
INTRODUCTION
Genetically engineered mouse models (GEMM) provide a powerful in vivo system to test the role of genes mutated or overexpressed in human tumors [1]. Typically by overexpressing an oncogene or inactivating a tumor suppressor gene, normal murine cells can be transformed with histopathology similar to human cancers [2]. Despite the myriad of successful examples, mouse models have been frequently scrutinized with regards to their validity [3]. One key question is whether murine cancers and their human counterparts share similar tumorigenic processes which can be evaluated by comparing somatic mutational landscapes. Indeed, certain GEMMs have been found to harbor mutations in genes also mutated in human cancers [4, 5]. Next-generation sequencing (NGS) can comprehensively characterize the mutation landscape of cancers. However, NGS-based somatic mutation analysis in mice can be challenging due to complex mouse genetic background, especially when the matched normal DNA is not available [4].
Medulloblastoma (MB) is the most common malignant pediatric brain tumor that arises within the posterior fossa [6, 7]. Expression profiling of human MBs subdivides this cancer into four major molecularly distinct subgroups: Wingless (WNT), Sonic Hedgehog (SHH), Group 3 (G3) and Group 4 G4 [8-12]. Genetically engineered MB mouse models have been developed for Wnt [13], Shh [14-18] and G3 [19-22]. These subgroup-specific mouse models recapitulate the histopathology and gene expression profiles of their human counterparts [23] and can be utilized for screening and preclinical testing of therapeutics [24], which have been shown to increase the cure-rate of MB and quality of life of surviving patients [25-27]. However, very little is known about the role of additional somatic mutations involved in the process of tumorigenesis. To evaluate this in MB, we performed whole-exome sequencing (WES) to identify somatic mutations in 12 mouse tumors from 3 MB subgroup-specific murine models established at St. Jude Children’s Research Hospital: five Shh (MBS) [17], three Wnt (MBW) [13] and four G3 (MBM) [19]. A total of 64 somatic mutations were identified and experimentally validated, including 40 non-silent mutations predicted to cause amino acid changes. Survival data were available for the MB subgroup G3 mouse model - one mouse had a much shortened life than the other mice in the cohort. This mouse tumor had four genes with non-silent mutations, Lrfn2, Smyd1, Ubn2 and Wdr11. We hypothesized that the mutations to these four genes may have facilitated tumor growth, and evaluated the function of each of these genes on G3 MB progression using in vivo systems.
RESULTS
Whole exome sequencing of tumor samples identified putative somatic mutations
The murine MB models of three different subgroups were established as described previously [13, 14, 19, 28]. After whole-exome sequencing (WES), the reads were mapped to mouse genome (mm9) with at least 90% mapping rates across all samples (Supplementary Table 1). Most (11 of 12) samples had at least 80% coding bases covered by a minimum of 20x coverage. We did not retrospectively bank germline DNA needed for somatic mutation calling. To overcome this challenge, we went through extensive germline polymorphisms filtering process that removed both common mouse polymorphisms and rare variants found in other raw sequencing data of common laboratory mouse strains. The entire process of single nucleotide variations (SNVs) identification, filtering and validation is illustrated in Figure 1a. Specifically, the initial SNV calling was performed using Bambino [29] by comparing each tumor with publicly available normal sequences of C57BL/6 mouse strain [30]. With filtering and manual curation, 213 putative mutations remained. Amplicon sequencing by Sanger or MiSeq using tumor specimen and all available mice of common ancestors as control validated 62 somatic SNVs and two indels (Supplementary Table 2).
The overall mutation burden, 64 mutations in 12 mouse tumors or 5.3 mutations per tumor, is lower than previously reported for human MBs [8]. The number of somatic mutations per tumor ranged from 0 to 31, where the Wnt subgroup had higher burden than the Shh (p-value = 0.03) and G3 (p-value = 0.10) subgroups (Figure 1b). The final missense-to-silent mutation ratio was 1.7, and 40 mutations were predicted to cause changes in amino acid sequence. Due to the limited numbers of mice (3-5 per subgroup), no gene was recurrently mutated.
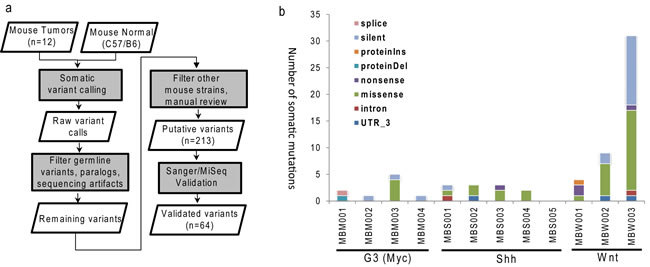
Figure 1: Identification of somatic mutations in MB mouse model. a. Schematic workflow for somatic single nucleotide variations identification and validation; b. Numbers of mutations and events of validated somatic mutations in each MB mouse tumor. G3 (MBM), Shh (MBS), Wnt (MBW).
Common non-synonymous mutations shared by MB mouse models and human MBs
To identify common mutations shared by mouse and human MBs, we compared the mutations identified from 12 mouse MBs to four independent human MB studies, consisting of a total of 366 MBs [8, 10, 11, 31]. Of the 40 mouse genes that carry amino-acid-changing mutations, 16 genes’ human orthologs were found to be mutated in human MBs. In seven genes, the exact type of mutation was found in the same subgroup of MB, including BAI3, DOCK7, LRFN2, MLL2, MLL3, PIWIL4 and WRD11 (Table 1). The most common mutations occurred in well-known MB genes KMT2D and KMT2C which contained 30 and 10 mutations in human MBs, respectively (Table 1). Remarkably, Kmt2d harbored a nonsense SNV predicted to cause loss of the SET domain which confers the methyltransferase activity [32, 33]. For comparison, the 366 human MBs harbored 30 KMT2D mutations, including 21 loss-of-function mutations (nonsense SNVs or frameshift indels) and nine missense mutations confirming the validity of the mouse models of MB. Specifically, 4 out of 48 KMT2D mutations occurred in WNT human MB subgroup, whereas one out of three WNT mice has a Kmt2d mutation (p = 0.015, hypergeometric test).
Table 1: Common non-synonymous mutations shared by mouse models and human MBs from four previous publications
gene |
MB mouse (n= 12) |
Lichter et al (n= 146) |
Parsons et al (n= 53) |
Pugh, Cho et al (n= 92) |
Robinson et al (n= 75) |
BAI3 |
WNT (1: M) |
- |
- |
G3 (1: M) |
WNT (1: M) |
DOCK7 |
WNT (1: M) |
WNT (1: M) |
- |
- |
- |
GAPVD1 |
WNT (1: M) |
- |
- |
G4 (1: N) |
- |
KIF14 |
WNT (1: M) |
G3 (1: M) |
- |
G3 (1: M) |
- |
LRFN2 |
G3 (1: M) |
- |
- |
- |
G3 (1: M) |
KMT2D |
WNT (1: N) |
WNT (3: M2,N) |
SHH,U,WNT (12: F6,M4,N2) |
G3,G4,SHH (10: N5,F3,M2) |
G3,SHH,U (5: F3,M,N) |
KMT2C |
WNT (1: M) |
G3,G4 (2: M,N) |
U,WNT (3: N3) |
G3,G4 (4: M3,S) |
U (1: N) |
MYO3A |
G3 (1: S) |
- |
- |
SHH (1: M) |
- |
OBSL1 |
WNT (1: M) |
- |
- |
SHH (1: M) |
- |
OR2K2 |
WNT (1: M) |
- |
- |
- |
SHH (1: N) |
PIWIL4 |
G3 (1: D) |
SHH (1: M) |
- |
- |
G3 (1: M) |
SLCO5A1 |
WNT (1: M) |
- |
- |
G4 (1: M) |
- |
SMYD1 |
G3 (1: M) |
- |
- |
- |
WNT (1: M) |
TNXB |
WNT (1: M) |
G3 (1: M) |
- |
G3 (1: M) |
G3 (1: M) |
UBN2 |
G3 (1: M) |
- |
- |
- |
SHH (1: M) |
WDR11 |
G3 (1: M) |
- |
- |
G3 (1: M) |
- |
First column - gene symbols; Columns 2 to 5 - mutations detected in the current MB mouse cohort, and in four previous publications. Detailed information includes: MB subgroup(s) where the mutations were detected; inside parenthesis ( ) represents the total number of mutations, and the breakdown by mutation class. M: missense, N: nonsense, S: splice variants, F: frameshift, D: in-frame deletion: a trailing number was added where more than one mutation were found in the medulloblastoma subgroup.
Potential secondary driver mutation suggested by survival data
We addressed the overall survival of mice with G3 MBs (Supplementary Table 3). Among four mice in this group, MBM003, which had the highest mutation burden (Figure 1b), succumbed to tumor-burden at least 15 days earlier than the other mice. This led to the hypothesis that this mouse may harbor deleterious mutations which accelerated tumor progression. There were four non-silent mutations in this tumor, all missense SNVs: Lrfn2 R656H, Smyd1 R237Q, Ubn2 Q549K and Wdr11 I46T.
To test the potential tumor suppressor function of the four candidate genes, we used lentiviral transduction to enforce the expression of each of these wild type genes or an empty vector as control, in the mouse G3 tumorsphere line # 19568 prior to implantation of 100,000 unsorted tumor cells into the cortices of CD1 nu/nu mice (Figure 2). Enforced expression of LRFN2 did not alter the survival of tumor-bearing mice (Figure 2a, upper left) despite its expression in resulting tumors (Figure 2b, upper left). Similarly, mice bearing mouse G3 MB tumor cells over-expressing Smyd1 or Ubn2 survived with similar latency compared to tumor cells transduced with empty vector (Figure 2a, upper right and lower left). In contrast, enforced expression of WDR11 led to a significant increase in survival of tumor-bearing mice compared to mouse G3 tumorspheres infected with an empty vector (Figure 2a, lower right). Specifically, enforced expression of WDR11 led to a 6-day increase in median survival of tumor-bearing mice. Analysis of these tumors showed increased WDR11 transcript levels (Figure 2b, lower right). Protein expression was observed in tumorspheres from secondary tumors, measured by immunofluorescence (Figure 2c). These data suggested that in murine G3 MB, WDR11 had tumor suppressive activity since its enforced expression delayed tumor progression. To confirm these results, we over-expressed WDR11 in two additional mouse G3 MB tumorsphere lines, # 9728 and # 19251. In these tumorsphere lines, over-expression of WDR11 gene (Figure 3b) and protein (Figure 3c) also resulted in a significant increase in survival of WDR11-expressing tumor-bearing mice compared to controls (p < 0.05) (Figure 3a). These data identify WDR11 or its downstream effectors as potential targets for therapy in G3 MB.
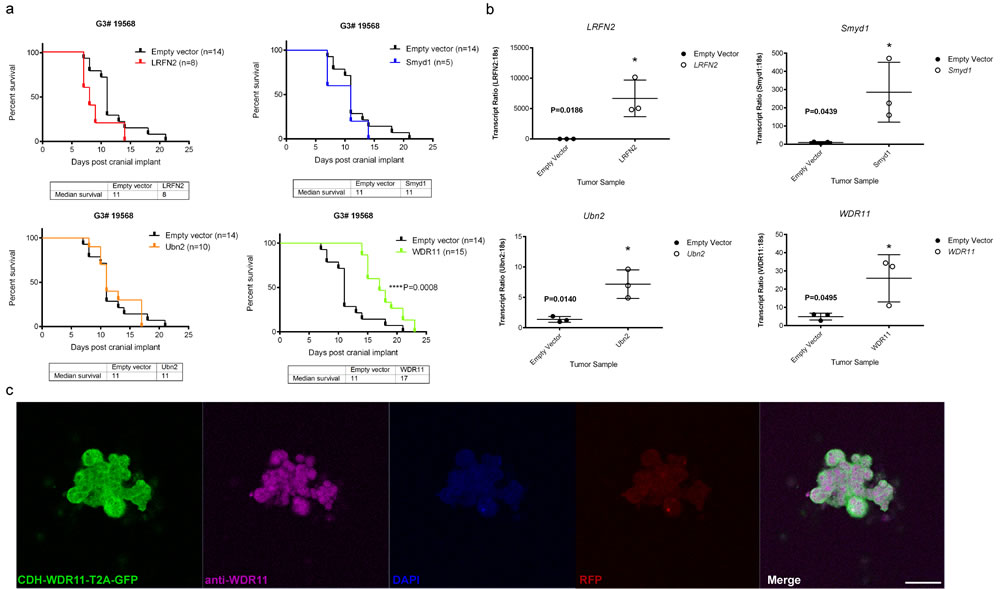
Figure 2: Functional study of genes mutated in mouse G3 MB. a. Survival of mice bearing mouse G3 MB tumorsphere line # 19568 overexpressing each of the four wild type genes; LRFN2 (red curve, top left panel), Smyd1 (blue curve, top right panel), Ubn2 (orange curve, bottom left panel), WRD11 (green curve, bottom right panel), or an empty vector as control for each panel (black curves); b. Relative qRT-PCR transcript ratios of LRFN2 (top left panel), Smyd1 (top right panel), Ubn2 (bottom left panel), and WDR11 (bottom right panel) in G3 MBs overexpressing each wild type gene (o) or an empty vector (•); c. Protein expression detected by immunofluorescence in tumorspheres derived from secondary tumors following intracranial implants of tumorsphere line #19568 (myc-IRES-RFP) transduced with lentiviruses encoding WDR11. Tumorspheres were immunostained with anti-GFP (Green) to visualize CDH-WDR11-T2A-GFP, anti-WDR11 detected with AlexaFluor 647 (purple), anti-RFP (red) to visualize Myc expression and nuclei counterstained with DAPI (blue). Scale bar = 20 µm.
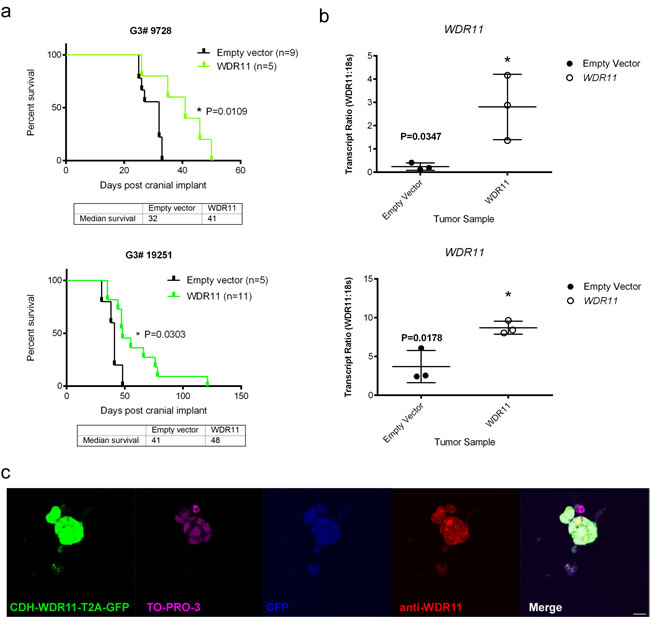
Figure 3: Survival of mice bearing mouse G3 MB overexpressing WDR11 in two additional G3 MB tumorsphere lines. a. Survival of mice carrying mouse G3 MB tumorspheres # 9728 (top panel, green curve) or # 19251 (bottom panel, green curve) with enforced expression of WDR11 compared to mouse G3 MB infected with an empty vector (black curves); b. Relative qRT-PCR transcript ratios of WDR11 in secondary tumors overexpressing WDR11 (o) or infected with an empty vector (•) c. Protein expression detected by immunofluorescence in tumorspheres derived from secondary tumors following intracranial implants of tumorsphere line # 9728 (Myc-IRES-CFP) transduced with lentiviruses encoding WRD11. Tumorspheres were immunostained with anti-GFP (green) to visualize CDH-WDR11-T2A-GFP, anti-WDR11 detected by AlexaFluor 594 (red), anti-RFP and nuclei counterstained with TO-PRO®-3 (purple). Endogenous CFP (blue) was imaged to detect expression in Myc-IRES-CFP. Scale bar = 20 µm.
WDR11 overexpression leads to down-regulation of the WNT signaling pathway
To further understand the underlying mechanism by which WDR11 overexpression contributed to the increased survival, we performed the whole transcriptome analysis on the mouse G3 tumors with and without WDR11 overexpression (Figure 4). While tumors with enforced WDR11 expression induced a different overall gene expression pattern compared to G3 MBs, the biological replicates showed good concordance with some degree of variability (Supplementary Figure 1). Differential expression analysis identifies 624 genes down-regulated and 346 genes up-regulated at least 2-fold (Figure 4a, 4b, Supplementary Table 4, FDR < 0.05). Gene set enrichment analysis showed that genes in the KEGG WNT signaling pathways were down-regulated in tumor cells overexpressing WDR11 (Figure 4c, 4d, Supplementary Figure 2, Supplementary Table 5). The genes in the leading edge of the GSEA included Myc, Ccnd1 and Ccnd2 (Figure 4e), consistent with the delay in tumor development in mice harboring G3 MB overexpressing WDR11.
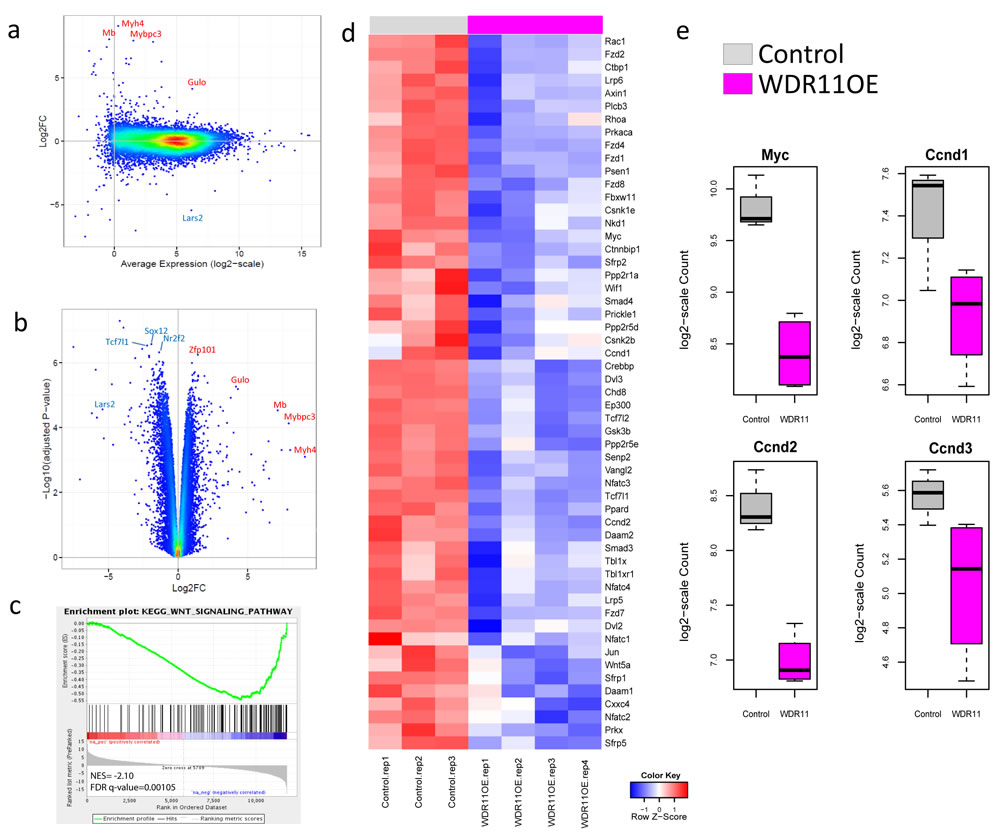
Figure 4: Over-expression of WDR11 induces down-regulation of WNT signaling. a. and b. M-A (log2FC vs Average Expression) plot and volcano plot (-log10 adjusted p-value against log2FC) for the comparison of WDR11 overexpressing mouse G3 MB cells vs those only carrying the empty vector; c., WNT signaling pathway is down-regulated in the WDR11 overexpressing cells through GSEA; d., heatmap showing the expression of genes in the leading edge of the WNT signaling pathway; e., expression of Myc, Ccnd1, Ccnd2 and Ccnd3 in mouse Group3 MB with and without WDR11 over-expression.
DISCUSSION
In the past few years, human MBs have been extensively characterized at the molecular level by whole genome sequencing, exome sequencing, gene expression profiling and DNA methylation array [8, 10, 11, 31, 34]. These studies highlighted the paucity of somatic mutations found in MB and the complete absence of mutations in a large proportion of tumors, particularly in G3 and G4 MBs that represent the majority of tumors. The mutation burden from the current MB mouse models was even lower than that reported in human MBs. A possible explanation for the lower mutation burden is that mouse tumors are established in a Trp53-null background and either MYC overexpression in G3 MBs, PTCH1 mutation in SHH MBs or B-CATENIN mutation in WNT MBs, all of which are found in corresponding human MB subgroups and sufficient to induce tumor development thus negating the requirement for additional driver mutations. Nevertheless, spontaneous mouse models of WNT and SHH and orthotopic models of G3 MB take 2-3 months to develop suggesting that additional mutations might be required to induce a full blown tumor.
One further question is whether the identified secondary mutations in mouse MBs match to the human MBs with similar background? In this study, we identified one Kmt2d nonsense mutation in the WNT subgroup. Remarkably, it has been reported that human WNT MBs also frequently harbor KMT2D nonsense or frameshift mutations, accounting for 16% of MB patients [30]. However, due to the limited sample size, we didn’t observe any shared mutations between human and murine SHH MBs. We previously analyzed and published chromosomal anomalies in orthotopic mouse SHH medulloblastoma derived from primary granule progenitors and found similar genomic alterations between mouse and human SHH MBs suggesting that these mouse models accurately model the human tumors [17, 35]. In future studies with additional samples and different types of data such as copy number, it will be interesting to evaluate other hallmark mutations.
Among the mutations identified in the mouse models of MB, epigenetic regulators appeared enriched compare to mutations in other biological pathways. In addition to Kmt2d, another mutated epigenetic histone modification gene was Smyd1, a histone methyltransferase that contains two known functional domains: MYND domain (myeloid translocation protein 8, Nervy, DEAF1) responsible for histone deacetylase-dependent transcriptional repression, and a SET domain [Su(var)3-9,enhancer-of-zeste,trithorax] that confers histone methyltransferase activity [32, 33]. Smyd1 has been reported to regulate endothelial cells [36] and skeletal muscle [37] and to be mutated in an indolent B cell non-Hodgkin lymphoma [38] but not in brain tumors or in cerebellar development. This suggests that either Smyd1 has a redundant function with other histone methyltransferases that compensate for its loss of function, or that this mutation is a passenger rather than a driver event. Indeed, in human MBs, inactivating mutations have been observed for KTM2B, KTM2C, KTM2D and SETD2, but not for other histone methyltransferases including SMYD1 [8, 10, 11, 31, 34].
Interestingly, the murine G3 MB carrying a mutant Smyd1 had a shortened life-span (Supplementary Table 3). In addition to Smyd1 mutation, this MB also carries mutation in three other genes (Lrfn2, Ubn2 and Wdr11). These genes belong to families that were previously found to be involved in neuronal development or chromatin-remodeling [39-42]. Since these mutations were expected to be deleterious, this suggested that they may have tumor suppressor activity and that enforced expression of the wild type gene would suppress G3 MB progression. Indeed, enforced expression of wild-type WDR11 (but not LRFN2, Ubn2, Smyd1) hindered G3 MB development, which is in-line with our hypothesis about its tumor suppressor activity. Over-expression of WDR11 resulted in the down-regulation of WNT signaling pathway and regulators of G1 progression including Myc and D-type cyclins, Ccnd1,2 and 3 consistent with the delay in tumor development and increased survival.
WDR11, previously named BRWD2 and then revised to WDR11 due to the lack of bromodomains, is co-localized with FGFR2 in tail-to-tail manner on chromosome 10q26 [43]. WDR11 is a member of the WD-repeat gene family and ubiquitously expressed in normal brain and glial tumors [43]. The WD-repeat superfamily of proteins are found in all eukaryotes and implicated in a wide variety of functions including apoptosis, its loss of function would increase proliferation [44, 45]. While the exact function of WDR11 function is unknown, the proteins in that family have been found to be involved in many biological processes. Structural analysis revealed 12 WD domains in WDR11, nine of them form two β propellers. WDR11 co-localizes and interacts with EMX1, a homeodomain transcription factor that participates in the development of the central nervous system during early development of the brain [46], and missense mutations disrupting EMX1-binding in WDR have led to Kallman Syndrome, a genetic condition causing puberty failure [39]. The MB mouse mutation is a missense SNV that changes codon 46 from isoleucine to threonine. The site is evolutionarily conservative across mammals. WDR11 was previously found to be mutated in one human MB [11] (Table 1). Another study also found WDR11 to be a tumor suppressor in glioblastoma (GBM) in which WRD11 located on chromosome 10q26 [43], is inactivated in a balanced reciprocal translocation t(10;19) in a region that show frequent loss of heterozygocity (LOH) in GBMs. As the cost decreases, sequencing genetically engineered mouse tumors is becoming an effective approach to identify additionally acquired driver mutations, which might be shared by human and mouse tumors and help unveil the common molecular mechanisms of cancer [5]. However, somatic mutation calling remains a challenge, especially when the matched normal DNA is unavailable. Even for inbred CD1 nu/nu mouse strains, the real genetic background are often found to be surprisingly complex [4], which could not be resolved by simply filtering out common SNPs. In the current analysis, we developed a robust germline filtering pipeline using publicly available whole-genome sequencing data of 18 common laboratory strains. In addition to filtering out SNPs identified from these strains, we also checked the raw sequencing data for the weak evidence of any mutation calls being present, which helped remove germline SNPs in difficult regions such as the ones with low coverage or poor quality. Our extensive germline filtering process allowed us to reduce the mutation list to a manageable size with good validation rate. However, it is also possible that real mutations might have been accidentally excluded due to potential over-filtering. In the future, it is still preferred to have a matched normal sample available for somatic mutation calling.
In conclusion, our experiments highlight the differences in number and identify of the somatic mutations found between human and mouse tumors despite their similarity in pathology and gene expression pattern. We also identified Wdr11 as a putative tumor suppressor in Group3 MB.
MATERIALS AND METHODS
Animal husbandry
[Ptch1+/-;Trp53-/-, Cdkn2c-/-], [Ptch1+/-;Trp53-/-, Cdkn2c-/-] (Uziel et al., 2005) and [Blbp-Cre;Ctnnb1+/lox(Ex3); Tp53flx/flx] (Gibson et al., 2010) mice develop spontaneous Shh and Wnt MBs. Mouse G3 MBs were generated by orthotopic transplantation of granule neuron progenitors (GNPs) purified from cerebella of 7 days old, P7, Trp53-/-; Cdkn2c-/- mice infected with Myc-encoding retroviruses into the cortices of 6-8- week-old CD-1 nu/nu mice (Charles River Laboratories), as previously described [19]. Mice were housed in an accredited facility of the Association for Assessment of Laboratory Animal Care in accordance with the NIH guidelines. The Institutional Animal Care and Use Committee of SJCRH approved all procedures in this study.
Exome-capture and illumina sequencing
Genomic DNA was extracted from mouse Shh and Wnt spontaneously occurring tumors and G3 orthotopic MBs using the Qiagen DNAeasy kit. DNA samples were submitted to the Pediatric Cancer Genome Project Validation Lab for exome sequencing. Paired end sequencing reads have been mapped to mouse reference genome mm9 assembly. The mapping statistics was obtained from ‘samtools’ and the coverage statistics was obtained via an in-house coverage analysis pipeline (covsum) based on the RefSeq annotation of gene coding region.
Identification of somatic mutations in mouse and mapping to human
Putative sequence variants including SNVs and indels were initially detected by running paired analyses using variation detection module of Bambino [29] with the following parameters: -min-flanking-quality 15 -min-alt-allele-count 2 -min-minor-frequency 0 -broad-min-quality 10 -mmf-max-hq-mismatches 15 -mmf-min-quality 15 -mmf-max-any-mismatches 20 -unique-filter-coverage 2 -min-mapq 1. A putative somatic sequence mutation was collected based on the following criteria: (1) Fisher’s exact test P value indicates that the number of reads harbouring the non-reference allele is significantly higher in tumour; (2) the non-reference allele frequency in tumour is > = 10%; Substitution variants are classified into four categories based on combination of their P value and sequence quality scores: High quality, high P value; high quality, low P value; low quality, high P value; low quality, low P value. P value refers to the P value of Fisher’s exact test comparing the distribution of the alternative allele in tumour and normal. High P value, P < 0.05; low P value, 0.05 < P < 0.10. A final review process re-maps and re-aligns the reads harbouring the non-reference allele to the reference genome to filter potential false positive calls introduced by mapping in repetitive regions and alignment artefacts. For putative somatic indels, the review process re-aligns all reads in tumour and normal at the indel site to a mutant allele template sequence constructed by substituting the wild-type allele with the indel. Presence of reads in normal sample that cover the mutant allele is considered a germline variant. Mouse gene symbols were converted to human ortholog gene symbols by using table “Orthology - Human vs. Mouse” from Mouse Genome Informatics (MGI), The Jackson Laboratory [47]. Gene symbols were mapped to geneID and RefSeq accession using “gene_info” and “gene2refseq” downloaded from NCBI (downloaded 04/29/2011). Afterwards, each mouse mutant variant was mapped to its human ortholog mRNA by running a local Blast to determine corresponding human mutation location.
To filter out germline SNPs, we first excluded any SNPs in public mouse germline SNP databases including: dbSNP build 128 [48], 18 common laboratory strains SNPs by The Wellcome Trust Sanger Institute [30], and Center for Genome Dynamics Mouse SNP Database from The Jackson Laboratory [49]. Afterwards, the remaining SNV calls were directly compared with the normal BAM files of the eighteen common laboratory strains by the Sanger Institute [30]. Putative mutations present in any of the eighteen strains were considered as non-somatic and excluded from further analyses. Remaining predictions were manually reviewed. Small insertions and deletions (indels) were identified in a similar manner. Overall, a total of 213 putative mutations, consisting of 207 SNVs and six Indels, were predicted and were sent for experimental validation.
Validating the somatic mutations with non-tumor mice
Putative somatic SNVs and indels went through experimental validation using either amplicon-based MiSeq or Sanger sequencing (Figure 1a). Since the matched germline DNA was not available, we used all available mice of common ancestors for the validation experiment. Mutations present in any of these control mice were considered as non-somatic. After excluding one uncovered mutation in the validation experiment, the validation rate for the remaining 212 predictions was 30% (64/212). The highest cause of false predictions was germline events, which accounted for 41% (88/212). After all, we identified and validated 62 somatic SNVs and two indels (Supplementary Table 2).
Subcloning of cDNAs and lentivirus production
The available murine and human cDNAs for the four genes of interest were purchased, amplified by polymerase chain reaction, and subcloned into pCDH cDNA cloning lentivectors (Systems Biosciences): human LRFN2 (Accession No. BC142616, Clone ID 40147341; GE Dharmacon); mouse Smyd1 (Accession No. BC076601, Clone ID 30609878; GE Dharmacon); mouse Ubn2 (Accession No. BC051458, Clone ID 6468017; GE Dharmacon) and human WDR11 (NM_018117; OriGene, SC113680). The homology between the human and mouse amino acid sequences of these genes were 95, 94, 90 and 94 percent, respectively. The cDNAs were amplified by PCR using the following primer sequences:. LRFN2_XbaI_F-TTCTAGAGTGACCAGACCATGGAGAC; LRFN2_NotI_R-TTGCGGCCGCGACCGTGCTCTCC ATCACCC; Smyd1_XbaI_F-ATTCTAGAGACTC TGAGATGACAATAGG; Smyd1_Not_R-TCGCGGCCGCGCTCCCCAGCCAC;
Ubn2_XbaI_F-TTTCTAGACAGAACAGTGGGGATGGCGGAGCCGCGC; Ubn2_NotI_R-TTGCGGCCGCCTGAGGTTTCCGTGGTAACTTAG; WDR11_XbaI_F-TACTCTAGAGCCACCGGGATGTTGCCCTACACA; WDR11_NotI_R-TTGCGGCCGCCTCTTCAATGGGTT
Wild type cDNAs of interest were subcloned into pCDH-EF1-MCS-T2A-copGFP (CD521A-1) and/or pCDH1-CMV-MCS-EF1-RFP (CD512B-1) using XbaI and NotI. The resulting lentiviral vectors were named: pCDH-EF1-LRFN2-T2A-copGFP, pCDH-EF1-Smyd1-T2A-copGFP, pCDH-EF1-Ubn2-T2A-copGFP, pCDH-EF1-WDR11-T2A-copGFP; pCDH1-CMV-LRFN2-EF1-RFP and pCDH1-CMV-Smyd1-EF1-RFP. High titer lentiviruses were produced transiently in 293T as previously described [50].
Tumor sphere cultures, lentivirus infection, orthotopic transplants and tumor harvest
Mouse G3 MBs were grown as tumorspheres as previously described [19]. G3 MB tumorspheres # 19568 and #19251 were derived from tumors that developed in CD1nu/nu mice implanted with cerebellar granule neuronal progenitors transduced with MSCV retroviruses expressing Myc and the green fluorescent protein (GFP) from an internal ribosomal entry site (IRES) (Myc-IRES-GFP). G3 MB tumorsphere # 9728 was derived from tumors explanted from mice implanted with cerebellar granule neuronal progenitors transduced with MSCV retroviruses expressing Myc and the cyan fluorescent protein (CFP) from an internal ribosomal entry site (IRES) (Myc-IRES-CFP). Tumorspheres were derived from each of the three tumors and grown in N2, B27, EGF and FGF supplemented neurobasal medium (“complete tumorsphere medium”0, as previously described [19]. Tumorspheres were dissociated and infected six times over a 2-day period in virus collection media supplemented with N2, B27, EGF and FGF. The following day, infected tumorspheres were suspended in Matrigel (BD Bioscience, San Jose) at a concentration of 100,000 tumorspheres/5µl Matrigel and transplanted into the cortices of CD1-nu/nu mice, as described previously [19, 51]. Infection efficiency was analyzed using flow cytometry for RFP or GFP expression. In all cases, lentiviral infection efficiency ranged from 40-80%. After transplant of virus-infected tumorspheres, mice were examined daily for symptoms of sickness (doming of the head, ataxia or reduced activity). When mice became moribund, tumors were isolated and grown on a coverslip embedded in a 1:2 ratio (Matrigel: complete tumor sphere media) for 48 hours to allow for sphere formation and fixed in 4% paraformaldehyde for 15 minutes at room temperature followed by 3 rinses in PBS prior to immunofluorescence staining. Tumor chunks or cell pellets were also collected and stored at -80◦C.
RNA extraction and quantitative real-time PCR
Total RNAs from tumorspheres and tumor chunks were extracted, reverse transcribed, and quantitative real-time PCR (qRT-PCR) for the following transcripts was conducted in accordance with manufacturer’s instructions using TaqMan RNA-to-Ct 1-step kit (ABI, 4392938) and TaqMan Gene Expression Assays [ABI, 43321182 with assay ID numbers of 4310893E (18s), Hs00608584_m1 (LRFN2), Mm00477663_m1 (Smyd1), Mm00723981_m1 (Ubn2), Hs00608584_m1 (WDR11)]. Relative levels of transcripts were quantified in triplicate. Ct values were normalized to 18s and to parent tumor sphere culture (relative value = 1).
Transcriptome sequencing and analysis
Total stranded RNA sequencing data were generated and mapped against mouse genome assembly NCBIM37.67 using the StrongArm pipeline described previously. Four murine G3 tumor samples overexpressing human WDR11 and three mouse G3 MB tumors expressing the empty vector control were characterized. The gene level quantification values were obtained with HT-seq [52] based on GENCODE annotation and normalized by TMM method with ‘EdgeR’ package [53]. Differential expression analysis was performed in ‘voom’ method in R ‘limma’ package [54]. Gene set enrichment analysis was carried out using GSEA with MSigDB [55].
Immunofluorescence and confocal microscopy
Tumorspheres were stained with antibodies to LRFN2 (Millipore, 1:250), SMYD1 (abcam, 1:250), UBN2 (Abgent, 1:250) or WDR11 (abcam, 1:250) and GFP or RFP (Abcam, 1:500). Tumorspheres were nuclear counterstained using DAPI or TO-PRO®-3 and immunostained cells were imaged using confocal microscopy. Images were acquired using a Zeiss 510 Meta point scanning confocal/multiphoton microscope equipped with 20X dry [0.8 numerical aperture (NA)], 40X oil immersion (1.3 NA) and 60X oil immersion (1.4 NA) objectives. The Argon single photon excitation laser was used to detect GFP at 488 nm, while the Helium/Neon laser detected RFP and AF594 at 543 nm and AF647 and TO-PRO®-3 at 633 nm. A femtosecond pulsed laser was used to excite and detect DAPI and CFP. 1024 x 1024 pixels optical sections were acquired (averaged four times) using the Zen 2009 software (version 5.5.0.433).
Statistical analysis
The Kaplan-Meier method was used to calculate the significance of mouse survival. Statistical analyses were performed in the GraphPad Prism software version 6.0.
Abbreviations
DNA, Deoxyribonucleic Acid; WNT, Wingless; SHH, Sonic Hedgehog; GSEA, Gene Set Enrichment Analysis; MB, medulloblastoma; G3, Group 3; GEMM, genetically engineered mouse models; NGS, Next-generation sequencing; WES, Whole-exome sequencing; MBS, Sonic hedgehog subgroup-specific murine medulloblastoma model; MBW, Wnt subgroup-specific murine medulloblastoma model; MBM, G3 subgroup-specific murine medulloblastoma model; mm9, University of California Santa Cruz mouse genome browser mm9 assembly; SNVs, Single nucleotide variations; cDNA, Complementary DNA; RFP, Red fluorescent protein; CFP, Cyan fluorescent protein; GFP, Green fluorescent protein; N2, N-2 supplement; B27, B-27 serum-free supplement; EGF, Epidermal growth factor; FGF, Fibroblast growth factor; PBS, Phosphate buffered saline; RNA, Ribonucleic acid; qRT-PCR, Quantitative real-time polymerase chain reaction; DAPI, 4’,6-Diamidino-2-Phenylindole (CAS name: 1H-Indole-6-carboximidamide, 2- [4-(aminoiminomethyl)phenyl]-, dihydrochloride); TO-PRO®-3, CAS name: Quinolinium,4- [3-(3-methyl-2(3H)-benzothiazolylidene)-1-propenyl]-1- [3-(trimethylammonio)propyl]-, diiodide; NA, Numerical aperture
Author contributions
Conception and design: L. Wei, M. Parker, G. Wu, J. Zhang, B.L. Murphy, M.F. Roussel
Development of methodology: L. Wei, M. Parker, G. Wu, J. Zhang, J. Easton, B.L. Murphy, M.F. Roussel
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): L. Wei, M. Parker, G. Wu, J. Zhang, B.L. Murphy, M.F. Roussel
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, Computational analysis): L. Wei, M. Parker, G. Wu, J. Zhang, B.L.Murphy
Writing, review, and/or revision of the manuscript: L. Wei, G. Wu, J. Zhang, B.L. Murphy, M.F. Roussel. All authors reviewed and commented on the manuscript.
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): L. Wei, M. Parker, G. Wu, J. Zhang, B.L. Murphy, M.F. Roussel
Study supervision: J. Zhang, M.F. Roussel, G. Wu
Data access
The RNAseq raw data and processed read counts files have been submitted to the Gene Expression Omnibus (GEO) under accession no. GSE101767.
ACKNOWLEDGMENTS
The authors thank Dr. Daisuke Kawauchi, Jose Grenet and Dana Farmer for Smyd1 and LRFN2 plasmid purification; Jose Grenet for RNA extraction of tumors; Dr. Richard Ashmun and staff for flow cytometric analysis; Dr. Christopher Calabrese and Shantel Brown for cranial implants and Dr. Yannan Ouyang for confocal imaging assistance.
CONFLICTs OF INTEREST
All authors have no conflict of interest to disclose.
FUNDING
This work was funded in part by the Pediatric Cancer Genome Project, including Kay Jewelers, a lead partner; NIH grant CA-096832; a core grant CA02165-29 (M.F. Roussel); Children’s Brain Tumor Foundation Award (M.F. Roussel); the George J. Mitchell Endowed Fellowship (B.L. Murphy); and the American Lebanese-Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital.
REFERENCES
1. Walrath JC, Hawes JJ, Van Dyke T, Reilly KM. Genetically engineered mouse models in cancer research. Adv Cancer Res. 2010; 106:113-64.
2. Politi K, Pao W. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol. 2011; 29:2273-81.
3. Klausner RD. Studying cancer in the mouse. Oncogene. 1999; 18:5249-52.
4. Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L, Cahan P, Klco JM, Welch JS, Li C, Payton JE, Uy GL, Varghese N, et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. The Journal of clinical investigation. 2011; 121:1445-55.
5. Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG, Payne-Turner D, Cheng J, Ma J, Qu C, Wu G, Song G, Huether RG, et al. Pax5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. 2015; 125:3609-17.
6. Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, Taylor RE, Bailey S, Clifford SC, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011; 121:381-96.
7. Ellison DW. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010; 120:305-16.
8. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012; 488:43-8.
9. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012; 123:465-72.
10. Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, Rausch T, Warnatz HJ, Ryzhova M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012; 488:100-5.
11. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012; 488:106-10.
12. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012; 488:49-56.
13. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, Finkelstein D, Pounds S, Weiss A, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010; 468:1095-9.
14. Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001; 61:513-6.
15. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002; 31:306-10.
16. Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, Olson JM. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004; 64:7794-800.
17. Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, Sherr SE, Plimmer S, Clifford SC, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005; 19:2656-67.
18. Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res. 2009; 7:33-40.
19. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW, Gilbertson RJ, Roussel MF. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012; 21:168-80.
20. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, Schmitt EM, Miller CR, Buckley AF, et al. An animal model of MYC-driven medulloblastoma. Cancer Cell. 2012; 21:155-67.
21. Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, Shih DJ, Hovestadt V, Zapatka M, Sturm D, Jones DT, Kool M, Remke M, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014; 511:428-34.
22. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, Lau J, Masic S, Nguyen K, Yakovenko S, Zhe XN, Gilmer HC, Collins R, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010; 24:1059-72.
23. Poschl J, Stark S, Neumann P, Grobner S, Kawauchi D, Jones DT, Northcott PA, Lichter P, Pfister SM, Kool M, Schuller U. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014; 128:123-36.
24. Morfouace M, Shelat A, Jacus M, Freeman BB 3rd, Turner D, Robinson S, Zindy F, Wang YD, Finkelstein D, Ayrault O, Bihannic L, Puget S, Li XN, et al. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell. 2014; 25:516-29.
25. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006; 7:813-20.
26. Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer. 2012; 118:1313-22.
27. Mabbott DJ, Spiegler BJ, Greenberg ML, Rutka JT, Hyder DJ, Bouffet E. Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. J Clin Oncol. 2005; 23:2256-63.
28. Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C, Zindy F, Roussel MF, Curran T, Gilbertson RJ, McKinnon PJ. A molecular fingerprint for medulloblastoma. Cancer Res. 2003; 63:5428-37.
29. Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics. 2011; 27:865-6.
30. Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellaker C, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011; 477:289-94.
31. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011; 331:435-9.
32. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000; 406:593-9.
33. Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, Lis JT, Allis CD, Reinberg D. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell. 2002; 9:1201-13.
34. Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, Milde T, Bourdeaut F, Ryzhova M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014; 25:393-405.
35. Zindy F, Uziel T, Ayrault O, Calabrese C, Valentine M, Rehg JE, Gilbertson RJ, Sherr CJ, Roussel MF. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007; 67:2676-84.
36. Ye X, Qian Y, Wang Q, Yuan W, Mo X, Li Y, Jiang Z, Xu W, Deng Y, Wan Y, Fan X, Wu X, Wang Y. SMYD1, an SRF-Interacting Partner, Is Involved in Angiogenesis. PLoS One. 2016; 11:e0146468.
37. Tan X, Rotllant J, Li H, De Deyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci U S A. 2006; 103:2713-8.
38. Peveling-Oberhag J, Wolters F, Doring C, Walter D, Sellmann L, Scholtysik R, Lucioni M, Schubach M, Paulli M, Biskup S, Zeuzem S, Kuppers R, Hansmann ML. Whole exome sequencing of microdissected splenic marginal zone lymphoma: a study to discover novel tumor-specific mutations. BMC Cancer. 2015; 15:773.
39. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, Ha KS, Itokawa Y, Meliciani I, Wenzel W, Lee D, Rosenberger G, Ozata M, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2010; 87:465-79.
40. Morimura N, Inoue T, Katayama K, Aruga J. Comparative analysis of structure, expression and PSD95-binding capacity of Lrfn, a novel family of neuronal transmembrane proteins. Gene. 2006; 380:72-83.
41. Wang CY, Chang K, Petralia RS, Wang YX, Seabold GK, Wenthold RJ. A novel family of adhesion-like molecules that interacts with the NMDA receptor. J Neurosci. 2006; 26:2174-83.
42. Banumathy G, Somaiah N, Zhang R, Tang Y, Hoffmann J, Andrake M, Ceulemans H, Schultz D, Marmorstein R, Adams PD. Human UBN1 is an ortholog of yeast Hpc2p and has an essential role in the HIRA/ASF1a chromatin-remodeling pathway in senescent cells. Mol Cell Biol. 2009; 29:758-70.
43. Katoh M, Katoh M. FGFR2 and WDR11 are neighboring oncogene and tumor suppressor gene on human chromosome 10q26. Int J Oncol. 2003; 22:1155-9.
44. Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994; 371:297-300.
45. Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci. 1999; 24:181-5.
46. Lichtneckert R, Nobs L, Reichert H. Empty spiracles is required for the development of olfactory projection neuron circuitry in Drosophila. Development. 2008; 135:2415-24.
47. Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic acids research. 2011; 39:D842-8.
48. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308-11.
49. Szatkiewicz JP, Beane GL, Ding Y, Hutchins L, Pardo-Manuel de Villena F, Churchill GA. An imputed genotype resource for the laboratory mouse. Mammalian genome. 2008; 19:199-208.
50. Vo BT, Wolf E, Kawauchi D, Gebhardt A, Rehg JE, Finkelstein D, Walz S, Murphy BL, Youn YH, Han YG, Eilers M, Roussel MF. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell. 2016; 29:5-16.
51. Ayrault O, Zhao H, Zindy F, Qu C, Sherr CJ, Roussel MF. Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer Res. 2010; 70:5618-27.
52. Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166-9.
53. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11:R25.
54. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47.
55. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545-50.