was still effective in inducing tumor regression in tumor bearing mice. Thus, dual HER2/PIK3CA blockade may represent a novel therapeutic option for EC patients harboring tumors with HER2 gene amplification and mutated PIK3CA.
WNT pathway
WNT signaling functions predominantly through both CTNNB1-dependent, and independent pathways, often termed canonical and non-canonical WNT signaling, respectively [97–99]. The WNT pathway is typically activated by one of the WNT family members binding to the frizzled (FZD) receptor to activate the disheveled (DVL) protein (Figure 3). This pathway is deregulated in many human tumors. Under normal conditions, CTNNB1 is phosphorylated by WNT pathway members and targeted for proteasomal degradation through ubiquitination, leading to active repression of CTNNB1 target genes. When the pathway is disrupted, such as somatic mutations in pathway members or CTNNB1, CTNNB1 is not targeted for degradation, avoids phosphorylation, and accumulates in the cytoplasm, ultimately entering the nucleus and leading to CTNNB1-mediated transcription. Secreted WNT antagonists include members of the Dickkopf (Dkk) family that prevent Wnt signaling through the low-density lipoprotein receptor-related protein (LRP). Dkk3 has been shown to be downregulated in EC and correlated with advanced stage and high-risk clinicopathologic factors. Forced Dkk3 expression in vitro reduced proliferation, anchorage-independent growth, and invasion [100].
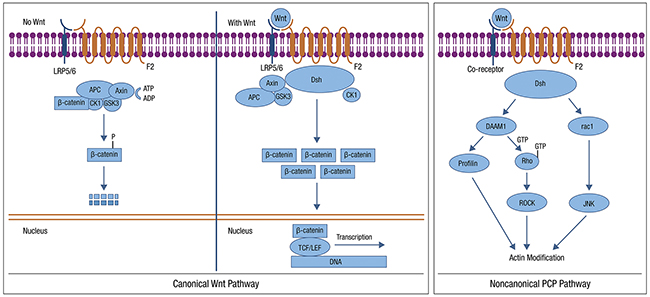
Figure 3: WNT Pathway: Most WNT signaling in EC occurs via the CTNNB1-dependent pathway. Possible targets include: use of WNT antagonists, reduced WNT ligand secretion, increased degradation of WNT and inhibition of CDK4/6 Canonical (left) and non-canonical (right) WNT signaling are shown. This file is licensed under the Creative Commons Attribution-Share Alike 3.0 Unported license. https://en.wikipedia.org/wiki/Wnt_signaling_pathway.
WNT signaling in EC predominantly involves the CTNNB1-dependent, or canonical, signaling pathway. CTNNB1 mutations occur in 52% of the CNL EC subgroup identified in the TCGA [7]. Alterations are uncommon among serous tumors and are present at low frequency in MSI+ tumors. Several studies have suggested that CTNNB1 mutations are associated with a worse prognosis in early-stage low grade EC [101]. The accumulation of nuclear CTNNB1 has been demonstrated to be more common in high-grade EC and associated with loss of CDH1, suggesting more aggressive behavior [102]. There are few WNT pathway targeted agents available currently [103]; such agents would be of interest to explore in EC.
Ubiquitin-ligase complexes
The proper regulation of cellular protein levels by the ubiquitin proteosome system is an important facet of cell biology. Dysregulation of ubiquitin mediated proteosomal degradation of cellular proteins is often observed in human cancers [104, 105]. Recent whole exome sequencing studies in EC have uncovered frequent somatic alterations in the FBXW7 and SPOP genes, which encode ubiquitin ligase adaptor proteins and are more commonly seen in the CNH TCGA subgroup. This pathway is thus a novel direction for consideration. The SKP1-CUL1-FBXW7 (SCFFBXW7) complex is an E3 ubiquitin ligase complex that regulates the degradation of a large number of protein substrates, many of which are transcriptional regulators [106]. Several proteins that are regulated by the SCFFBXW7 complex, such as cyclin E, cMYC, mTOR, and MCL1 promote oncogenesis in solid tumors. FBXW7 is a haplo-insufficient tumor suppressor [107], and is somatically mutated and/or deleted across a wide range of human cancers, with EC and T cell acute lymphocytic leukemias most frequently mutated [7, 14, 106, 108, 109]. Comprehensive sequencing studies in EC have shown that FBXW7 mutations are more abundant in serous (15%-29%) [7, 14, 16, 109] and serous-like (21%) [7] cancers, than in either clear cell (7%-13%) [14, 110] or endometrioid (10%-27%) ECs [7, 14]. The presence of FBXW7 mutations in concurrent cases of serous EC and serous endometrial intraepithelial carcinoma suggests these are early genetic events for this subtype [109]. In addition, FBXW7 is also mutated in 20% of undifferentiated uterine carcinomas and in 23% of uterine carcinosarcomas [111–113].
The SPOP-CUL3-RBX1 ubiquitin ligase complex also regulates protein turnover via ubiquitin-mediated proteasomal degradation. Thus far, the repertoire of proteins that are regulated by the SPOP-CUL3-RBX1 complex appears to be largely distinct from those that are regulated by the SCFFBXW7 complex. Although somatic mutations in SPOP are rare in most human cancers, they occur at higher rates in EC [7, 14, 16], and prostate cancer [114]. SPOP mutations have been documented in 7%-8% of serous ECs [7, 14, 16], 5% of serous-like ECs [7], 0-9% of endometrioid ECs [7, 16], and 8% of clear cell ECs [14]. The majority of SPOP mutations in EC and in prostate cancer localize to the MATH domain, which binds proteins that are targeted for ubiquitination and proteasomal degradation. The MATH domain of SPOP functions in an analogous manner to the WD repeats of FBXW7, leading to the speculation that missense mutations in the MATH domain are likely to be dominant-negative or loss-of-function mutants that disrupt the binding of SPOP to one or more of its protein substrates. Functional studies of the SPOP mutations that have been found in EC are at a very early stage, but thus far indicate that a subset of SPOP mutants have an impaired ability to regulate ERα [115]. Recent work by Barberi et al., 2015 indicates that SPOP mutations in prostate cancer lead to defects in HR and confer sensitivity to PARP inhibition [114]. Whether this phenomenon also holds true for EC, remains to be elucidated.
Chromatin-remodeling
Mutated chromatin-remodeling genes have been reported in primary serous EC. Whole-exome sequencing analysis of a small number of primary serous endometrial tumors (n=13), followed by targeted gene sequencing in a larger cohort of serous ECs, identified frequent somatic mutations in CHD4 (17%), which encodes a subunit of the NuRD-chromatin-remodeling complex [14, 16]. A confirmatory study also noted frequent deletion of a small segment of chromosome 19 containing MBD3, another subunit of the NuRD-chromatin-modification complex, and frequent mutations in TAF1. The TAF1 protein has histone acetyltransferase activity and is an element of the TFIID basal transcription factor complex. Le Gallo et al., have reported TAF1 mutations in nearly 10% of clear cell EC [116]. Lower frequency mutations in several other chromatin remodeling genes including EP300 (8%), and ARID1A (6%) have been noted in serous ECs [14]. ARID1a, a component of the SWI/SNF chromatin remodeling complex, is also frequently mutated in endometrioid ECs including all TCGA subgroups except CNH, up to 40% by different databases [7, 117]. Overactivity/overexpression of EZH2, a histone methyl transferase, downregulates the suppressor ARID1a, creating a loss of suppressor function and resulting in increased proliferation, migration, and invasion of target malignant cells. Therapeutic inhibition of EZH2 is now in early evaluation and could be targeted to ARID1a mutant EC [118]. The expansion of the histone deacetylase inhibitor group and development of demethylating agents may be of use in this subset of patients.
THE PATH FORWARD FOR IDENTIFYING FUTURE ACTIONABLE OPPORTUNITIES
Clearly the emerging molecular data provide us with multiple potential avenues to pursue in terms of early phase clinical trial design for EC. In addition, TCGA identified molecular subgroups provide a potential framework for developing new risk stratification models. Actionable opportunities are those for which abnormalities have been identified and demonstrated to be biologically necessary and/or sufficient. We are now beyond the point of mutations being actionable by definition alone and require more than simple biologic plausibility. Refinement of the molecular subgroup model by combining conventional parameters, and potentially other biomarkers will add value and enhance potential for patient selection clinical trial designs. Incorporation of high quality, validated, correlative studies into our clinical trials with appropriate collection of tumor and surrogate samples will be essential to capitalize on the wealth of information that can be gained from positive and negative studies. Collaborative data sharing and access to samples for biomarker identification, and perhaps more importantly, validation will speed discovery. Lastly, translating opportunity and hypothesis into the clinic always requires consideration of other unique issues that may be found in patient populations. Endometrial cancer is now affecting younger women, though still remains a cancer predominantly of older women, many of whom have other medical comorbidities. Such clinical variables may affect the complexity of some of the combination treatment ideas that could be evaluated in EC. The clinical opportunities identified in this review illustrate the complexity of EC, and also provide us with a framework for leveraging combination strategies targeting the many interrelated pathways implicated in EC biology.
Author contributions
All of the authors wrote sections for this manuscript and reviewed and made commentary on the final drafts.
ACKNOWLEDGMENTS
The authors acknowledge all those who participated in the US National Cancer Institute (NCI) Gynecologic Cancer Steering Committee (GCSC), and all individuals who participated and contributed to the Uterine Cancer Clinical Trials Planning Meeting (UCTPM), and its associated working groups in January 2016.
CONFLICTS OF INTEREST
The following authors have no conflicts of interests to disclose: Helen J MacKay, Douglas A Levine, Victoria L Bae-Jump, Jessica N McAlpine, Alexandro Santin, Gini F Fleming, David G Mutch, Kenneth P Nephew, Nicholas Wentzensen, Paul J Goodfellow, Oliver Dorigo, Hans W Nijman, Russell Broaddus and Elise C Kohn.
Daphne W Bell receives royalties as a co-inventor on U.S. Patent No.7,294,468 “Method to Determine Responsiveness of Cancer to Epidermal Growth Factor Receptor Targeting Treatments”, which is licensed to Esoterix Genetic Laboratories LLC.
REFERENCES
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74: 2913-21. https://doi.org/10.1158/0008-5472.CAN-14-0155.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7-30. https://doi.org/10.3322/caac.21332.
3. Lheureux S, Wilson M, Mackay HJ. Recent and current Phase II clinical trials in endometrial cancer: review of the state of art. Expert Opin Investig Drugs. 2014; 23: 773-92. https://doi.org/10.1517/13543784.2014.907272.
4. Bendifallah S, Darai E, Ballester M. Predictive modeling: a new paradigm for managing endometrial cancer. Ann Surg Oncol. 2016; 23: 975-88. https://doi.org/10.1245/s10434-015-4924-2.
5. Gilks CB, Oliva E, Soslow RA. Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. Am J Surg Pathol. 2013; 37: 874-81. https://doi.org/10.1097/PAS.0b013e31827f576a.
6. Hoang LN, McConechy MK, Kobel M, Han G, Rouzbahman M, Davidson B, Irving J, Ali RH, Leung S, McAlpine JN, Oliva E, Nucci MR, Soslow RA, et al. Histotype-genotype correlation in 36 high-grade endometrial carcinomas. Am J Surg Pathol. 2013; 37: 1421-32. https://doi.org/10.1097/PAS.0b013e31828c63ed.
7. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497: 67-73. https://doi.org/10.1038/nature12113.
8. Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, Stover E, Strickland KC, D’Andrea AD, Wu CJ, Matulonis UA, Konstantinopoulos PA. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015; 1: 1319-23. https://doi.org/10.1001/jamaoncol.2015.2151.
9. Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, Leary A, Edmondson RJ, Powell ME, Crosbie EJ, Kitchener HC, Mileshkin L, Pollock PM, et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod Pathol. 2015; 28: 836-44. https://doi.org/10.1038/modpathol.2015.43.
10. Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, Yang W, Senz J, Boyd N, Karnezis AN, Huntsman DG, Gilks CB, McAlpine JN. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer. 2015; 113: 299-310. https://doi.org/10.1038/bjc.2015.190.
11. Bashir S, Jiang G, Joshi A, Miller C Jr, Matrai C, Yemelyanova A, Caputo TA, Holcomb KM, Ellenson LH, Gupta D. Molecular alterations of PIK3CA in uterine carcinosarcoma, clear cell, and serous tumors. Int J Gynecol Cancer. 2014; 24: 1262-7. https://doi.org/10.1097/IGC.0000000000000183.
12. Jones NL, Xiu J, Reddy SK, Burke WM, Tergas AI, Wright JD, Hou JY. Identification of potential therapeutic targets by molecular profiling of 628 cases of uterine serous carcinoma. Gynecol Oncol. 2015; 138: 620-6. https://doi.org/10.1016/j.ygyno.2015.06.034.
13. Kuhn E, Ayhan A, Bahadirli-Talbott A, Zhao C, Shih Ie M. Molecular characterization of undifferentiated carcinoma associated with endometrioid carcinoma. Am J Surg Pathol. 2014; 38: 660-5. https://doi.org/10.1097/PAS.0000000000000166.
14. Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC, Hieter P, Mullikin JC, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012; 44: 1310-5. https://doi.org/10.1038/ng.2455.
15. Urick ME, Rudd ML, Godwin AK, Sgroi D, Merino M, Bell DW. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011; 71: 4061-7. https://doi.org/10.1158/0008-5472.CAN-11-0549.
16. Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, Bortolomai I, Buza N, Hui P, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013; 110: 2916-21. https://doi.org/10.1073/pnas.1222577110.
17. Buza N, Roque DM, Santin AD. HER2/neu in endometrial cancer: a promising therapeutic target with diagnostic challenges. Arch Pathol Lab Med. 2014; 138: 343-50. https://doi.org/10.5858/arpa.2012-0416-RA.
18. Diver EJ, Foster R, Rueda BR, Growdon WB. The therapeutic challenge of targeting HER2 in endometrial cancer. Oncologist. 2015; 20: 1058-68. https://doi.org/10.1634/theoncologist.2015-0149.
19. O’Hara AJ, Bell DW. The genomics and genetics of endometrial cancer. Adv Genomics Genet. 2012; 2012: 33-47. https://doi.org/10.2147/AGG.S28953.
20. Ivy SP, de Bono J, Kohn EC. The ‘Pushmi-Pullyu’ of DNA REPAIR: clinical synthetic lethality. Trends Cancer. 2: 646-56. https://doi.org/10.1016/j.trecan.2016.10.014.
21. Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A, Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010; 2: 53ra75. https://doi.org/10.1126/scitranslmed.3001538.
22. Miyasaka A, Oda K, Ikeda Y, Wada-Hiraike O, Kashiyama T, Enomoto A, Hosoya N, Koso T, Fukuda T, Inaba K, Sone K, Uehara Y, Kurikawa R, et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer. 2014; 14: 179. https://doi.org/10.1186/1471-2407-14-179.
23. Giannini G, Rinaldi C, Ristori E, Ambrosini MI, Cerignoli F, Viel A, Bidoli E, Berni S, D’Amati G, Scambia G, Frati L, Screpanti I, Gulino A. Mutations of an intronic repeat induce impaired MRE11 expression in primary human cancer with microsatellite instability. Oncogene. 2004; 23: 2640-7. https://doi.org/10.1038/sj.onc.12074091207409.
24. Miquel C, Jacob S, Grandjouan S, Aime A, Viguier J, Sabourin JC, Sarasin A, Duval A, Praz F. Frequent alteration of DNA damage signalling and repair pathways in human colorectal cancers with microsatellite instability. Oncogene. 2007; 26: 5919-26. https://doi.org/10.1038/sj.onc.1210419.
25. Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, Mukherjee B, Iniesta MD, Morgan MA, Rennert G, Gruber SB. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011; 71: 2632-42. https://doi.org/10.1158/0008-5472.CAN-10-1120.
26. Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, Kapoor P, Ju Z, Mo Q, Shih Ie M, Uray IP, Wu X, Brown PH, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015; 5: 752-67. https://doi.org/10.1158/2159-8290.CD-14-0849.
27. Teng PN, Bateman NW, Darcy KM, Hamilton CA, Maxwell GL, Bakkenist CJ, Conrads TP. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol Oncol. 2015; 136: 554-61. https://doi.org/10.1016/j.ygyno.2014.12.035.
28. Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994; 6: 877-82.
29. Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012; 40: 477-86. https://doi.org/10.1093/nar/gkr697.
30. Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001; 61: 8211-7.
31. Kokka F, Brockbank E, Oram D, Gallagher C, Bryant A. Hormonal therapy in advanced or recurrent endometrial cancer. Cochrane Database Syst Rev. 2010: CD007926. https://doi.org/10.1002/14651858.CD007926.pub2.
32. Markman M. Hormonal therapy of endometrial cancer. Eur J Cancer. 2005; 41: 673-5. https://doi.org/10.1016/j.ejca.2004.12.008.
33. Singh M, Zaino RJ, Filiaci VJ, Leslie KK. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol. 2007; 106: 325-33. https://doi.org/10.1016/j.ygyno.2007.03.042.
34. Temkin SM, Fleming G. Current treatment of metastatic endometrial cancer. Cancer Control. 2009; 16: 38-45.
35. Covens AL, Filiaci V, Gersell D, Lutman CV, Bonebrake A, Lee YC. Phase II study of fulvestrant in recurrent/metastatic endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2011; 120: 185-8. https://doi.org/10.1016/j.ygyno.2010.10.015.
36. Emons G, Gunthert A, Thiel FC, Camara O, Strauss HG, Breitbach GP, Kolbl H, Reimer T, Finas D, Rensing K. Phase II study of fulvestrant 250 mg/month in patients with recurrent or metastatic endometrial cancer: a study of the Arbeitsgemeinschaft Gynakologische Onkologie. Gynecol Oncol. 2013; 129: 495-9. https://doi.org/10.1016/j.ygyno.2013.02.039.
37. Ma BB, Oza A, Eisenhauer E, Stanimir G, Carey M, Chapman W, Latta E, Sidhu K, Powers J, Walsh W, Fyles A. The activity of letrozole in patients with advanced or recurrent endometrial cancer and correlation with biological markers--a study of the National Cancer Institute of Canada Clinical Trials Group. Int J Gynecol Cancer. 2004; 14: 650-8. https://doi.org/10.1111/j.1048-891X.2004.14419.x.
38. Rose PG, Brunetto VL, VanLe L, Bell J, Walker JL, Lee RB. A phase II trial of anastrozole in advanced recurrent or persistent endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2000; 78: 212-6. https://doi.org/10.1006/gyno.2000.5865.
39. Fiorica JV, Brunetto VL, Hanjani P, Lentz SS, Mannel R, Andersen W. Phase II trial of alternating courses of megestrol acetate and tamoxifen in advanced endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2004; 92: 10-4. doi: S009082580300787X.
40. Kolbe DL, DeLoia JA, Porter-Gill P, Strange M, Petrykowska HM, Guirguis A, Krivak TC, Brody LC, Elnitski L. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS One. 2012; 7: e32941. https://doi.org/10.1371/journal.pone.0032941.
41. Tao MH, Freudenheim JL. DNA methylation in endometrial cancer. Epigenetics. 2010; 5: 491-8.
42. Whitcomb BP, Mutch DG, Herzog TJ, Rader JS, Gibb RK, Goodfellow PJ. Frequent HOXA11 and THBS2 promoter methylation, and a methylator phenotype in endometrial adenocarcinoma. Clin Cancer Res. 2003; 9: 2277-87.
43. Zhang B, Xing X, Li J, Lowdon RF, Zhou Y, Lin N, Zhang B, Sundaram V, Chiappinelli KB, Hagemann IS, Mutch DG, Goodfellow PJ, Wang T. Comparative DNA methylome analysis of endometrial carcinoma reveals complex and distinct deregulation of cancer promoters and enhancers. BMC Genomics. 2014; 15: 868. https://doi.org/10.1186/1471-2164-15-868.
44. Sasaki M, Kaneuchi M, Fujimoto S, Tanaka Y, Dahiya R. Hypermethylation can selectively silence multiple promoters of steroid receptors in cancers. Mol Cell Endocrinol. 2003; 202: 201-7.
45. Yang S, Xiao X, Jia Y, Liu X, Zhang Y, Wang X, Winters CJ, Devor EJ, Meng X, Thiel KW, Leslie KK. Epigenetic modification restores functional PR expression in endometrial cancer cells. Curr Pharm Des. 2014; 20: 1874-80. doi: CPD-EPUB-54351.
46. Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011; 1: 170-85. https://doi.org/10.1158/2159-8290.CD-11-0039.
47. Slomovitz BM, Jiang Y, Yates MS, Soliman PT, Johnston T, Nowakowski M, Levenback C, Zhang Q, Ring K, Munsell MF, Gershenson DM, Lu KH, Coleman RL. Phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J Clin Oncol. 2015; 33: 930-6. doi: JCO.2014.58.3401.
48. Soliman PT, Neville Westin S, Iglesias DA, Munsell MF, Slomovitz BM, Lu KH, Coleman RL. Phase II study of everolimus, letrozole, and metformin in women with advanced/recurrent endometrial cancer. ASCO Meeting Abstracts. 2016; 34.
49. Fleming GF, Filiaci VL, Marzullo B, Zaino RJ, Davidson SA, Pearl M, Makker V, Burke JJ 2nd, Zweizig SL, Van Le L, Hanjani P, Downey G, Walker JL, et al. Temsirolimus with or without megestrol acetate and tamoxifen for endometrial cancer: a gynecologic oncology group study. Gynecol Oncol. 2014; 132: 585-92. doi: S0090-8258(14)00026-2.
50. Myers AP, Filiaci VL, Zhang Y, Pearl M, Behbakht K, Makker V, Hanjani P, Zweizig S, Burke JJ 2nd, Downey G, Leslie KK, Van Hummelen P, Birrer MJ, et al. Tumor mutational analysis of GOG248, a phase II study of temsirolimus or temsirolimus and alternating megestrol acetate and tamoxifen for advanced endometrial cancer (EC): An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2016; 141: 43-8. https://doi.org/10.1016/j.ygyno.2016.02.025.
51. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015; 16: 25-35. https://doi.org/10.1016/S1470-2045(14)71159-3.
52. Shih HC, Shiozawa T, Kato K, Imai T, Miyamoto T, Uchikawa J, Nikaido T, Konishi I. Immunohistochemical expression of cyclins, cyclin-dependent kinases, tumor-suppressor gene products, Ki-67, and sex steroid receptors in endometrial carcinoma: positive staining for cyclin A as a poor prognostic indicator. Hum Pathol. 2003; 34: 471-8.
53. Tsuda H, Yamamoto K, Inoue T, Uchiyama I, Umesaki N. The role of p16-cyclin d/CDK-pRb pathway in the tumorigenesis of endometrioid-type endometrial carcinoma. Br J Cancer. 2000; 82: 675-82. https://doi.org/10.1054/bjoc.1999.0980.
54. Rudd ML, Price JC, Fogoros S, Godwin AK, Sgroi DC, Merino MJ, Bell DW. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011; 17: 1331-40. https://doi.org/10.1158/1078-0432.CCR-10-0540.
55. Colombo N, McMeekin DS, Schwartz PE, Sessa C, Gehrig PA, Holloway R, Braly P, Matei D, Morosky A, Dodion PF, Einstein MH, Haluska F. Ridaforolimus as a single agent in advanced endometrial cancer: results of a single-arm, phase 2 trial. Br J Cancer. 2013; 108: 1021-6. https://doi.org/10.1038/bjc.2013.59.
56. Oza AM, Pignata S, Poveda A, McCormack M, Clamp A, Schwartz B, Cheng J, Li X, Campbell K, Dodion P, Haluska FG. Randomized phase II trial of ridaforolimus in advanced endometrial carcinoma. J Clin Oncol. 2015; 33: 3576-82. https://doi.org/10.1200/JCO.2014.58.8871.
57. Ray-Coquard I, Favier L, Weber B, Roemer-Becuwe C, Bougnoux P, Fabbro M, Floquet A, Joly F, Plantade A, Paraiso D, Pujade-Lauraine E. Everolimus as second- or third-line treatment of advanced endometrial cancer: ENDORAD, a phase II trial of GINECO. Br J Cancer. 2013; 108: 1771-7. https://doi.org/10.1038/bjc.2013.183.
58. Myers AP. New strategies in endometrial cancer: targeting the PI3K/mTOR pathway--the devil is in the details. Clin Cancer Res. 2013; 19: 5264-74. https://doi.org/10.1158/1078-0432.CCR-13-0615.
59. Mackay HJ, Eisenhauer EA, Kamel-Reid S, Tsao M, Clarke B, Karakasis K, Werner HM, Trovik J, Akslen LA, Salvesen HB, Tu D, Oza AM. Molecular determinants of outcome with mammalian target of rapamycin inhibition in endometrial cancer. Cancer. 2014; 120: 603-10. https://doi.org/10.1002/cncr.28414.
60. Makker V, Recio FO, Ma L, Matulonis UA, Lauchle JO, Parmar H, Gilbert HN, Ware JA, Zhu R, Lu S, Huw LY, Wang Y, Koeppen H, et al. A multicenter, single-arm, open-label, phase 2 study of apitolisib (GDC-0980) for the treatment of recurrent or persistent endometrial carcinoma (MAGGIE study). Cancer. 2016. https://doi.org/10.1002/cncr.30286.
61. de Jong RA, Leffers N, Boezen HM, ten Hoor KA, van der Zee AG, Hollema H, Nijman HW. Presence of tumor-infiltrating lymphocytes is an independent prognostic factor in type I and II endometrial cancer. Gynecol Oncol. 2009; 114: 105-10. https://doi.org/10.1016/j.ygyno.2009.03.022.
62. Cermakova P, Melichar B, Tomsova M, Zoul Z, Kalabova H, Spacek J, Dolezel M. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in patients with endometrial carcinoma. Anticancer Res. 2014; 34: 5555-61.
63. Kubler K, Ayub TH, Weber SK, Zivanovic O, Abramian A, Keyver-Paik MD, Mallmann MR, Kaiser C, Serce NB, Kuhn W, Rudlowski C. Prognostic significance of tumor-associated macrophages in endometrial adenocarcinoma. Gynecol Oncol. 2014; 135: 176-83. https://doi.org/10.1016/j.ygyno.2014.08.028.
64. Chang WC, Li CH, Huang SC, Chang DY, Chou LY, Sheu BC. Clinical significance of regulatory T cells and CD8+ effector populations in patients with human endometrial carcinoma. Cancer. 2010; 116: 5777-88. https://doi.org/10.1002/cncr.25371.
65. Yamagami W, Susumu N, Tanaka H, Hirasawa A, Banno K, Suzuki N, Tsuda H, Tsukazaki K, Aoki D. Immunofluorescence-detected infiltration of CD4+FOXP3+ regulatory T cells is relevant to the prognosis of patients with endometrial cancer. Int J Gynecol Cancer. 2011; 21: 1628-34. https://doi.org/10.1097/IGC.0b013e31822c271f.
66. van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, de Bruyn M, Palles C, Nout RA, de Kroon CD, Osse EM, Klenerman P, Creutzberg CL, Tomlinson IP, et al. POLE proofreading mutations rlicit an antitumor immune response in endometrial cancer. Clin Cancer Res. 2015; 21: 3347-55. https://doi.org/10.1158/1078-0432.CCR-15-0057.
67. Garg K, Soslow RA. Lynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinoma. J Clin Pathol. 2009; 62: 679-84. https://doi.org/10.1136/jcp.2009.064949.
68. Santin AD, Bellone S, Buza N, Choi J, Schwartz PE, Schlessinger J, Lifton RP. Regression of chemotherapy-resistant polymerase epsilon (POLE) ultra-mutated and MSH6 hyper-mutated endometrial tumors with nivolumab. Clin Cancer Res. 2016. https://doi.org/10.1158/1078-0432.CCR-16-1031.
69. Vanderstraeten A, Everaert T, Van Bree R, Verbist G, Luyten C, Amant F, Tuyaerts S. In vitro validation of survivin as target tumor-associated antigen for immunotherapy in uterine cancer. J Immunother. 2015; 38: 239-49. https://doi.org/10.1097/CJI.000000000000008500002371-201507000-00003.
70. Coosemans A, Vanderstraeten A, Tuyaerts S, Verschuere T, Moerman P, Berneman ZN, Vergote I, Amant F, VAN Gool SW. Wilms’ Tumor Gene 1 (WT1)--loaded dendritic cell immunotherapy in patients with uterine tumors: a phase I/II clinical trial. Anticancer Res. 2013; 33: 5495-500.
71. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003; 348: 1625-38. https://doi.org/10.1056/NEJMoa021423348/17/1625.
72. Chia VM, Newcomb PA, Trentham-Dietz A, Hampton JM. Obesity, diabetes, and other factors in relation to survival after endometrial cancer diagnosis. Int J Gynecol Cancer. 2007; 17: 441-6. https://doi.org/10.1111/j.1525-1438.2007.00790.x.
73. Steiner E, Plata K, Interthal C, Schmidt M, Faldum A, Hengstler JG, Sakuragi N, Watari H, Yamamoto R, Kolbl H. Diabetes mellitus is a multivariate independent prognostic factor in endometrial carcinoma: a clinicopathologic study on 313 patients. Eur J Gynaecol Oncol. 2007; 28: 95-7.
74. Ko EM, Walter P, Jackson A, Clark L, Franasiak J, Bolac C, Havrilesky LJ, Secord AA, Moore DT, Gehrig PA, Bae-Jump V. Metformin is associated with improved survival in endometrial cancer. Gynecol Oncol. 2014; 132: 438-42. https://doi.org/10.1016/j.ygyno.2013.11.021.
75. Trabert B, Wentzensen N, Felix AS, Yang HP, Sherman ME, Brinton LA. Metabolic syndrome and risk of endometrial cancer in the united states: a study in the SEER-medicare linked database. Cancer Epidemiol Biomarkers Prev. 2015; 24: 261-7. https://doi.org/10.1158/1055-9965.EPI-14-0923.
76. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006; 29: 254-8.
77. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005; 330: 1304-5. https://doi.org/10.1136/bmj.38415.708634.F7.
78. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009; 32: 1620-5. https://doi.org/10.2337/dc08-2175.
79. Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation--implications for a novel treatment strategy. Gynecol Oncol. 2010; 116: 92-8. https://doi.org/10.1016/j.ygyno.2009.09.024.
80. Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014; 10: 143-56. https://doi.org/10.1038/nrendo.2013.256.
81. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014; 3: e02242.
82. Laskov I, Drudi L, Beauchamp MC, Yasmeen A, Ferenczy A, Pollak M, Gotlieb WH. Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol Oncol. 2014; 134: 607-14. https://doi.org/10.1016/j.ygyno.2014.06.014.
83. Mitsuhashi A, Kiyokawa T, Sato Y, Shozu M. Effects of metformin on endometrial cancer cell growth in vivo: a preoperative prospective trial. Cancer. 2014; 120: 2986-95. https://doi.org/10.1002/cncr.28853.
84. Schuler KM, Rambally BS, DiFurio MJ, Sampey BP, Gehrig PA, Makowski L, Bae-Jump VL. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015; 4: 161-73. https://doi.org/10.1002/cam4.353.
85. Sivalingam VN, Kitson S, McVey R, Roberts C, Pemberton P, Gilmour K, Ali S, Renehan AG, Kitchener HC, Crosbie EJ. Measuring the biological effect of presurgical metformin treatment in endometrial cancer. Br J Cancer. 2016; 114: 281-9. https://doi.org/10.1038/bjc.2015.453.
86. English DP, Roque DM, Santin AD. HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Mol Diagn Ther. 2013; 17: 85-99. https://doi.org/10.1007/s40291-013-0024-9.
87. Mentrikoski MJ, Stoler MH. HER2 immunohistochemistry significantly overestimates HER2 amplification in uterine papillary serous carcinomas. Am J Surg Pathol. 2014; 38: 844-51. https://doi.org/10.1097/PAS.0000000000000182.
88. Jewell E, Secord AA, Brotherton T, Berchuck A. Use of trastuzumab in the treatment of metastatic endometrial cancer. Int J Gynecol Cancer. 2006; 16: 1370-3. https://doi.org/10.1111/j.1525-1438.2006.00543.x.
89. Santin AD. Letter to the Editor referring to the manuscript entitled: “Phase II trial of trastuzumab in women with advanced or recurrent HER-positive endometrial carcinoma: a Gynecologic Oncology Group study” recently reported by Fleming et al., (Gynecol Oncol., 116;15-20;2010). Gynecol Oncol. 2010; 118: 95-6; author reply 6-7. https://doi.org/10.1016/j.ygyno.2010.01.043.
90. Fleming GF, Sill MW, Darcy KM, McMeekin DS, Thigpen JT, Adler LM, Berek JS, Chapman JA, DiSilvestro PA, Horowitz IR, Fiorica JV. Phase II trial of trastuzumab in women with advanced or recurrent, HER2-positive endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2010; 116: 15-20. https://doi.org/10.1016/j.ygyno.2009.09.025.
91. Growdon WB, Groeneweg J, Byron V, DiGloria C, Borger DR, Tambouret R, Foster R, Chenna A, Sperinde J, Winslow J, Rueda BR. HER2 over-expressing high grade endometrial cancer expresses high levels of p95HER2 variant. Gynecol Oncol. 2015; 137: 160-6. https://doi.org/10.1016/j.ygyno.2015.01.533.
92. Todeschini P, Cocco E, Bellone S, Varughese J, Lin K, Carrara L, Guzzo F, Buza N, Hui P, Silasi DA, Ratner E, Azodi M, Schwartz PE, et al. Her2/neu extracellular domain shedding in uterine serous carcinoma: implications for immunotherapy with trastuzumab. Br J Cancer. 2011; 105: 1176-82. https://doi.org/10.1038/bjc.2011.369.
93. Black JD, Lopez S, Cocco E, Bellone S, Altwerger G, Schwab CL, English DP, Bonazzoli E, Predolini F, Ferrari F, Ratner E, Silasi DA, Azodi M, et al. PIK3CA oncogenic mutations represent a major mechanism of resistance to trastuzumab in HER2/neu overexpressing uterine serous carcinomas. Br J Cancer. 2015; 113: 1020-6. https://doi.org/10.1038/bjc.2015.306.
94. Leslie KK, Sill MW, Lankes HA, Fischer EG, Godwin AK, Gray H, Schilder RJ, Walker JL, Tewari K, Hanjani P, Abulafia O, Rose PG. Lapatinib and potential prognostic value of EGFR mutations in a Gynecologic Oncology Group phase II trial of persistent or recurrent endometrial cancer. Gynecol Oncol. 2012; 127: 345-50. https://doi.org/10.1016/j.ygyno.2012.07.127.
95. Janiszewska M, Liu L, Almendro V, Kuang Y, Paweletz C, Sakr RA, Weigelt B, Hanker AB, Chandarlapaty S, King TA, Reis-Filho JS, Arteaga CL, Park SY, et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet. 2015; 47: 1212-9. https://doi.org/10.1038/ng.3391.
96. Lopez S, Cocco E, Black J, Bellone S, Bonazzoli E, Predolini F, Ferrari F, Schwab CL, English DP, Ratner E, Silasi DA, Azodi M, Schwartz PE, et al. Dual HER2/PIK3CA targeting overcomes single-agent acquired resistance in HER2-amplified uterine serous carcinoma cell lines in vitro and in vivo. Mol Cancer Ther. 2015; 14: 2519-26. https://doi.org/10.1158/1535-7163.MCT-15-0383.
97. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008; 8: 387-98. https://doi.org/10.1038/nrc2389.
98. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275: 1784-7.
99. Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988; 55: 619-25.
100. Dellinger TH, Planutis K, Jandial DD, Eskander RN, Martinez ME, Zi X, Monk BJ, Holcombe RF. Expression of the Wnt antagonist Dickkopf-3 is associated with prognostic clinicopathologic characteristics and impairs proliferation and invasion in endometrial cancer. Gynecol Oncol. 2012; 126: 259-67. https://doi.org/10.1016/j.ygyno.2012.04.026.
101. Myers A, Barry WT, Hirsch MS, Matulonis U, Lee L. beta-Catenin mutations in recurrent FIGO IA grade I endometrioid endometrial cancers. Gynecol Oncol. 2014; 134: 426-7. https://doi.org/10.1016/j.ygyno.2014.06.010.
102. Shih HC, Shiozawa T, Miyamoto T, Kashima H, Feng YZ, Kurai M, Konishi I. Immunohistochemical expression of E-cadherin and beta-catenin in the normal and malignant human endometrium: an inverse correlation between E-cadherin and nuclear beta-catenin expression. Anticancer Res. 2004; 24: 3843-50.
103. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013; 13: 11-26. https://doi.org/10.1038/nrc3419.
104. Sahasrabuddhe AA, Elenitoba-Johnson KS. Role of the ubiquitin proteasome system in hematologic malignancies. Immunol Rev. 2015; 263: 224-39. https://doi.org/10.1111/imr.12236.
105. Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014; 13: 889-903. https://doi.org/10.1038/nrd4432.
106. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014; 26: 455-64. https://doi.org/10.1016/j.ccell.2014.09.013.
107. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004; 432: 775-9. https://doi.org/10.1038/nature03155.
108. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007; 67: 9006-12. https://doi.org/10.1158/0008-5472.CAN-07-1320.
109. Kuhn E, Wu RC, Guan B, Wu G, Zhang J, Wang Y, Song L, Yuan X, Wei L, Roden RB, Kuo KT, Nakayama K, Clarke B, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst. 2012; 104: 1503-13. https://doi.org/10.1093/jnci/djs345.
110. Hoang LN, McConechy MK, Meng B, McIntyre JB, Ewanowich C, Gilks CB, Huntsman DG, Kobel M, Lee CH. Targeted mutation analysis of endometrial clear cell carcinoma. Histopathology. 2015; 66: 664-74. https://doi.org/10.1111/his.12581.
111. Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M, Makar R, Wood LD, Diaz LA Jr, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun. 2014; 5: 5006. https://doi.org/10.1038/ncomms6006.
112. Zhao S, Bellone S, Lopez S, Thakral D, Schwab C, English DP, Black J, Cocco E, Choi J, Zammataro L, Predolini F, Bonazzoli E, Bi M, et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2016; 113: 12238-43. https://doi.org/10.1073/pnas.1614120113.
113. McConechy MK, Hoang LN, Chui MH, Senz J, Yang W, Rozenberg N, Mackenzie R, McAlpine JN, Huntsman DG, Clarke BA, Gilks CB, Lee CH. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J Pathol Clin Res. 2015; 1: 173-85. https://doi.org/10.1002/cjp2.18.
114. Barbieri CE, Boysen G, Prandi D, Chae SS, Dahiya A, Nataraj S, Blattner M, Marotz C, Xu L, Huang J, Lecca P, Chhangawala S, Zhou P, et al. Abstract NG01: SPOP mutation is associated with genomic instability in prostate cancer. Cancer Research. 2015; 75: NG01-NG. https://doi.org/10.1158/1538-7445.am2015-ng01.
115. Zhang P, Gao K, Jin X, Ma J, Peng J, Wumaier R, Tang Y, Zhang Y, An J, Yan Q, Dong Y, Huang H, Yu L, et al. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-alpha protein turnover. Cell Death Dis. 2015; 6: e1687. https://doi.org/10.1038/cddis.2015.47.
116. Le Gallo M, Rudd M, Urick ME, Hansen N, Zhang S, Lozy F, Vidal Bel A, Batias-Guiu X, Boraddus R, Lu R, Levine K, Mutch D, Goodfellow P, et al. Somatic mutation profiles of clear cell endometrial tumors revealed by whole exome and targeted gene sequencing. Cancer. 2017; 123: 3261-8.
117. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363: 1532-43. https://doi.org/10.1056/NEJMoa1008433.
118. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016; 22: 128-34. https://doi.org/10.1038/nm.4036.