
INTRODUCTION
Colorectal cancer (CRC), with an estimated 1,360,602 newly diagnosed cases and 693,933 deaths in 2012, is the third-most common cancer in men and the second-most in women worldwide [1]. In China, CRC is the fifth-most common cancer in men and the fourth-most in women, with an estimated 376,300 newly diagnosed cases and 191,000 deaths in 2015 [2]. The cancerization of colon mucosal epithelial cells is a complex and multifactorial, gradual process, characterized by the accumulation of cancer-specific genetic and epigenetic alterations. Epigenetic DNA modifications, including aberrant DNA methylation, are recognized as major and causal epigenetic events that occur during CRC initiation [3]. Accumulating evidence suggests that the global hypomethylation of DNA might induce chromosomal instability and gene-specific hypermethylation can silence tumor suppressor genes, all of which might contribute to CRC formation.
Until now, studies have mainly focused on tumor-derived DNA methylation changes, and studied the relationship between DNA methylation status in tumor tissue and prognosis of various cancers including CRC. The aberrant methylated genes in tumor tissue are commonly involved in aspects of cell function such as cell cycle regulation (CDKN2A (also known as p16) and IGF2), DNA mismatch repair (MLH1 and MGMT), apoptosis (DAPK1), cell adhesion (CDH1) and signal transduction (APC and WIF1) in CRC [4–10]. It is clear that tumors do not develop as an isolated phenomenon in their target tissue, other organ systems including the immune system (such as peripheral blood leukocytes) are also involved in tumor initiation [11]. In addition, altered DNA methylation in peripheral blood leukocytes is associated with environmental exposures encountered throughout life [12, 13]. Meanwhile, several experiments in vitro showed that environmental factors may disrupt epigenetic balance and may cause methylation abnormalities, and these aberrations have been associated with cancer susceptibility [14, 15]. Therefore, DNA methylation alterations in leukocytes may reflect epigenetic modifications, environmental exposures, or interactions between these factors that increase disease susceptibility. However, only a few studies have assessed the association between DNA methylation in leukocytes and the risk of CRC or colorectal adenomas. Some studies focused on genomic methylation of leukocyte DNA in relation to the risk CRC [16–19] or colorectal adenomas [20, 21], and other studies have reported an association between gene-specific methylation in leukocytes and the risk of CRC [22–26] or colorectal adenomas [27–29]. In addition, recently, several studies have reported gene-specific methylation alterations in leukocytes from patients with various cancers, including bladder, breast, renal, and head and neck cancer [30–34]. The results of these studies did imply aberrant methylation of multiple genes in leukocytes might predispose toward susceptibility to CRC, like genetic variants of germline DNA. Therefore, we carried out this case-control study to investigate the associations between leukocyte-derived DNA methylation in a panel of 10 genes and the risk of CRC.
RESULTS
Main characteristics of the participants
Table 1 shows the basic characteristics of the cases and their matched controls. The CRC cases exhibited an overall lower body mass index than the controls. Of the 914 eligible DNA samples, methylation status of the 10 genes was successfully assessed in 856 samples (93.65%), which were included in the final analysis. The characteristics of demographic variables and questionnaire-derived variables before and after multiple imputations are listed in Supplementary Table 1.
Table 1: Main characteristics of colorectal cancer cases and controls
Characteristics |
Number of cases (%) |
Number of controls (%) |
P |
|
|---|---|---|---|---|
Total number |
428 |
428 |
||
Age |
Mean (SD) |
59.37 (10.30) |
59.36 (10.35) |
0.99 |
Gender |
Male |
266 (62.15) |
266 (62.15) |
1.00 |
Female |
162 (37.85) |
162 (37.85) |
||
BMI |
<18.50 |
28 (6.54) |
26 (6.07) |
<0.0001 |
18.5-24.00 |
219 (51.17) |
146 (34.11) |
||
24.0-28.00 |
150 (35.05) |
139 (32.48) |
||
≥28.00 |
31 (7.24) |
117(27.34) |
||
Tumour site |
Colon |
177 (41.35) |
- |
- |
Rectum |
251 (58.65) |
- |
- |
|
Pathological morphology |
Protruding type |
237 (55.37) |
- |
- |
Ulcerative type |
135 (31.54) |
- |
- |
|
Other types |
56 (13.09) |
- |
- |
|
Degree of differentiation |
Low |
43 (10.05) |
- |
- |
Medium |
350 (81.78) |
- |
- |
|
High |
34 (7.94) |
- |
- |
|
Unknown |
1 (0.23) |
- |
- |
|
Histological classification |
Adenocarcinoma |
395 (92.29) |
- |
- |
Other types |
33 (7.71) |
- |
- |
|
Dukes stage |
A-B |
231 (53.97) |
- |
- |
C-D |
157 (36.68) |
- |
- |
|
Unknown |
40 (9.35) |
- |
- |
BMI: body mass index; SD: standard deviation.
DNA methylation and CRC risk
CRC risk was significantly associated with the methylation of DAPK1, IGF2, MINT31, NEUROG1 and WIF1 (Table 2). A marginally significant association was observed for MGMT. For the methylation of APC, CDH1, p16 and MLH1, there were no significant differences between the cases and the controls regardless of adjustment. For MRS_10, compared with the subjects in the MRS-Low group (57.36% of participants), the subjects in MRS-High (11.80% of participants) and MRS-Medium groups (30.84% of participants) exhibited a 6.51-fold (95% CI, 3.77 to 11.27, P<0.0001) and 3.85-fold (95% CI, 2.72 to 5.45, P<0.0001) increased risk for CRC, respectively. Moreover, the CRC risk increased with increasing MRS_10 (Ptrend < 0.0001).
Table 2: Associations between methylation at individual genes, MRS_10 and the risk of CRC
DNA methylation statusa |
Cases (%)b |
Controls (%)b |
Crude OR |
95% CI |
P |
ORadjustedc |
95% CI |
P |
ORadjustedd |
95% CI |
P |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
APC |
Negative |
412 (96.26) |
419 (97.90) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
16 (3.74) |
9 (2.10) |
1.81 |
0.79-4.14 |
0.16 |
1.77 |
0.76-4.11 |
0.19 |
1.82 |
0.74-4.49 |
0.19 |
|
CDH1 |
Negative |
405 (94.63) |
392 (91.59) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
23 (5.37) |
36 (8.41) |
0.62 |
0.36-1.06 |
0.08 |
0.59 |
0.34-1.02 |
0.06 |
0.65 |
0.36-1.19 |
0.16 |
|
CDKN2A |
Negative |
419 (97.90) |
424 (99.07) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
9 (2.10) |
4 (0.93) |
2.28 |
0.70-7.45 |
0.17 |
2.26 |
0.68-7.51 |
0.18 |
1.99 |
0.56-7.04 |
0.29 |
|
DAPK1 |
Negative |
322 (75.23) |
385 (89.95) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
106 (24.77) |
43 (10.05) |
2.95 |
2.01-4.33 |
<0.0001 |
2.93 |
1.98-4.33 |
<0.0001 |
2.95 |
1.94-4.49 |
<0.0001 |
|
IGF2 |
Negative |
334 (78.04) |
389 (90.89) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
94 (21.96) |
39 (9.11) |
2.81 |
1.88-4.19 |
<0.0001 |
2.63 |
1.75-3.96 |
<0.0001 |
2.54 |
1.65-3.92 |
<0.0001 |
|
MGMT |
Negative |
365 (85.28) |
389 (90.89) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
63 (14.72) |
39 (9.11) |
1.72 |
1.13-2.63 |
0.01 |
1.48 |
0.96-2.27 |
0.08 |
1.82 |
1.00-2.60 |
0.05 |
|
MINT31 |
Negative |
409 (95.56) |
423 (98.83) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
19 (4.44) |
5 (1.17) |
3.93 |
1.45-10.62 |
0.01 |
4.02 |
1.47-10.99 |
0.01 |
4.27 |
1.52-12.05 |
0.01 |
|
MLH1 |
Negative |
412 (96.26) |
420 (98.13) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
16 (3.74) |
8 (1.87) |
2.04 |
0.86-4.82 |
0.11 |
1.94 |
0.81-4.65 |
0.14 |
1.72 |
0.68-4.34 |
0.25 |
|
NEUROG1 |
Negative |
363 (84.81) |
401 (93.69) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
65 (15.19) |
27 (6.31) |
2.66 |
1.66-4.26 |
<0.0001 |
2.61 |
1.62-4.21 |
<0.0001 |
2.57 |
1.55-4.25 |
<0.0001 |
|
WIF1 |
Negative |
354 (82.71) |
392 (91.59) |
1.00 |
1.00 |
1.00 |
||||||
Positive |
74 (17.29) |
36 (8.41) |
2.28 |
1.49-3.48 |
<0.0001 |
2.26 |
1.47-3.48 |
<0.0001 |
2.44 |
1.53-3.87 |
<0.0001 |
|
MRS_10 |
Low |
172 (40.18) |
319 (74.53) |
1.00 |
1.00 |
1.00 |
||||||
Medium |
177 (41.36) |
87 (20.33) |
3.77 |
2.75-5.18 |
<0.0001 |
3.66 |
2.66-5.05 |
<0.0001 |
3.85 |
2.72-5.45 |
<0.0001 |
|
High |
79 (18.46) |
22 (5.14) |
6.67 |
4.01-11.06 |
<0.0001 |
6.41 |
3.84-10.71 |
<0.0001 |
6.51 |
3.77-11.27 |
<0.0001 |
|
Ptrend |
<0.0001 |
|||||||||||
Medium or High |
256 (59.81) |
109 (25.47) |
4.36 |
3.26-5.83 |
<0.0001 |
4.22 |
3.14-5.66 |
<0.0001 |
4.39 |
3.19-6.05 |
<0.0001 |
CI: confidence interval; CRC: colorectal cancer; MRS: methylation risk score; OR: odds ratio.
a According to the Youden index, a 1% level of methylation was used as the cut-off value for APC, CDH1 and IGF2, and a 0% level of methylation was used as the cut-off value for CDKN2A, DAPK1, MGMT, MINT31, MLH1, NEUROG1 and WIF1. The positive indicates methylated status and the negative indicates unmethylated status. b The rates represent the percentages of all cases and controls, respectively. c ORs adjusted for age, gender and BMI. d ORs adjusted for age, gender, BMI, occupational physical activity, smoking, and consumption of coarse grains, fish stewed with brown sauce, fried food, leftovers and pork.
P values < 0.05 are in bold.
Subgroup analysis
For MRS_10, the MRS_High and the MRS-Medium groups conferred an increased risk of CRC among both men and women, although the effect was attenuated in women compared with men (Supplementary Table 2). Additionally, significant associations were observed between the methylation of DAPK1, IGF2, NEUROG1 and WIF1 and CRC risk in both men and women. However, the methylation of MGMT and MINT31 only displayed statistically significant associations with CRC in men.
The methylation of DAPK1, IGF2, NEUROG1 and WIF1 was associated with CRC risk in both the younger (<60 years) and older groups (≥60 years) (Supplementary Table 3), whereas the associations between the methylation of MGMT and MINT31 and CRC risk were significant only in the younger participants. Clearly significant associations were observed between the MRS-High and the MRS_Medium groups and CRC risk among both the younger and the older participants.
According to body-mass index (BMI), the MRS_High and the MRS-Medium groups conferred a similarly increased risk of CRC in both normal weight or underweight subjects (<24) and overweight or obese subjects. For individual gene-specific methylation, the methylation of DAPK1, IGF2, NEUROG1 and WIF1 was associated with CRC risk in both of the normal weight or underweight group (<24) and the overweight or obese group (≥24) (Supplementary Table 4), whereas the associations between the methylation of CDH1, MGMT and MINT31 and CRC risk were significant only in the overweight or obesity participants.
Interactions between EF and DNA methylation
The significant interaction between increased intake of fruit and the methylation of IGF2 displayed an antagonistic effect on the risk of CRC. Alternatively, the significant interaction between increased consumption of fish stewed with brown sauce and the methylation of CDH1 displayed a synergistic effect on the risk of CRC. Furthermore, the interactions between increased consumption of coarse grains and the methylation of IGF2 and between increased consumption of pork and the methylation of NEUROG1 demonstrated a marginally significant antagonistic effect on the risk of CRC (Table 3).
Table 3: Effects of interactions between environmental factors and gene methylation on the risk of colorectal cancer
Gene methylation |
Environmental factor |
Gene methylation |
Environmental factor |
||||||
|---|---|---|---|---|---|---|---|---|---|
Consumption of fruits (times/week) |
Consumption of coarse grains (g/week) |
||||||||
<2 |
≥2 |
Interaction |
<200 |
≥200 |
Interaction |
||||
IGF2 |
OReg (95% CI) |
ORia (95% CI) |
P |
IGF2 |
OReg (95% CI) |
ORia (95% CI) |
P |
||
Negative |
1 |
0.91(0.74-1.11) |
Negative |
1 |
0.69(0.57-0.84) |
||||
Positive |
3.72(2.08-6.66) |
1.64(1.00-2.71) |
0.35(0.14-0.88) |
0.0248 |
Positive |
5.44(2.67-11.08) |
1.50(0.95-2.38) |
0.44(0.18-1.10) |
0.0787 |
Consumption of stewed fish with brown sauce (times/week) |
Consumption of pork (g/week) |
||||||||
<1 |
≥1 |
Interaction |
<250 |
≥250 |
Interaction |
||||
CDH1 |
OReg (95% CI) |
ORia (95% CI) |
P |
NEUROG1 |
OReg (95% CI) |
ORia (95% CI) |
P |
||
Negative |
1 |
1.50(1.15-1.95) |
Negative |
1 |
1.13(0.91-1.41) |
||||
Positive |
0.31(0.15-0.66) |
2.00(0.81-4.96) |
3.82(1.11-13.15) |
0.0333 |
Positive |
3.33(1.62-6.86) |
1.90(1.06-3.39) |
0.39(0.14-1.07) |
0.0671 |
CI: confidence interval; OReg: odds ratio for the combined effect of the gene methylation pattern and the environmental factor; ORi: odds ratio for the interaction between the gene methylation pattern and the environmental factor.
a The ORi was adjusted for age, gender and BMI.
P values < 0.05 are in bold.
Pyrosequencing verification
For DAPK1 and MLH1, the methylation status obtained via MS-HRM was compared with the mean methylation level based on quantitative pyrosequencing and the results indicated that the MS-HRM results were well confirmed by the pyrosequencing results (Supplementary Figure 1). The Spearman correlation coefficients between these two methylation assessment techniques were high (r=0.7147, 95% CI, 0.6145 to 0.8077, P<0.0001 for DAPK1; r=0.6089, 95% CI, 0.3972 to 0.7392, P<0.0001 for MLH1), and the AUC was 0.8699 (95% CI, 0.7919 to 0.9479, P<0.0001) for DAPK1 and 0.9113 (95% CI, 0.8445 to 0.9782, P<0.0001) for MLH1. Additional results about the Bland-Altman plots are provided in the Supplementary Materials.
Performance of the MRS_10 model
We developed two basic models (including the MRS_10 model and the EF-only model) and a combined model (the combined EF and MRS_10 model). The AUC for the MRS_10 model was 69.04% (95% CI, 65.57 to 72.66%, P<0.0001), which represented significantly higher discrimination accuracy than any individual gene methylation (Figure 1). The AUC for the EF-only model was 72.94% (95% CI, 69.60 to 76.26%, P<0.0001). Notably, the combined EF and MRS_10 model yielded an AUC of 79.12% (95% CI, 76.22 to 82.15%, P<0.0001), representing an increase of 6.18% (95% CI, 3.99 to 8.54%, P<0.0001) compared to the EF-only model. Based on the NRI, the IDI and the AUC difference, the improvement of adding MRS_10 to the EF-only model was statistically significant (Supplementary Table 5). The NRI and the IDI for addition of the MRS_10 to the EF-only model was 14.72% (95% CI, 8.92 to 20.52%, P<0.0001) and 9.30% (95% CI, 7.36 to 11.23%, P<0.0001), respectively.
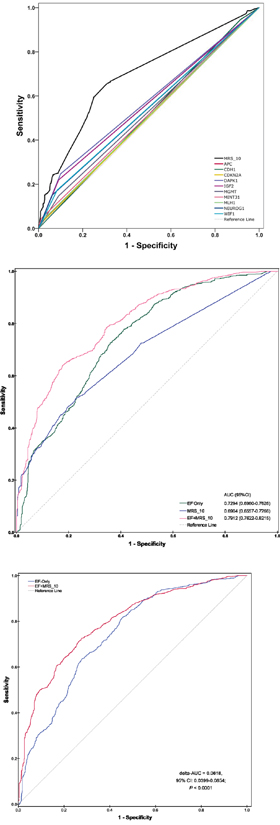
Figure 1: Receiver operating characteristic (ROC) curves and the corresponding area under the curves (AUC) analyses of prediction models of CRC risk. (A) The MRS_10 model versus any single gene methylation pattern. (B) Comparisons of the prediction accuracy by the EF-only model, the MRS_10 model and the combined EF and MRS_10 model. The straight dotted line (reference line), corresponding to an AUC of 0.5, indicates that the model is no more accurate than random classification in predicting CRC risk. (C) The combined EF and MRS_10 model versus the EF-only model. ROC curves and AUC analyses were computed for a reference prediction model (blue) and for an extended model (red) including additional risk predictors. Indicated are the increases in AUC (delta-AUC) obtained by adding the additional predictors.
Relationships between EF and DNA methylation
Based on comparisons of the higher MRS group (combining the MRS-Medium and MRS-High group) with the MRS-Low group, smoking increased the risk of MRS_10 in all the subjects and cases but not in the controls (Supplementary Table 6). Considering the methylation of individual genes, we found that smoking and high consumption of leftover were significantly associated with DAPK1 hypermethylation in all the subjects; the associations remained marginally significant in the cases but not in the controls. High consumption of pork was significantly associated with NEUROG1 hypermethylation in all the subjects and controls but not in the CRC cases. High consumption of fish stewed with brown sauce was significantly associated with CDH1 hypermethylation only in the CRC cases.
DISCUSSION
This study found that MRS_10 were strongly associated with the risk of CRC, indicating a positive relationship between leukocyte-derived DNA methylation of the panel of 10 genes and CRC risk. By using of MRS_10, compared with the MRS-Low group, subjects with MRS-High and MRS-Medium showed 6.51-fold and 3.85-fold increased risk of CRC, respectively. Stratified analyses demonstrated that these significant associations retained in both men and women, younger and older, and normal weight or underweight and overweight or obese subjects.
The findings demonstrated that the DNA methylation pattern in peripheral blood leukocytes is a detectable biomarker for CRC risk assessment. This observation is similar to those results for other cancers reported in previous studies [31–34] suggesting that DNA methylation alterations in peripheral blood are potential biomarkers for risk prediction.
The gene involved in the study illustrated a wide range of cancer related cellular events, for example, CDH1 encoded a member of the family of cell adhesion, and abrogation in its expression have been involved in unregulated growth and invasion of adjacent tissues in carcinogenesis [35]. Likewise, as a tumor suppressor gene, CDKN2A played an important control role in cell cycle regulating during the G1 phase [36]. DAPK1 was a positive mediator of apoptosis which executed juncture of cell death signaling, and its loss or inactivation has been linked to human tumor [37]. WIF1 was a secreted inhibitory factor of Wnt pathway, which played a pivotal role in blockade of Wnt signaling and induced apoptosis in colorectal cancer cells [10]. A growing body of evidence suggested that aberrant hypermethylation of the promoter can lead to reduced expression of these genes including APC [9], CDH1 [38], CDKN2A [39], DAPK1 [40], IGF2 [41], MGMT [42], MLH1 [43], NEUROG1 [44] and WIF1 [45]. In contrast to the methylation status of a single gene, the MRS, which summarized the data for the methylation of multiple genes, might demonstrate systematically altered gene methylation profiles in subjects and might more comprehensively represent the susceptibility of an individual to CRC. In addition, sensitivity analyses by omitting each individual locus showed a robust association between the MRS and the risk of CRC (Supplementary Table 7).
The exact mechanism underlying the alterations in the methylation of peripheral blood-derived DNA among individuals who are susceptible to CRC remains unclear. Alterations in leukocyte-derived DNA methylation may reflect a response of the hematopoietic system to tumorigenesis and may be partially explained by systemic differences in the methylation signatures of leukocyte subpopulations during tumorigenesis [46]. The leukocyte-derived DNA methylation profiles represented the overall methylation status; nevertheless, isolation of specific cell subpopulations is difficult in epidemiologic studies [47]. Additionally, in the present case-control study, it is not possible to determine the etiologically relevant time windows of DNA methylation or confirm the temporal sequence of DNA methylation and CRC occurrence. Although we cannot definitely determine whether these methylation alterations in peripheral blood-derived DNA represent an early response of the hematologic system to the presence of tumor cells or appear before tumor development [31], the observed significant association between gene-specific DNA methylation in leukocytes and CRC risk is likely to be useful in identifying the population at high risk of CRC.
Importantly, circulating tumor cell DNA is unlikely to interfere with the results of leukocyte-derived DNA methylation because the number of circulating tumor cells is negligible and the effects of their aberrant methylation can be excluded [32, 48]. Another potential source of support of our findings is that accumulating studies have shown no significant correlation between tissue and blood DNA methylation [49, 50]. We also compared the DNA methylation levels of six genes in peripheral-blood leukocytes and colorectal tumor tissues from 217 CRC patients; no statistically significant correlations were observed (Supplementary Table 8).
The potential biological mechanism for EF interfering with DNA methylation processes in carcinogenesis is complex and variable. Several researchers have suggested the plausible mechanisms, including that DNA methylation requires methyl group donors such as S-adenosylmethionine (SAM), while environmental factors may affect SAM synthesis and alter DNA methyltransferase (DNMT) activity [51]. In this study, we found that smoking was associated with DNA hypermethylation, which was consistent with several previous studies [52, 53]. Smoking can induce DNMT1 overexpression and subsequently result in hypermethylation of the promoter of tumor suppressor genes, which could lead to tumorigenesis [54]. The molecular mechanism underlying the link between consumption of pork or stewed fish with brown sauce and DNA methylation remains unclear. Saturated fatty acids and heterocyclic amines, which form during the high-temperature cooking of pork and fish, may partly account for the aberrant hypermethylation [55, 56]. Alteration of DNA methylation is a gradual and reversible process, thus, there is a critical window of opportunity through which we might inhibit or reverse the process and counteract cancer by making changes in diet and lifestyle [57].
We initially used MS-HRM to assess DNA methylation status. MS-HRM technology has been shown to reliably and accurately evaluate low-level methylation [58]. By comparing the estimates of methylation of DAPK1 and MLH1 based on MS-HRM and pyrosequencing, we found that the MS-HRM results were highly consistent with the pyrosequencing results. This consistency was also supported by a previous study that analyzed APC and CDKN2A methylation and found a high correlation between the results from these two techniques [59]. Only a small subset of genes and participants were analyzed via pyrosequencing because of its high cost. From a cost perspective, the assessment of peripheral blood-derived DNA methylation via MS-HRM might be an efficient strategy for the early detection of individuals who are at high risk for CRC.
In this study, we developed the weighted MRS_10 that combined these 10 genes in a leukocyte-derived DNA methylation marker panel and found that the AUCs for the MRS_10 model and the combined EF and MRS_10 model reached 0.6904 and 0.7912, respectively. Compared to the EF-only model with a similar AUC to those reported in previous studies (AUCs ranging from 0.61 to 0.76) [60–69], adding the MRS_10 significantly improved the discriminatory performance, as demonstrated by a 0.0618 units of AUC gain. However, the combined EF and MRS model is needed to be further validated in future prospective studies. To evaluate whether adding the MRS_10 to the EF-only model improved risk prediction performance, we assessed the accuracy of the combined model in classifying individuals as cases or controls based on the NRI and the IDI, which are useful statistics that have gained increasing acceptance for the evaluation of new biomarkers and risk models [70, 71]. Adding MRS_10 to the EF-only model resulted in the reclassification of 14.72% of the subjects into more accurate risk categories. This advancement might improve the selection of those who require more frequent screening and shorter follow-up intervals. However, NRI is sensitive to arbitrary cut-off values [72]. Therefore, we reset the cut-off points to 0.3, 0.4, 0.6 or 0.7 and found similar NRIs (Supplementary Table 9), which implied that the discriminatory improvement was robust across the cut-off values used in our study.
Our study has several additional limitations. First, the current panel of gene methylation sites incorporated into the calculation of MRS_10 might not be ideal because we have identified the genes from published studies rather than next-generation sequencing, which would likely reveal additional methylation biomarkers. Therefore, the present MRS_10 models in this study must be updated. Another potential limitation of this study is the restriction of EFs included in the combined EF and MRS_10 model. The age and gender matching design inherent to our present study removes two CRC-associated variables. Additionally, the discriminatory performance of the combined model might be improved by including additional factors such as colonoscopy or sigmoidoscopy findings.
In summary, the MRS_10 combining a panel of 10 gene-specific DNA methylation in leukocytes seemed to be a promising and robust risk prediction tool and might be a useful marker of susceptibility for CRC. The MRS_10 may be useful for the identification of individuals who are at high risk of developing CRC. However, our results should be further validated in future studies.
MATERIALS AND METHODS
Study population
We included primary sporadic CRC cases diagnosed at the Third Affiliated Hospital (from June 2004 to May 2005 and May 2007 to January 2008) and the Second Affiliated Hospital of Harbin Medical University (from October 2010 to December 2011) in Harbin, China. Cancer-free controls were selected contemporaneously from the Second Affiliated Hospital of Harbin Medical University by individual matching each case according to gender and age (±2 years). All participants were Chinese. All CRC cases were newly diagnosed, histologically confirmed, and alive at the time of initial contact; and the exclusion criteria included subjects with metastatic colorectal carcinoma, adenomatous polyposis coli, or a family history of CRC in first-degree relatives according to the Amsterdam criteria [73]. The participants were interviewed face-to-face to complete a structured standard questionnaire, which was partially adopted from the report by Shu et al [74]. The questionnaire queried information on demographic characteristics and potential risk factors for CRC, including family history, smoking, alcohol drinking, occupational physical activity, and diet consumption. Dietary consumption over the past year was assessed using a validated food frequency questionnaire (FFQ) [74]. The FFQ included 9 major food groups, which represent most of the common foods in Northeast China. The food items included coarse grains, dairy products, fish stewed with brown sauce, fried food, fresh fruits, green vegetables, leftover, pork, and soybean products.
Through professional training, the investigators made efforts to reduce recall and investigation bias. All study subjects gave informed consent, and appropriate ethical approval for sample collection was obtained from the Ethics Committee of Harbin Medical University prior to the study. Peripheral blood (5 milliliters) was donated before chemotherapy or adjuvant radiotherapy and was stored in a freezer. The sample size estimation was presented in the Supplementary Materials. In total, 553 eligible patients were recruited; 32 did not complete the questionnaire, and another 19 did not provide sufficient blood samples. Therefore, 502 extracted DNA samples were available for the CRC cases, with a response rate of 90.78%. Similarly, 1,210 subjects were eligible as controls, and 1,083 provided complete questionnaire data and blood samples with a response rate of 89.50%. Because eligible matched controls could not be found for 45 cases, 457 controls were ultimately enrolled.
Genomic DNA extraction and bisulfite modification
DNA was extracted from buffy coats using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and was then bisulfite-modified using the EpiTect Plus DNA Bisulfite Kit (Qiagen) according to the manufacturer’s protocols. Detailed methods are provided in the Supplementary Materials.
Methylation-sensitive high-resolution melting (MS-HRM) assays
Fourteen CRC-related genes, including tumor suppressor genes such as APC, CDH1, CDKN2A (also known as p16), CDKN2B (also known as p15), DAPK1, GSTP1, MGMT, MLH1, PTEN and WIF1 and CpG island methylator phenotype-related markers such as IGF2, APBA1 (also known as MINT1), MINT31 and NEUROG1, were selected according to review of literatures. Eight primer pairs were selected from previously published studies [75–81]; the remaining primers were designed using Methprimer software [82]. The primer sequences used are listed in Supplementary Table 10. The PCR mixture consisted of a total volume of 10 μl containing 2× LightCycler 480 High Resolution Melting Master Mix (Roche Applied Science, Mannheim, Germany), 3 mM MgCl2, 0.2-0.4 μM of each primer and approximately 10 ng of bisulfite-modified template DNA. PCR amplification and MS-HRM analyses were performed using the LightCycler 480 platform (Roche), and the resulting data were analyzed using software module of Gene Scanning (Roche). A set of methylation standards (100, 25, 10, 5, 2, 1, and 0% methylated DNA) were prepared by mixing commercially available methylated and unmethylated DNA (Zymo Research); these standards were used to semi-quantitatively measure the DNA methylation level in the samples. Normalized melting curves of MS-HRM assays for each gene were shown in Supplementary Figure 2. In addition, a blank control (non-template control) sample was included in each batch, and all reactions were performed in duplicate. A third trial was conducted for the samples that presented inconsistent results between the two trials.
Two investigators (Y.L. and Y.W.) blinded to outcome and other predictive variables assessed the MS-HRM results, and discrepancies were resolved by discussion and consensus with another investigator (Y.Z.). The DNA methylation pattern was first tested in 167 cases and their matched controls. Four genes displaying no abnormal methylation (PTEN, GSTP1, APBA1 and CDKN2B) were tested no further and were excluded from the analysis. Finally, the other 10 genes were assessed in all 457 cases and their matched controls.
Pyrosequencing verification
To verify the results of MS-HRM, we performed pyrosequencing in a subset of the samples (further details are provided in the Supplementary Materials and the primer sets are listed in Supplementary Table 11).
Methylation risk score (MRS) computation
First, we coded each gene methylation status as 0 or 1 for non-methylation or methylation, respectively, according to the optimal cut-off value as determined by the Youden index (J = max {sensitivity + specificity – 1}) using the receiver operating characteristic (ROC) curve analysis. Then, we computed the weighted MRS for each individual by multiplying the methylation status by the β-coefficients for each gene included in the model and then dividing by the number of genes included in the model. Simply, the weighted MRS was computed using the following equation:
Where βi is the β-coefficient for gene i, xi is the methylation status of the same gene i (0 or 1), and k is the total number of genes included in the model. According to the formulation listed above, we generated MRS_10 comprehensively considering the methylation status of all the 10 genes. Using the MRS_10 model, the subjects were classified as follows: MRS-Low, predicted probability ≤0.5; MRS-Medium, 0.5< predicted probability ≤0.7; or MRS-High, predicted probability >0.7.
Missing data analysis and imputation
Definitions of questionnaire-derived variables are provided in the Supplementary Materials. All questionnaire-derived variables were analyzed via missing value analysis and were imputed via multiple imputation (further details are provided in the Supplementary Materials).
Model development and performance
We selected CRC-associated risk factors by using of multivariable logistic regression model with backward conditional selection method (P values of 0.05 and 0.10 were specified as the thresholds for entry and removal of variables, respectively), and we further included gender and age in the final model. Interactions between environmental factors (EF) and gene methylation were also explored, but we did not included interaction terms in the models because interaction terms rarely add to the predictive ability of the model [83, 84]. A multivariable logistic regression model was used to fit the CRC risk prediction models: Model 1 was the MRS_10 model; Model 2 was an EF-only model, which exclusively contained questionnaire-derived risk factors; and Model 3 was a combined model incorporating EF and MRS_10. For each logistic regression model, the regression coefficient estimates were corrected according to the pooled imputed datasets. The estimates of regression coefficients for each model were listed in Supplementary Table 12.
To assess the discriminatory accuracy of each model, we generated ROC curves and calculated the areas under ROC curves (AUCs). The 95% CI for the AUC were estimated using the bootstrapping method (1000 replicates). We used the method of DeLong [85] to assess the differences in the AUC between the models. We further calculated the categorical net reclassification improvement (NRI), the integrated discrimination improvement (IDI) to evaluate the improvement in the discriminatory accuracy of the model (considering 0.5 as the cut-off point).
Statistical analysis
The differences in characteristics between the cases and the controls were tested using Student’s t-test for continuous variables or the χ2 test for categorical variables. We used both univariable and multivariable logistic regression models to estimate odds ratios (ORs) and 95% confidence interval (CI) for the associations between gene-specific DNA methylation or EF and CRC risk. The effects of interactions between EF and the methylation status at individual genes or MRS_10 on the risk of CRC were evaluated on a multiplicative scale with a product-term coefficient using multivariable logistic regression models. Additionally, to assess the associations between EF and DNA methylation among all the subjects, we used multivariable logistic regression models that included age, gender, BMI and case-control status that allowed us to adjust for the potential influence from case-control status. We also assessed the associations by case and control separately. All statistical tests were two-sided except for the test of the increase in AUC (one-sided) and statistical significance was defined as P values of less than 0.05. Missing value analysis, multiple imputations and multivariable logistic regression analysis were performed using SPSS Statistics version 19.0 (IBM, Inc., USA). The 95% CIs for the AUC and the NRI and the IDI were estimated using the pROC package and the PredictABEL package in R software version 2.15.3, respectively. All other statistical analyses were performed using SAS software version 9.1 (SAS Institute Inc., USA).
Author contributions
Y.S.Z., Y.P.L. and Y.B.W. contributed to study conception and design; Y.P.L., Y.B.W., F.L.H., H.R.S. Z.M.Z., X.W., and X.Luo. contributed to DNA preparation and bisulfite modification; Y.P.L, Y.B.W., F.L.H., H.R.S. Z.M.Z., X.W., Y.L., X.Li., R.H. and S.Q.L. contributed to MS-HRM and pyrosequencing;, Y.P.L., Y.B.W. and Y.S.Z. contributed to collection and assembly of data; Y.P.L., Y.B.W., F.L.H., H.R.S., L.Z., G.X.L., X.Li., F.W., Y.H.L., J.S.R., H.P.Y., and Y.S.Z. contributed to the analysis and interpretation of data; Y.P.L., Y.B.W., Y.H.L., J.S.R., H.P.Y. and Y.S.Z. contributed to sample collection; Y.P.L., Y.B.W. and Y.S.Z. contributed to the drafting of the initial versions of the manuscript; all authors contributed to the review and final approval of the manuscript; Y.S.Z. was responsible for study supervision.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant numbers 81473055, 30972539).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016; 66:115-132.
3. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007; 128:683-692.
4. Park SJ, Rashid A, Lee JH, Kim SG, Hamilton SR, Wu TT. Frequent CpG island methylation in serrated adenomas of the colorectum. Am J Pathol. 2003; 162:815-822.
5. Cheng YW, Idrees K, Shattock R, Khan SA, Zeng Z, Brennan CW, Paty P, Barany F. Loss of imprinting and marked gene elevation are 2 forms of aberrant IGF2 expression in colorectal cancer. Int J Cancer. 2010; 127:568-577.
6. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006; 38:787-793.
7. Lee S, Hwang KS, Lee HJ, Kim JS, Kang GH. Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab Invest. 2004; 84:884-893.
8. Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 2001; 48:367-371.
9. Galamb O, Kalmar A, Peterfia B, Csabai I, Bodor A, Ribli D, Krenacs T, Patai AV, Wichmann B, Bartak BK, Toth K, Valcz G, Spisak S, et al. Aberrant DNA methylation of WNT pathway genes in the development and progression of CIMP-negative colorectal cancer. Epigenetics. 2016; 11:588-602.
10. He B, Reguart N, You L, Mazieres J, Xu Z, Lee AY, Mikami I, McCormick F, Jablons DM. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene. 2005; 24:3054-3058.
11. Marsit C, Christensen B. Blood-derived DNA methylation markers of cancer risk. Adv Exp Med Biol. 2013; 754:233-252.
12. Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, Sugarbaker DJ, Yeh RF, Wiencke JK, Kelsey KT. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009; 5:e1000602.
13. Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010; 20:440-446.
14. Huang YC, Hung WC, Chen WT, Yu HS, Chai CY. Sodium arsenite-induced DAPK promoter hypermethylation and autophagy via ERK1/2 phosphorylation in human uroepithelial cells. Chem Biol Interact. 2009; 181:254-262.
15. Qin W, Zhu W, Shi H, Hewett JE, Ruhlen RL, MacDonald RS, Rottinghaus GE, Chen YC, Sauter ER. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr Cancer. 2009; 61:238-244.
16. Kitkumthorn N, Tuangsintanakul T, Rattanatanyong P, Tiwawech D, Mutirangura A. LINE-1 methylation in the peripheral blood mononuclear cells of cancer patients. Clin Chim Acta. 2012; 413:869-874.
17. Huang WY, Su LJ, Hayes RB, Moore LE, Katki HA, Berndt SI, Weissfeld JL, Yegnasubramanian S, Purdue MP. Prospective study of genomic hypomethylation of leukocyte DNA and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2012; 21:2014-2021.
18. Walters RJ, Williamson EJ, English DR, Young JP, Rosty C, Clendenning M, Walsh MD, Parry S, Ahnen DJ, Baron JA, Win AK, Giles GG, Hopper JL, et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Epigenetics: official journal of the DNA Methylation Society. 2013; 8:748-755.
19. Nan H, Giovannucci EL, Wu K, Selhub J, Paul L, Rosner B, Fuchs CS, Cho E. Pre-diagnostic leukocyte genomic DNA methylation and the risk of colorectal cancer in women. PLoS One. 2013; 8:e59455.
20. Lim U, Flood A, Choi SW, Albanes D, Cross AJ, Schatzkin A, Sinha R, Katki HA, Cash B, Schoenfeld P, Stolzenberg-Solomon R. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women. Gastroenterology. 2008; 134:47-55.
21. King WD, Ashbury JE, Taylor SA, Tse MY, Pang SC, Louw JA, Vanner SJ. A cross-sectional study of global DNA methylation and risk of colorectal adenoma. BMC Cancer. 2014; 14:488.
22. Ally MS, Al-Ghnaniem R, Pufulete M. The relationship between gene-specific DNA methylation in leukocytes and normal colorectal mucosa in subjects with and without colorectal tumors. Cancer Epidemiol Biomarkers Prev. 2009; 18:922-928.
23. Kaaks R, Stattin P, Villar S, Poetsch AR, Dossus L, Nieters A, Riboli E, Palmqvist R, Hallmans G, Plass C, Friesen MD. Insulin-like growth factor-II methylation status in lymphocyte DNA and colon cancer risk in the Northern Sweden Health and Disease cohort. Cancer Res. 2009; 69:5400-5405.
24. Miroglio A, Jammes H, Tost J, Ponger L, Gut IG, El Abdalaoui H, Coste J, Chaussade S, Arimondo PB, Lamarque D, Dandolo L. Specific hypomethylated CpGs at the IGF2 locus act as an epigenetic biomarker for familial adenomatous polyposis colorectal cancer. Epigenomics. 2010; 2:365-375.
25. Gao Y, Killian K, Zhang H, Yu K, Li QZ, Weinstein S, Virtamo J, Tucker M, Taylor P, Albanes D, Meltzer P, Caporaso N. Leukocyte DNA methylation and colorectal cancer among male smokers. World J Gastrointest Oncol. 2012; 4:193-201.
26. Luo X, Huang R, Sun H, Liu Y, Bi H, Li J, Yu H, Sun J, Lin S, Cui B, Zhao Y. Methylation of a panel of genes in peripheral blood leukocytes is associated with colorectal cancer. Sci Rep. 2016; 6:29922.
27. Ashktorab H, Begum R, Akhgar A, Smoot DT, Elbedawi M, Daremipouran M, Zhao A, Momen B, Giardiello FM. Folate status and risk of colorectal polyps in African Americans. Dig Dis Sci. 2007; 52:1462-1470.
28. Ho V, Ashbury JE, Taylor S, Vanner S, King WD. Genetic and epigenetic variation in the DNMT3B and MTHFR genes and colorectal adenoma risk. Environ Mol Mutagen. 2016; 57:261-268.
29. Alexander M, Burch JB, Steck SE, Chen CF, Hurley TG, Cavicchia P, Shivappa N, Guess J, Zhang H, Youngstedt SD, Creek KE, Lloyd S, Jones K, Hebert JR. Case-control study of candidate gene methylation and adenomatous polyp formation. Int J Colorectal Dis. 2017; 32:183-192.
30. Vineis P, Chuang SC, Vaissiere T, Cuenin C, Ricceri F, Genair EC, Johansson M, Ueland P, Brennan P, Herceg Z. DNA methylation changes associated with cancer risk factors and blood levels of vitamin metabolites in a prospective study. Epigenetics. 2011; 6:195-201.
31. Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol. 2011; 29:1133-1139.
32. Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y, Noguchi S. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res Treat. 2011; 129:69-77.
33. Liao LM, Brennan P, van Bemmel DM, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Rothman N, Boffetta P, et al. LINE-1 methylation levels in leukocyte DNA and risk of renal cell cancer. PLoS One. 2011; 6:e27361.
34. Langevin SM, Koestler DC, Christensen BC, Butler RA, Wiencke JK, Nelson HH, Houseman EA, Marsit CJ, Kelsey KT. Peripheral blood DNA methylation profiles are indicative of head and neck squamous cell carcinoma: an epigenome-wide association study. Epigenetics. 2012; 7:291-299.
35. Graziano F, Humar B, Guilford P. The role of the E-cadherin gene (CDH1) in diffuse gastric cancer susceptibility: from the laboratory to clinical practice. Ann Oncol. 2003; 14:1705-1713.
36. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998; 92:725-734.
37. Bialik S, Kimchi A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin Cancer Biol. 2004; 14:283-294.
38. Michailidi C, Theocharis S, Tsourouflis G, Pletsa V, Kouraklis G, Patsouris E, Papavassiliou AG, Troungos C. Expression and promoter methylation status of hMLH1, MGMT, APC, and CDH1 genes in patients with colon adenocarcinoma. Exp Biol Med (Maywood). 2015; 240:1599-1605.
39. Shima K, Nosho K, Baba Y, Cantor M, Meyerhardt JA, Giovannucci EL, Fuchs CS, Ogino S. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: cohort study and literature review. Int J Cancer. 2011; 128:1080-1094.
40. Ye M, Li D, Zhou F, Guo Q, Xia B. Epigenetic regulation of death-associated protein kinase expression in primary gastric cancers from Chinese patients. Eur J Cancer Prev. 2012; 21:241-246.
41. Murata A, Baba Y, Watanabe M, Shigaki H, Miyake K, Ishimoto T, Iwatsuki M, Iwagami S, Yoshida N, Oki E, Morita M, Nakao M, Baba H. IGF2 DMR0 methylation, loss of imprinting, and patient prognosis in esophageal squamous cell carcinoma. Ann Surg Oncol. 2014; 21:1166-1174.
42. Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000; 60:2368-2371.
43. Nakagawa H, Nuovo GJ, Zervos EE, Martin EW Jr, Salovaara R, Aaltonen LA, de la Chapelle A. Age-related hypermethylation of the 5' region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001; 61:6991-6995.
44. Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, Saw CL, Cheung KL, Kong AN. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011; 13:606-614.
45. Abdelmaksoud-Dammak R, Miladi-Abdennadher I, Saadallah-Kallel A, Khabir A, Sellami-Boudawara T, Frikha M, Daoud J, Mokdad-Gargouri R. Downregulation of WIF-1 and Wnt5a in patients with colorectal carcinoma: clinical significance. Tumour Biol. 2014; 35:7975-7982.
46. Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, Nelson HH, Karagas MR, Wiencke JK, Kelsey KT. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomarkers Prev. 2012; 21:1293-1302.
47. Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011; 6:828-837.
48. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004; 10:6897-6904.
49. Dauksa A, Gulbinas A, Barauskas G, Pundzius J, Oldenburg J, El-Maarri O. Whole blood DNA aberrant methylation in pancreatic adenocarcinoma shows association with the course of the disease: a pilot study. PLoS One. 2012; 7:e37509.
50. Barault L, Ellsworth RE, Harris HR, Valente AL, Shriver CD, Michels KB. Leukocyte DNA as surrogate for the evaluation of imprinted Loci methylation in mammary tissue DNA. PLoS One. 2013; 8:e55896.
51. Ross SA. Diet and DNA methylation interactions in cancer prevention. Ann N Y Acad Sci. 2003; 983:197-207.
52. Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, Moreno-Macias H, Smith JA, Brody JA, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016; 9:436-447.
53. Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016; 354:618-622.
54. Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, Wang YC. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest. 2010; 120:521-532.
55. Sinha R, Kulldorff M, Chow WH, Denobile J, Rothman N. Dietary intake of heterocyclic amines, meat-derived mutagenic activity, and risk of colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 2001; 10:559-562.
56. Vo AT, Millis RM. Epigenetics and breast cancers. Obstet Gynecol Int. 2012; 2012:602720.
57. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007; 8:253-262.
58. Wojdacz TK, Windelov JA, Thestrup BB, Damsgaard TE, Overgaard J, Hansen L. Identification and characterization of locus-specific methylation patterns within novel loci undergoing hypermethylation during breast cancer pathogenesis. Breast Cancer Res. 2014; 16:R17.
59. Migheli F, Stoccoro A, Coppede F, Wan Omar WA, Failli A, Consolini R, Seccia M, Spisni R, Miccoli P, Mathers JC, Migliore L. Comparison study of MS-HRM and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS One. 2013; 8:e52501.
60. Park Y, Freedman AN, Gail MH, Pee D, Hollenbeck A, Schatzkin A, Pfeiffer RM. Validation of a colorectal cancer risk prediction model among white patients age 50 years and older. J Clin Oncol. 2009; 27:694-698.
61. Imperiale TF, Wagner DR, Lin CY, Larkin GN, Rogge JD, Ransohoff DF. Using risk for advanced proximal colonic neoplasia to tailor endoscopic screening for colorectal cancer. Ann Intern Med. 2003; 139:959-965.
62. Lin OS, Kozarek RA, Schembre DB, Ayub K, Gluck M, Cantone N, Soon MS, Dominitz JA. Risk stratification for colon neoplasia: screening strategies using colonoscopy and computerized tomographic colonography. Gastroenterology. 2006; 131:1011-1019.
63. Kastrinos F, Allen JI, Stockwell DH, Stoffel EM, Cook EF, Mutinga ML, Balmana J, Syngal S. Development and validation of a colon cancer risk assessment tool for patients undergoing colonoscopy. Am J Gastroenterol. 2009; 104:1508-1518.
64. Freedman AN, Slattery ML, Ballard-Barbash R, Willis G, Cann BJ, Pee D, Gail MH, Pfeiffer RM. Colorectal cancer risk prediction tool for white men and women without known susceptibility. J Clin Oncol. 2009; 27:686-693.
65. Wei EK, Colditz GA, Giovannucci EL, Fuchs CS, Rosner BA. Cumulative risk of colon cancer up to age 70 years by risk factor status using data from the Nurses' Health Study. Am J Epidemiol. 2009; 170:863-872.
66. Yeoh KG, Ho KY, Chiu HM, Zhu F, Ching JY, Wu DC, Matsuda T, Byeon JS, Lee SK, Goh KL, Sollano J, Rerknimitr R, Leong R, et al. The Asia-Pacific Colorectal Screening score: a validated tool that stratifies risk for colorectal advanced neoplasia in asymptomatic Asian subjects. Gut. 2011; 60:1236-1241.
67. Cai QC, Yu ED, Xiao Y, Bai WY, Chen X, He LP, Yang YX, Zhou PH, Jiang XL, Xu HM, Fan H, Ge ZZ, Lv NH, et al. Derivation and validation of a prediction rule for estimating advanced colorectal neoplasm risk in average-risk Chinese. Am J Epidemiol. 2012; 175:584-593.
68. Stegeman I, de Wijkerslooth TR, Stoop EM, van Leerdam ME, Dekker E, van Ballegooijen M, Kuipers EJ, Fockens P, Kraaijenhagen RA, Bossuyt PM. Combining risk factors with faecal immunochemical test outcome for selecting CRC screenees for colonoscopy. Gut. 2014; 63:466-471.
69. Wells BJ, Kattan MW, Cooper GS, Jackson L, Koroukian S. Colorectal cancer predicted risk online (CRC-PRO) calculator using data from the multi-ethnic cohort study. J Am Board Fam Med. 2014; 27:42-55.
70. Pencina MJ, D'Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011; 30:11-21.
71. McGeechan K, Macaskill P, Irwig L, Liew G, Wong TY. Assessing new biomarkers and predictive models for use in clinical practice: a clinician's guide. Arch Intern Med. 2008; 168:2304-2310.
72. Vickers AJ, Elkin EB, Steyerberg E. Net reclassification improvement and decision theory. Stat Med. 2009; 28:525-526; author reply 526-528.
73. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999; 116:1453-1456.
74. Shu XO, Yang G, Jin F, Liu D, Kushi L, Wen W, Gao YT, Zheng W. Validity and reproducibility of the food frequency questionnaire used in the Shanghai Women's Health Study. Eur J Clin Nutr. 2004; 58:17-23.
75. Balic M, Pichler M, Strutz J, Heitzer E, Ausch C, Samonigg H, Cote RJ, Dandachi N. High quality assessment of DNA methylation in archival tissues from colorectal cancer patients using quantitative high-resolution melting analysis. J Mol Diagn. 2009; 11:102-108.
76. Huang KT, Dobrovic A, Yan M, Karim RZ, Lee CS, Lakhani SR, Fox SB. DNA methylation profiling of phyllodes and fibroadenoma tumours of the breast. Breast Cancer Res Treat. 2010; 124:555-565.
77. Kristensen LS, Wojdacz TK, Thestrup BB, Wiuf C, Hager H, Hansen LL. Quality assessment of DNA derived from up to 30 years old formalin fixed paraffin embedded (FFPE) tissue for PCR-based methylation analysis using SMART-MSP and MS-HRM. BMC Cancer. 2009; 9:453.
78. Candiloro IL, Mikeska T, Hokland P, Dobrovic A. Rapid analysis of heterogeneously methylated DNA using digital methylation-sensitive high resolution melting: application to the CDKN2B (p15) gene. Epigenetics Chromatin. 2008; 1:7.
79. Wojdacz TK, Borgbo T, Hansen LL. Primer design versus PCR bias in methylation independent PCR amplifications. Epigenetics. 2009; 4:231-234.
80. Tserga A, Michalopoulos NV, Levidou G, Korkolopoulou P, Zografos G, Patsouris E, Saetta AA. Association of aberrant DNA methylation with clinicopathological features in breast cancer. Oncol Rep. 2012; 27:1630-1638.
81. Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007; 35:e41.
82. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18:1427-1431.
83. Moons KG, Altman DG, Reitsma JB, Ioannidis JP, Macaskill P, Steyerberg EW, Vickers AJ, Ransohoff DF, Collins GS. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): explanation and elaboration. Ann Intern Med. 2015; 162:W1-73.
84. Aschard H, Chen J, Cornelis MC, Chibnik LB, Karlson EW, Kraft P. Inclusion of gene-gene and gene-environment interactions unlikely to dramatically improve risk prediction for complex diseases. Am J Hum Genet. 2012; 90:962-972.
85. DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988; 44:837-845.