INTRODUCTION
Although gastric cancer is the third leading cause of cancer-related death worldwide [1–4], our basic understanding of gastric cancer falls behind that of many other cancer types. Clinically, gastric cancer is treated by surgical resection with chemotherapeutic interventions as major options [5]. Even with an increased enrollment rate for chemotherapy of gastric cancer, the overall median survival remains between 15 to 17 months [6]. Patients initially respond to chemotherapy but cancer eventually relapses. Therefore, chemotherapy resistance becomes a major barrier to achieve effective gastric cancer treatment. Thus, finding novel strategies to sensitize cancer cells to chemotherapy will significantly improve gastric cancer patient survival.
Chemotherapy in gastric cancer is generally used as multimodality treatment, such as perioperative and adjuvant chemotherapy [7, 8]. The common chemotherapeutical agents for gastric cancer include cisplatin (CDDP), 5-fluorouracil (5-FU) or its oral administered derivatives capecitabine and S-1. Common combinations include ECF (epirubixin, cisplatin and 5-FU), 5-FU plus either cisplatin or docetaxel (or irinotecan) with radiation [9, 10]. The mechanisms underlying chemoresistance in gastric cancer are not entirely known, but the following mechanisms have been reported: decreased intracellular drug accumulation and/or increased drug efflux, increased nucleotide excision-repair activity, evasion of apoptosis, activation of several signaling pathways and the existence of putative cancer stem cells. Another theory for chemosensitivity is the cancer stem cell hypothesis: a small percentage of cancer cells, the residual cancer cells or the putative cancer stem cells, are resistant to chemotherapy-mediated cell killing, and become the source for cancer relapse. If the regulatory mechanisms for maintaining this cell population are discovered, agents disrupting the mechanisms may be used to develop novel strategies to treat gastric cancer.
There are a number of signaling pathways involved in regulation of drug resistance, including the hedgehog pathway [11–15]. Hedgehog signaling is an important pathway for embryonic development, tissue patterning, cell differentiation, cancer development and drug resistance [16–18]. In the last few years, hedgehog signaling is reported in regulation of drug resistance in several types of cancer including ovarian, prostate and pancreatic cancer [11, 15, 19–21]. In hedgehog signaling active cells, the hedgehog ligands (Shh, Ihh or Dhh) bind to PTCH1, allowing smoothened to signal to the downstream GLI transcription factors, which then turn on the target genes [18]. Target genes of hedgehog signaling include GLI1, PTCH1 as well as other molecules involved in regulation of cancer cell function, such as ABCG2 [22]. ABCG2 is an important transportor and functions in maintenance of the stem cell population through side population regulation.
In this study, we report a novel mechanism underlying drug resistance in gastric cancer. We found that treatment of gastric cancer cells with the chemotherapeutical drug CDDP often results in elevated hedgehog (Hh) signaling which is associated with an increase in putative cancer stem cell markers. We further investigated the functional significance of Hh signaling and ABCG2 in cultured cells and in mouse models. The relevance of our findings to gastric cancer patients was further examined in human specimens. These findings may lead to novel strategies to improve the effectiveness of chemotherapy in gastric cancer.
RESULTS
Expression of Hh target genes following chemotherapeutical drug treatment
To delineate the molecular mechanisms underlying chemotherapy resistance in gastric cancer, we examined molecular changes following treatment with CDDP, the most common chemotherapeutical agent [23–25]. Two cancer cell lines N87 and AGS were used in our initial studies. We predicted that the residual cells with intrinsic drug resistance to chemotherapy will survive following drug treatment, and the responsible genes will be up-regulated. As a first step, we measured the IC50 of CDDP in gastric cell lines from the inhibition rate of cell viability by different concentrations of CDDP (Figure 1A and 1B). We then treated cells with CDDP at the IC50 dose for 48 hr and examined the candidate gene expression. Previous studies indicate many candidate genes involved in regulation of residual cancer cells or putative cancer stem cells, such as Hh, Wnt and Notch signaling molecules [26–28]. As shown in Figure 1C, we found that target genes for the Hh signaling pathway, GLI1, GLI2 and PTCH1, were significantly induced in CDDP-treated N87 cells. Similar results were also obtained from AGS cells (Figure 1D). In contrast, we did not observe significant gene expression changes in DKK1, JAG2 or CTGF, molecules involved in Wnt, Notch and Hippo/YAP signaling (Figure 1C and 1D, Supplementary Figure 1).
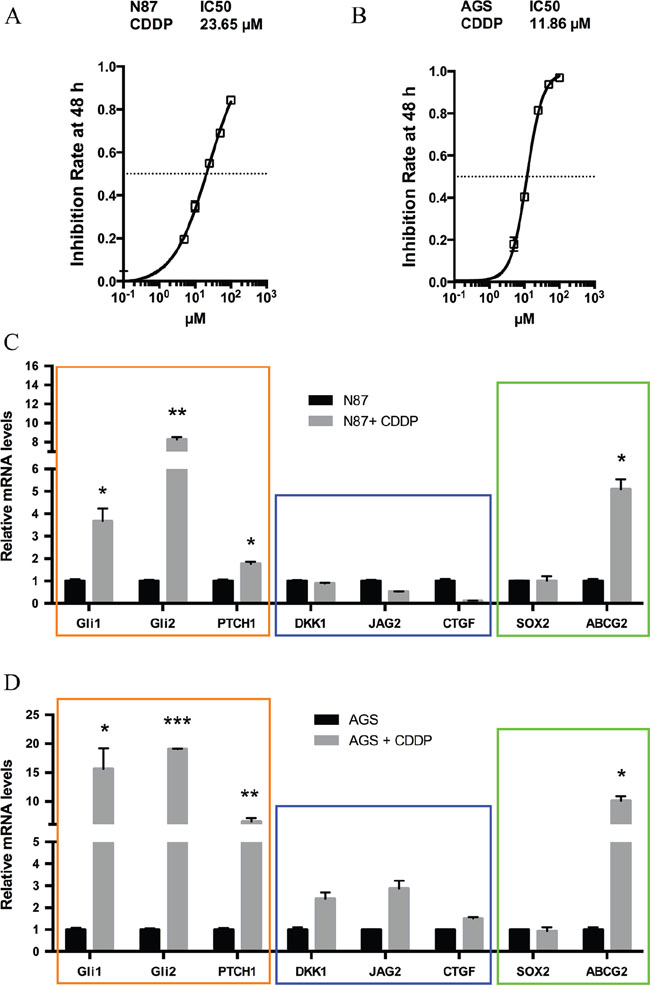
Figure 1: Elevated hedgehog signaling following CDDP treatment in gastric cancer cells. (A) The inhibition rate of cell viability by different concentrations of CDDP in N87 cells were determined by chemosensitivity assay (see Methods), and the IC50 was calculated from this experiment. Cells were treated with increasing concentrations of CDDP (5, 10, 25, 50, 100 μM) for 48 hr. (B) The rate of cell viability inhibition by CDDP in AGS cells were also determined by chemosensitivity assay (see Methods), and we calculated the IC50 from this experiment. (C) The transcript levels of GLI1, GLI2, PTCH1, DKK1, JAG2, CTGF, SOX2 and ABCG2 in N87 cells after treatment with 25 μM CDDP for 48 hr. (D) The transcript levels of above genes in AGS cells after 10 μM CDDP for 48 hr. Data are represents as mean ± SD from three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001.
Like Wnt and Notch signaling, Hh signaling plays an important role in embryonic development, and is also critical for maintenance of putative cancer stem cells or residual cancer cells [26, 29, 30]. We thus examined expression of several putative cancer stem cell markers [31–38] following CDDP treatment in N87 cells. There are a number of factors involved in regulation of putative cancer stem cells [39–43]. For example, the side population is often enriched in stem cells and cancer stem cells, and ABCG2 is the major gene regulating side population [44]. Sox2 is another important factor involved in regulation of putative cancer stem cells [45]. Through real-time PCR analysis, we found high expression of ABCG2 following drug treatment (Figure 1C). This phenomenon did not appear to be cell line specific because similar results were also observed in AGS cells (Figure 1D). In contrast, SOX2 expression was not significantly changed (Figure 1C and 1D).
These results indicate that elevated Hh signaling may be responsible for maintenance of residual cancer cells (or putative cancer stem cells or tumor initiating cells) following chemotherapeutic drug treatment in gastric cancer.
Significance of GLI1 expression for intrinsic drug resistance in gastric cancer cells
To evaluate the functional relevance of Hh signaling for the intrinsic drug resistance in N87 and AGS cells, we first knocked down GLI1 expression by expressing GLI1 shRNAs in both cell lines, and then determined the IC50 for CDDP. We found that down-regulation of GLI1 in N87 cells (Figure 2A) reduced the IC50 by nearly half (Figure 2B). The IC50 value was also reduced by GLI1 knockdown in AGS cells (Figure 2C and 2D). Additional experiments in IC50 measurement and tumor sphere formation indicate that knocking down both GLI1 and GLI2 has similar effect as GLI1 knockdown (Supplementary Figure 2 for IC50 value, and Supplementary Figure 3 for tumor sphere formation), suggesting that the feed-forward loop exerted by GLI1 is the major factor for regulation of putative cancer stem cells. Thus, GLI1, the focus for the rest of our study, appears to be critical for drug resistance in gastric cancer cells.
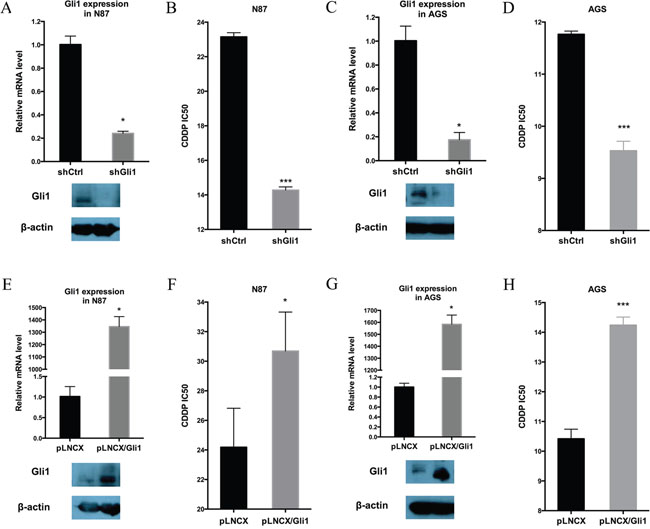
Figure 2: GLI1 expression is required and sufficient for intrinsic drug resistance in gastric cancer cells. (A) GLI1 transcript level and the protein level in N87/shCtrl and N87/shGLI1 cells as determined by real-time PCR and Western blot analysis, respectively. (B) IC50 dose of CDDP in N87/shCtrl and N87/shGLI1 cells determined by chemosensitivity assay. (C) GLI1 transcript and protein levels in AGS/shCtrl and AGS/shGLI1 cells. (D) The CDDP IC50 dose in AGS/shGLI1 cells compared with AGS/shCtrl cells. (E, F) The effect of ectopic Gli1 expression on the IC50 of CDDP in N87 cell. (E) shows GLI1 transcript (upper) and protein (low) levels in N87 with or without ectopic GLI1 expression (pLNCX indicates the vector control, and pLNCX-Gli1 indicates ectopic Gli1 expression). (F) shows the IC50 values from pLNCX and pLNCX-Gli1 N87 cells. (G, H) The effect of ectopic Gli1 expression on the IC50 value of CDDP in AGS cells. (G) shows GLI1 transcript (upper) and protein (low) levels in AGS cells with pLNCX as the vector control and pLNCX/GLI1 as the ectopic Gli1 expression. (H) shows the IC50 values from pLNCX and pLNCX/GLI1. Means ± SD from three independent experiments are shown. * P < 0.05, ** P < 0.01, *** P < 0.001.
Furthermore, we determined whether elevated Hh signaling is sufficient to drive drug resistance in gastric cancer cells by ectopic expression of GLI1 in N87 and AGS cells, and examining their IC50 values for CDDP. We discovered that ectopic GLI1 expression in both N87 and AGS cells significantly increased the IC50 value (Figure 2E-2H).
Taken together, we found that the GLI1 expression level is highly associated with chemosensitivity in gastric cancer cells. While down-regulation of GLI1 decreases the IC50, ectopic expression of GLI1 increases the CDDP IC50.
Regulation of the putative cancer stem cell population by GLI1
Previous studies have revealed heterogeneous cell populations even within the established cell lines [31–38], and our experiments with CDDP treatment studies also suggest that a subset of cells (e.g. putative cancer stem cells) are more resistant to drug treatment, and that GLI1 may play an important role in maintenance of this cell population. To directly test the role of GLI1 for putative cancer stem cell maintenance, we detected the putative cancer stem cell population in gastric cancer cells using three methods.
First, we measured tumor sphere forming efficiency from N87 cells with GLI1 shRNAs, ectopic GLI1 expression or the control cells. Tumor sphere formation efficiency is a known biological readout of cancer stem cells [46]. We found that GLI1 knockdown significantly reduced the size of tumor spheres (Figure 3A). GLI1 shRNA expression also reduced the tumor sphere forming efficiency (Figure 3A, with the control ~38 spheres /2000 cells and Gli1 knockdown cells 29 spheres/ 2000 cells, P = 0.032). Conversely, ectopic expression of GLI1 increased the tumor sphere forming efficiency (Figure 3C, with the control ~32.5 spheres/2000 cells and GLI1 expressing cells 80 spheres/ 2000 cells, P = 0.034). It thus appears that high GLI1 expression increases the tumor sphere forming efficiency.
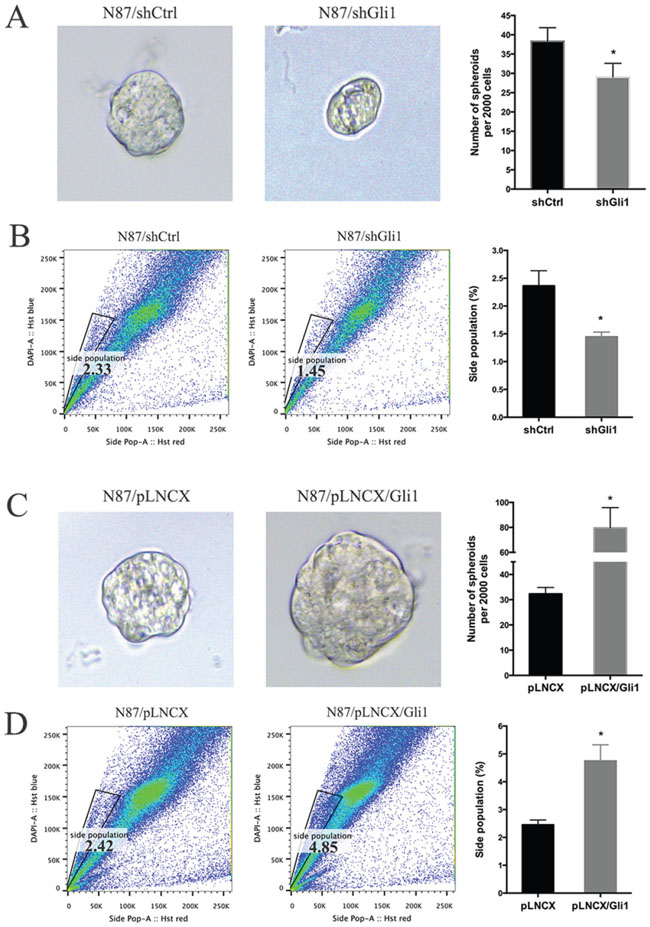
Figure 3: GLI1 regulates the putative cancer stem cell population in N87 cells. (A) Representative images and statistical graph of N87/shCtrl and N87/shGLI1 cells grown as tumor spheres. (B) Side population in N87/shCtrl and N87/shGLI1 cells was measured by flow cytometry. Representative flow cytometric analysis was shown. (C) Representative images of spheres from N87/pLNCX and N87/pLNCX/GLI1 cells. (D) The percentage of side population in N87/pLNCX/GLI1 cell and the control cells. All data are means ± SD of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001.
Second, we also detected side population of cancer cells after GLI1 alteration. Side population is a functional assay for the transporter ABCG2, and is a well-known readout for stem cells and putative cancer stem cells [47–49]. We found that GLI1 shRNA reduced the side population by 40% (Figure 3B) whereas ectopic GLI1 expression increased the side population by 100% (Figure 3D).
Third, we also detected cell surface marker expression following alteration of GLI1 levels. Since there are no consensus cell surface markers for putative cancer stem cells in gastric cancer, we examined changes in the following markers: CD24, CD33, CD44, CD90 and CD133 that are reported in the literature [31–38]. We did not see significant changes in CD24, CD44 and CD33 in our experiments. However, we found that GLI1 knockdown significantly reduced expression of CD90 and CD133 (Supplementary Figure 4A/B) whereas ectopic GLI1 expression increased expression of CD90 (Supplementary Figure 4C/D). Although CD44 was reduced by GLI1 shRNAs and was reportedly regulated by hedgehog signaling [50], the decrease in our experiments did not reach statistical significance.
Taken all the data together, we concluded that GLI1 expression is an important factor for maintenance of the putative cancer stem cell population as indicated by tumor sphere forming efficiency and side population. While GLI1 knocking down reduces the putative cancer stem cell population, ectopic expression of GLI1 increases this population.
The role of ABCG2 in drug sensitivity regulation in gastric cancer cells
Because ABCG2 is responsible for exclusion of Hoechst 33342 dye and the subsequent side population, we tested whether GLI1 directly regulate ABCG2 expression. Through real-time PCR analysis, we found that while GLI1 shRNAs significantly reduced ABCG2 expression by half, ectopic GLI1 expression induced ABCG2 transcript by 100% (Figure 4A). Similarly, treatment with CDDP increased expression of ABCG2 (Figure 1C and 1D) and the side population (Figure 4B).
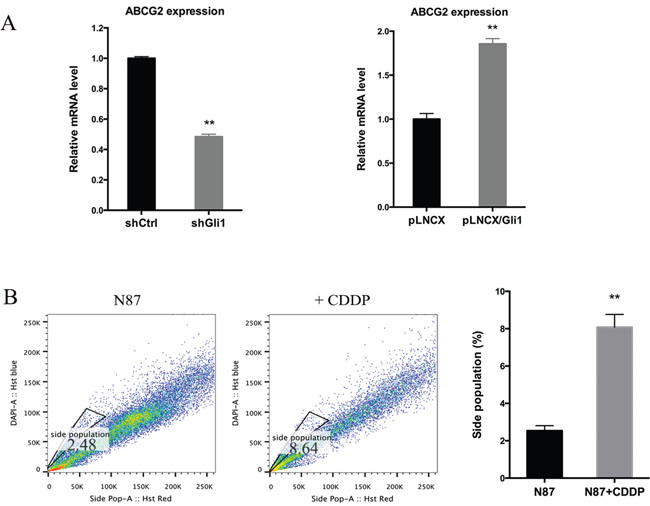
Figure 4: Regulation of ABCG2 by GLI1. (A) The ABCG2 transcript level in N87/shCtrl and N87/shGLI1 cells (left panel), N87/pLNCX and N87/pLNCX/GLI1 cells (right panel) as detected by real-time PCR. (B) Representative side population analysis and statistical graph of side population in N87 cells treated with 25 μM CDDP in comparison with control cells. Means ± SD are shown. ** P < 0.01.
After promoter analysis, we discovered a GLI1-binding consensus site [18] in the promoter of ABCG2 (Supplementary Figure 5C), suggesting that ABCG2 may be a transcriptional target of GLI1 molecules. We performed ChIP analysis [51] following ectopic expression of GLI1 in N87 and AGS cells (Supplementary Figure 5A and 5B). Supplementary Figure 5C showed the 9 base pairs sequence of the potential GLI1 binding site within the ABCG2 promoter. Our data indicate that ectopically expressed GLI1 protein (with MYC tag) can be pulled down with the ABCG2 promoter region encompassing the GLI1 binding site (Supplementary Figure 5D and 5E) whereas the control IgG does not pull down the fragment. Similarly, MYC antibodies did not pull down the fragment in cells without ectopic GLI1 expression. These results suggest that GLI1 may directly regulate ABCG2 expression by transcriptional regulation, which is consistent with a previous report in lymphomas [52].
Next, we evaluated the functional relevance of ABCG2 for regulating drug sensitivity. We used two methods to reduce ABCG2 function. First, we used a truncated form of ABCG2, which has a dominant negative effect on the endogenous ABCG2 gene function [53], and found that ectopic expression of this truncated form of ABCG2 reduced the IC50 of CDDP by half (Figure 5A), which was associated with reduced side population (Figure 5B and Supplementary Figure 6A) and a decrease in CD90 expression (Supplementary Figure 6B). Similarly, when an ABCG2 inhibitor FTC [54] was used, we observed a significant reduction in the IC50 (Figure 5C), sphere formation (Figure 5D) and side population (Figure 5E, Supplementary Figure 6C). Conversely, we found that overexpression of ABCG2 increased the IC50 for CDDP (Figure 6A), which was associated with an increase in side population (Figure 6B), sphere formation (Figure 6C and Supplementary Figure 6D) and CD90 expression (Supplementary Figure 6E).
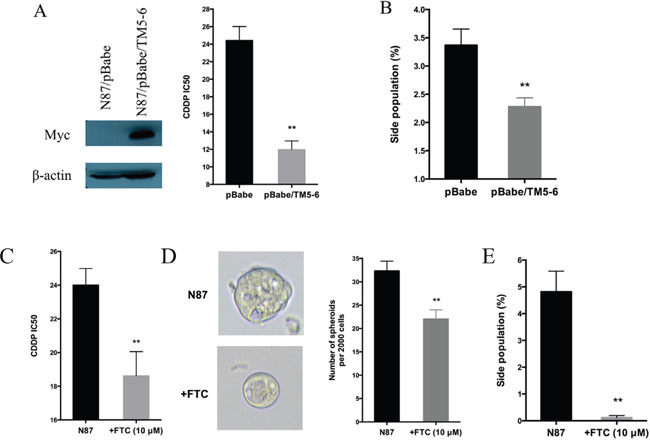
Figure 5: Regulation of drug sensitivity by ABCG2. (A) Effects of ABCG2-TM5-6, the truncated form of ABCG2 on drug sensitivity. ABCG2-TM5-6 was detected by the Myc-tag in Western blot analysis (left panel). The IC50 for CDDP in these two groups of cells were shown in the right panel. (B) Statistical graph of side population distribution in N87/pBabe and N87/pBabe/TM5-6 cells. (C) The IC50 of CDDP in N87 cells with or without FTC (10 μM). (D) Representative images and statistical graph of spheres in N87 cells treated or untreated with FTC. (E) Statistical graph of side population in N87 cells with or without FTC. The results are means of three independent experiments ± SD. * P < 0.05, ** P < 0.01.
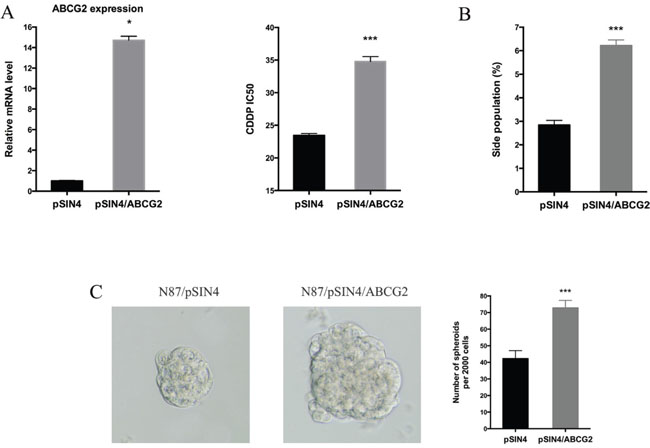
Figure 6: ABCG2 overexpression increases drug resistance. (A) The ABCG2 transcript level in N87/pSIN4 and N87/pSIN4/ABCG2 cells was detected by real-time PCR (left panel). The IC50 of CDDP was shown in the right panel. (B) Statistical graph of side population analysis in N87/pSIN4 and N87/pSIN4/ABCG2 cells. (C) Representative images of spheres (left two panels) and statistical graph of tumor spheres in N87/pSIN4 and N87/pSIN4/ABCG2 cells. Means ± SD are shown. * P < 0.05, *** P < 0.001.
The effect of ectopic ABCG2 expression on the IC50 of CDDP was very similar to that GLI1 expression, suggesting that ABCG2 may be the major mediator for regulating drug sensitivity in gastric cancer. In consistent with our hypothesis, ectopic expression of ABCG2 had no significant effects on expression of GLI1, GLI2 and PTCH1 in both N87 and AGS cells (Supplementary Figure 7), further confirming that hedgehog signaling is upstream of ABCG2 in our experiment system.
Therapeutic effects of CDDP in ectopic expression of GLI1 xenograft model
To determine the relevance of our in vitro studies to therapeutic implication, we tested the effects of CDDP on the subcutaneous mouse model using N87 cells with alterations in GLI1 and ABCG2. GLI1 was ectopically expressed in N87/pLNCX/GLI1 cells while GLI1 expression was knocked down in N87/shGLI1 cells. A truncated form of ABCG2 was expressed in N87/pBabe/TM5-6 cells. As shown in Figure 7A and 7B, CDDP treatment reduced the tumor significantly in the N87 control group. In contrast, ectopic GLI1 expression in N87 cells did not respond to CDDP treatment whereas GLI1 shRNA expression sensitized tumors to CDDP treatment. Blockage of ABCG2 function, via ectopic expression of a dominant negative construct ABCG2/TM5-6, made N87 gastric cancer cells more sensitive to CDDP treatment (Figure 7B). Tumors formed from the cells expressing shGLI1 or TM5-6 reduced the tumor size significantly following CDDP treatment (Figure 7D and 7E). In contrast, tumors formed from the cells with ectopic GLI1 expression were not significantly affected by CDDP (in compared with the N87 control group, Figure 7C). These results are consistent with the in vitro studies, further confirming that the levels of GLI1 and ABCG2 expression determine the sensitivity of gastric cancer cells to CDDP treatment.
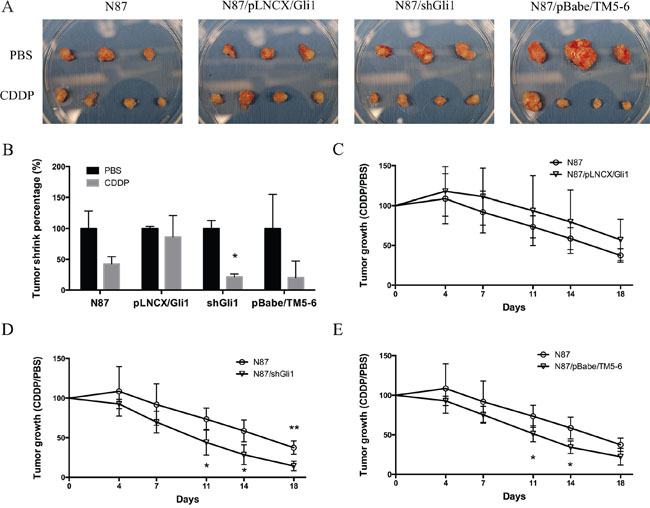
Figure 7: CDDP sensitivity in mouse models. (A) Photographs of tumor xenografts derived from N87, N87/pLNCX/GLI1, N87/shGLI1 or N87/pBabe/TM5-6 in NSG mice treated with PBS (top) or CDDP treatment (bottom). (B) Tumor weights shrink percentage after CDDP treatment compared with PBS control in four different xenografts groups. (C) The growth curves of tumor xenografts in mice during CDDP treatment in N87 and N87/pLNCX/GLI1 groups. (D) The tumor growth in N87/shGLI1 group and the N87 control group during CDDP treatment. (E) Tumor growth curves in N87 and N87/pBabe/TM5-6 groups. Means ± SD are shown. * P < 0.05, ** P < 0.01.
Higher ABCG2 expression in gastric cancer patients with poor prognosis
We went further to find the relevance of our data to gastric cancer patients by correlating ABCG2 expression in the tumor with the survival of patients who underwent chemotherapy using CDDP. The expression level of ABCG2 was analyzed by immunohistochemistry in 180 cases of gastric cancer patients who underwent chemotherapy. According to our criteria (see Materials and Methods), 62% (111/180) of gastric cancer specimens were positively stained with anti-ABCG2 antibodies (Figure 8A). We found that positivity of ABCG2 staining was significantly correlated with clinicopathological parameters. As shown in Table 1, the presence of ABCG2 was higher in poorly differentiated gastric cancer specimens in comparison with well-differentiated ones (Figure 8B, P = 0.0005). There was no difference in the correlation between ABCG2 expression and gender, age, tumor size and tumor location (see Table 1 for details). Among these 180 gastric cancer patients, 170 cases were available with the follow-up information. As Figure 8C shown, higher ABCG2 expression in gastric cancer was correlated with a shorter overall survival, indicating that high ABCG2 expression in the tumor predicts a poor outcome in chemotherapy in gastric cancer patients.
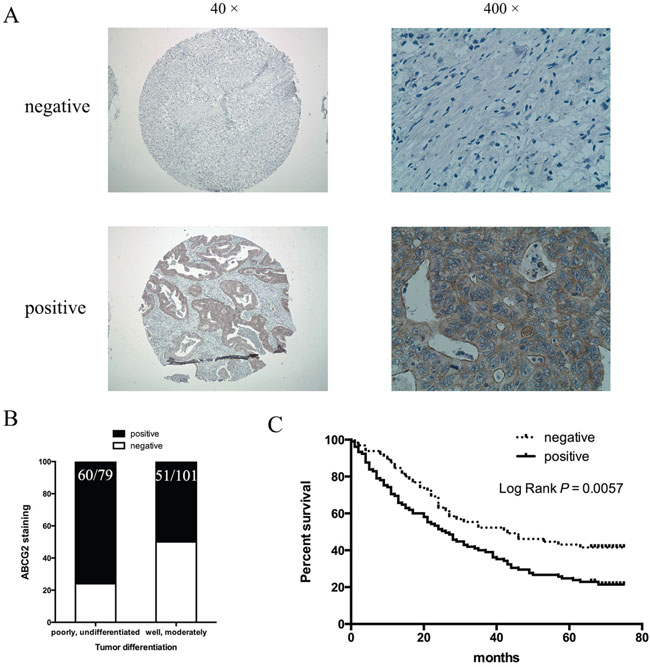
Figure 8: Correlation of ABCG2 expression in gastric cancer with tumor differentiation and patient survival. (A) Representative images of immunohistochemical staining of ABCG2 in gastric cancer specimens. Original magnification: 40× and 400×. (B) Staining of ABCG2 and correlation with tumor differentiation (P = 0.0005). (C) Kaplan-Meier analysis of the significance of ABCG2 in predicting gastric cancer patient overall survival. P = 0.0057 by log rank test.
Table 1: Relationship between ABCG2 membrane staining and clinicopathologic features in 180 gastric cancer tissues
Clinicopathologic parameters |
ABCG2 expression |
P-value |
|
|---|---|---|---|
negative (n = 69) |
positive (n = 111) |
||
Gender |
|||
Male |
46 |
77 |
0.7047 |
Female |
23 |
34 |
|
Age (years) |
|||
≤ 60 |
35 |
41 |
0.0686 |
>60 |
34 |
70 |
|
Tumor size (cm) |
|||
≤ 5 |
36 |
55 |
0.7321 |
>5 |
33 |
56 |
|
Location |
|||
Distal third |
41 |
59 |
0.4107 |
Middle third, proximal third |
28 |
52 |
|
Differentiation |
|||
Poorly, undifferentiated |
19 |
60 |
0.0005 |
Well, moderatelly |
50 |
51 |
|
Local invasion |
|||
T1, T2 |
9 |
16 |
0.7960 |
T3, T4 |
60 |
95 |
|
Lymph node metastasis |
|||
No |
19 |
27 |
0.6310 |
Yes |
50 |
84 |
|
TNM stage |
|||
I, II |
28 |
41 |
0.6250 |
III, IV |
41 |
70 |
|
Because there are no GLI1 antibodies suitable for immunohistochemistry in paraffin-embedded tissues, we took the advantage of the CTGA data to test whether higher GLI1 transcript level is correlated with cancer relapse or patient survival in those patients who underwent chemotherapy. In a cohort of 415 cancer specimens, we found that 7 out of 12 (>50%) patients with high GLI1 transcript had relapsed cancer whereas 102 out of 310 patients with low GLI1 expression had relapsed cancer. In addition, patients with high Gli1 expression had worse disease-free survival (Supplementary Figure 9). These results suggest that the GLI1-ABCG2 signaling axis is associated with poor outcomes of the gastric cancer patients.
From all these data, we conclude that activation of Hh signaling, through regulation of ABCG2, plays an important role in regulation of drug sensitivity in gastric cancer. We predict that the novel strategies aimed at reducing GLI1 expression or interrupting ABCG2 function, together with CDDP-based chemotherapy, should improve the survival of gastric cancer patients.
DISCUSSION
Gastric cancer remains a major contributor for cancer-related mortality. Most patients are diagnosed with advanced disease where the five-year survival rate is very low (< 5%) [1]. Although the regulatory mechanisms for chemotherapy resistance in gastric cancer have been reported in the last 10 years [55], very little data have been linked the mechanisms to patient survival. Our results indicate that the GLI1-mediated regulation of ABCG2 is an important mechanism responsible for maintenance of the residual cancer cells (or putative cancer stem cells) in gastric cancer following treatment with CDDP, a major chemotherapeutical drug. We have data to show that knocking down GLI1 (Figure 2 and Figure 3) or inhibiting ABCG2 functions (Figure 5), sensitizes cancer cells to chemotherapy in cultured cells (Figure 2-6) and in mice (Figure 7). More importantly, we have shown that higher GLI1/ ABCG2 expression is associated with poor survival of gastric cancer patients who underwent CDDP-based chemotherapy (Figure 8 and Supplementary Figure 9). Since inhibitors for GLI1 and ABCG2 are already available, we predict that these agents, together with CDDP-based chemotherapy, will improve gastric cancer patient survival.
Like Wnt and Notch signaling pathways, Hh signaling is known to be a critical regulator for embryonic development, and is involved in maintenance of the cancer cell stemness in a number of cancer types [18, 26, 29, 30, 56]. There are several reports on association of Hh signaling with drug resistance [13, 56, 57], but the molecular link is often not known. Yoon et al reported a link between Hh signaling and CD44 expression in a number of cell lines through sphere formation studies, which was shown to be associated with cisplatin resistance in gastric cancer [50]. While we have confirmed regulation of CD44 by GLI1 expression, we found that GLI1 affects ABCG2 more dramatically, with little effects on CD44 (Supplementary Figure 4). Furthermore, we demonstrated that inhibition of ABCG2 by expressing a truncated ABCG2 molecule or addition of a specific inhibitor, sensitizes cancer cells to CDDP treatment (Figure 5). We found that the ligands, Shh and Ihh, were not significantly altered by CDDP in N87 and AGS cells (Supplementary Figure 8), suggesting that up-regulation of GLI1 in these cells was not caused by ligand-dependent signaling. This implies that the SMO inhibitors, with two already approved by FDA for basal cell carcinoma treatment [58], will not be effective in sensitizing gastric cancer cells to chemotherapy. It will be interesting, though, to reveal the molecule mechanisms by which GLI1 expression is induced in cancer cells with intrinsic tolerance of CDDP, which may provide additional strategies to sensitize gastric cancer cells to chemotherapy. Targeting ABCG2 will be a more feasible strategy to improve the sensitivity of gastric cancer cells to chemotherapy. Although ABCG2 is known to be responsible for transporting several types of small molecules, such as ions, dyes and some chemotherapeutical drugs, CDDP transport in and out of cells is not affected by ABCG2 [59, 60], further confirming that ABCG2 expression may represent a subset of cell population (side population) with cancer cell stemness.
MATERIALS AND METHODS
Cell culture
Human gastric cancer cell lines AGS and N87, and HEK293T were purchased from ATCC. Gastric cancer cells were maintained in RPMI-1640, while HEK293T cells were cultured in DMEM, supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified environment containing 5% CO2. Exponentially growing cells were used for experiments.
Cell viability and chemosensitivity assay
Cells (2000/well for AGS, 4000/well for N87) were seeded into 96-well plates and stained at the indicated time point using alamarBlue (Thermo scientific, FL, USA) according to the manufacturer’s instructions. The optical density measured at 530/590 nm was used as an indicator of cell viability. For chemosensitivity assay, the medium was replaced by fresh medium with or without various concentrations of Cisplatin (CDDP, Calbiochem, CA, USA) (5, 10, 25, 50, 100 μM). Cell viability assay was performed after 48 hrs of treatment [61–63]. Six wells were counted for each drug concentration and at least three independent experiments were performed. The half maximal inhibitory concentration (IC50) value was defined as the concentration that resulted in a 50% reduction in cell growth compared with growth of the control.
Plasmids construction, lentiviral and retroviral infection
The plasmid pLNCX/GLI1 (with a Myc tag) was previously constructed in our laboratory [51] and pLKO.1/shGLI1 and their corresponding control plasmid pLKO.1/shCtrl were from the Broad Institute. Expression plasmid pSIN4/ABCG2 was purchased from Addgene (#25983). The ABCG2/TM5-6 plasmid was kindly provided by Dr. Jian-ting Zhang at Indiana University School of Medicine, and subcloned into pBabe using EcoRI and BamHI to obtain pBabe/TM5-6 (with Myc tag) [53].
To generate GLI1 knockdown, ABCG2 and TM5-6 overexpression cells, AGS and N87 cells were infected with lentivirus containing pLKO.1/shGLI1, pSIN4/ABCG2 and pBabe/TM5-6, respectively. Lentiviral packaging plasmids PRRE, RSV/REV and CMVG were co-transfected into HEK293T cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) for virus production and infection as previously described [64]. Twenty-four hours after infection, cells were selected with 1 μg/ml puromycin (Sigma, St Louis, MO, USA) or 1 mg/ml G418 (Invitrogen).
Reverse transcriptase and real-time PCR
Total RNA was isolated from cells using TRIzol reagent (Sigma) according to the manufacturer’s instructions. One microgram of total RNA was reverse transcribed into cDNAs using the First-Strand Synthesis Kit (Roche, Indianapolis, IN, USA). The abundance of transcripts in the cDNA samples was measured by real-time PCR with specific probes as instructed from the provider and previously described [65, 66]. All probes for real-time PCR were purchased from the Applied Biosystems.
Western blot analysis
Cells were lysed in cell lysis buffer (50 mM Hepes, pH 7.4, 2 mM EDTA, 100 mM NaCl, 1% Glycerol, 1% Triton X-100) containing protease and phosphatase inhibitors. After separation by SDS-polyacrylamide gel electrophoresis and protein transfer onto PVDF membrane. Antibodies were used to incubate with the membrane [Rabbit anti-GLI1 antibody diluted at 1:1000 (from the Cell Signaling Technology, MA, USA); mouse anti-Myc 9B11 antibody (diluted at 1:1000, the Cell Signaling Technology); mouse anti-β actin antibody (diluted at 1:10000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and goat anti-rabbit-HRP/goat anti-mouse-HRP (diluted at 1:10000, Thermo scientific)]. Detection was performed with ECL Western blotting detection kit (Thermo scientific) as described previously [64, 66].
Flow cytometry analyses
Single cells were dissociated using Accutase® (Gibco, CA, USA) and re-suspended in PBS containing 10% FBS. The cells were incubated with fluorescence-conjugated antibody against human CD24, CD33, CD44, CD90, CD133 and ABCG2 (all from Biolegend, CA, USA) for 30min at 4°C. After washing, cells were analyzed on FACSCalibur or FACSCanto II (Beckton Dickinson, Franklin Lakes, NJ, USA) [66].
For side population assay, single cells were re-suspended at 1 × 106/ml in DMEM with 2% FBS and 10 mM HEPES, stained with Hoechst 33342 dye (final concentration 5 μg/ml, Invitrogen) and incubated at 37°C for 90 min with shaking. The cells were washed with ice-cold HBSS with 2% FBS and 10 mM HEPES, spined down at 4°C and re-suspended in ice-cold HBSS containing 2 μg/ml propidium iodide (PI, Invitrogen). An aliquot of cells was used as a negative control by adding ABC transporter inhibitor fumitremorgin C (FTC, 10 μM, Calbiochem) and incubated at 37°C for 30 min before addition of Hoechst 33342.
Sphere formation assay
Cells were re-suspended in sphere formation medium (Neural Basal medium with 1× B-27, 20 ng/ml of EGF, 10 ng/ml of βFGF and 5 μg/ml of heparin), and plated on ultralow attachment 24-well plates with 2000 cells each well (Corning, Lowell, MA, USA) as described previously [67]. Sphere formation medium was changed every other day. After 7 days, spheres were counted and taken photos, or cells were dissociated with Accutase® and used for other experiments. The sphere forming efficiency was calculated by counting the number of spheres formed from 2000 cells.
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed as previously described [51]. In brief, cells at 80% confluence were cross-linked with formaldehyde (Sigma) at room temperature for 15 min. Formaldehyde was neutralized by addition of 125 mM glycine for 5 min. The cells were collected by centrifugation and rinsed in cold phosphate-buffered saline. The cell pellets were collected by centrifugation and then re-suspended in sonication buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 0.5 mM PMSF, and 100 ng of leupeptin and aprotinin/ml) and incubated on ice for 20 min. The samples were sonicated on ice with an Ultrasonics sonicator at setting 10 for 20-second pulses to yield an average length of 500-1000 bp for genomic DNA. The chromatin solution was pre-cleared with the addition of protein A/G plus agarose beads (from the Santa Cruz Biotechnology Inc.) for 30 min at 4°C. Prior to use, the protein A/G plus agarose beads were blocked with sheared herring sperm DNA (1 μg/μl) and bovine serum albumin (1 μg/μl) for at least 4 hr at 4°C. 10% of the supernatant was saved as total input, and the rest was immunoprecipitated with 5 μg of normal anti-mouse IgG (Biolegend), 5 μg of Myc-tag mouse monoclonal antibody (Cell signaling technology) or 5 μg of anti-Histone H3 polyclonal antibody (Biolegend) at 4°C overnight, and then incubated with protein A/G plus agarose beads for 1 hr. Immunoprecipitates were washed twice in TE buffer (10 mM Tris-Cl, pH 8.0, 1 mM EDTA, 1 mM PMSF), once with Low Salt Wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 1 mM PMSF), twice with High Salt Wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-Cl, pH 8.0, 500 mM NaCl, 1 mM PMSF), once with LiCl Wash buffer (0.25 M LiCl, 1% NP-40, 1% Sodium deoxycholate, 1 mM EDTA, 10 mM Tris-Cl, pH 8.0, 1 mM PMSF) and three times in TE buffer. Immunoprecipitates were eluted in Elution buffer (50 mM Tris-Cl, pH 8.0, 10 mM EDTA, 1% SDS, 200 mM NaCl) and incubated at 65°C for 30 min. DNA was purified by extraction with phenol/chloroform and precipitated with 1/10 volume of 3 M NaOAc (pH 5.3) and 2.5 volumes of ethanol. The DNA fragment was amplified by PCR using primers flanking the promoter region 1000 bp upstream. Primers used to PCR-amplify the ABCG2 gene chromatin were 5’-ATCCCATTCACCAGAAACCA-3’ and 5’–CGAACGGAATGAACCAGAGT-3’ resulting in a product size of 205 bp. Primers used to PCR-amplify 18s as an internal control were 5’-CAGCCACCCGAGATTGAGC-3’ and 5’-TAGTAGCGACGGGCGGTGTG-3’ resulting in a product size of 252 bp.
Tumor xenografts and anticancer chemotherapy in vivo
All animal procedures were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee in Indiana University (Indianapolis, IN). To generate subcutaneous tumors, 5 × 106 cells were re-suspended in 100 μl PBS and subcutaneously injected into NOD scid gamma (NSG) mice. Tumor volume (volume = 0.5 × length × width2) was measured with a caliper twice a week. When tumors reached 80-100 mm3 (as day 0 in this study), mice were injected intraperitoneally (i.p.) once a week with CDDP (4 mg/kg) or vehicle (PBS) control group for two weeks. On day 21, mice were sacrificed, and tumor xenografts were removed, weighted and fixed in 10% buffered formalin for further analysis.
Immunohistochemistry (IHC)
Two ninety-dot tissue arrays were purchased from Shanghai Outdo Biotech Company (Shanghai, China). Formalin-fixed, paraffin-embedded sections were deparaffinized by xylene, rehydrated in ethanol and boiled in 10 mM citrate buffer (pH 6.0) for 30 min for antigen retrieval. Endogenous peroxidase was blocked by 3% H2O2 for 10 min for immunoperoxidase labeling. Sections were then incubated at 4°C overnight with primary antibodies against rabbit anti-ABCG2 (1:100, Abcam, Cambridge, UK). Incubation with corresponding secondary antibody (ImmPRESS universal peroxidase reagent, Vector Lab, CA, USA) and the peroxidase-antoperoxidase complex was visualized by DAB kit (ImmPACT DAB Peroxidase Substrate, Vector Lab), as previously described [68]. Two individuals (B.Y. and J.X.), who had no prior knowledge of the clinical pathologic data of the patients, examined the stained sections independently. Positive membrane ABCG2 staining was assessed by the intensity of stained cells and determined in two categories (negative and positive). The staining intensity was classified from 0 to 3+ as follows: 0, no staining; 1+, < 25% of the section was stained; 2+, 26% - 50% of the section was stained; 3+, > 50% of the section was stained. Scores of 1+, 2+ and 3+ were considered to be positive.
Statistical analysis
Data are presented as mean ± SD. from at least three independent experiments. IC50 values of CDDP were calculated with the GraphPad Prism software. Survival data were analyzed using the Kaplan-Meier method with a log-rank test for comparison of survival curves. Statistical comparisons between two groups were performed using a two-tail unpaired t-test or the chi-square test, with P values of < 0.05 indicating statistically significant difference.
ACKNOWLEDGMENTS
This work was supported by National Cancer Institute R01CA155086, The Wells Center for Pediatric Research, Riley Children Foundation, Jeff Gordon Children’s Foundation and IU Simon Cancer Center. This work was also funded by grants from National Natural Science foundation of China (No. 91529302, 81472641), Key Projects in the National Science & Technology Pillar Program of China (No. 2014BAI09B03). B.Y. was supported by the Liu Haoqing Talent Fund from Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. We thank Dr. Zizheng Dong and Dr. Jian-ting Zhang for help with the truncated ABCG2 expressing plasmid.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015; 136:E359-386.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
3. Razzak M. Genetics: new molecular classification of gastric adenocarcinoma proposed by The Cancer Genome Atlas. Nature reviews Clinical oncology. 2014; 11:499.
4. McLean MH, El-Omar EM. Genetics of gastric cancer. Nature reviews Gastroenterology & hepatology. 2014; 11:664-674.
5. Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ, Falk SJ, Iveson TJ, Smith DB, Langley RE, Verma M, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. The New England journal of medicine. 2006; 355:11-20.
6. Bernards N, Creemers GJ, Nieuwenhuijzen GA, Bosscha K, Pruijt JF, Lemmens VE. No improvement in median survival for patients with metastatic gastric cancer despite increased use of chemotherapy. Annals of oncology. 2013; 24:3056-3060.
7. Orditura M, Galizia G, Sforza V, Gambardella V, Fabozzi A, Laterza MM, Andreozzi F, Ventriglia J, Savastano B, Mabilia A, Lieto E, Ciardiello F, De Vita F. Treatment of gastric cancer. World journal of gastroenterology. 2014; 20:1635-1649.
8. Proserpio I, Rausei S, Barzaghi S, Frattini F, Galli F, Iovino D, Rovera F, Boni L, Dionigi G, Pinotti G. Multimodal treatment of gastric cancer. World journal of gastrointestinal surgery. 2014; 6:55-58.
9. Ajani JA, Bentrem DJ, Besh S, D'Amico TA, Das P, Denlinger C, Fakih MG, Fuchs CS, Gerdes H, Glasgow RE, Hayman JA, Hofstetter WL, Ilson DH, et al. Gastric cancer, version 2.2013: featured updates to the NCCN Guidelines. Journal of the National Comprehensive Cancer Network. 2013; 11:531-546.
10. Yuan M, Yang Y, Lv W, Song Z, Zhong H. Paclitaxel combined with capecitabine as first-line chemotherapy for advanced or recurrent gastric cancer. Oncology letters. 2014; 8:351-354.
11. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, Bonal DM, Charytonowicz E, Gladoun N, de la Iglesia-Vicente J, Petrylak DP, Benson MC, Silva JM, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012; 22:373-388.
12. Keysar SB, Le PN, Anderson RT, Morton JJ, Bowles DW, Paylor JJ, Vogler BW, Thorburn J, Fernandez P, Glogowska MJ, Takimoto SM, Sehrt DB, Gan GN, et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer research. 2013; 73:3381-3392.
13. Della Corte CM, Bellevicine C, Vicidomini G, Vitagliano D, Malapelle U, Accardo M, Fabozzi A, Fiorelli A, Fasano M, Papaccio F, Martinelli E, Troiani T, Troncone G, et al. SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer. Clinical cancer research. 2015; 21:4686-4697.
14. Alonso S, Hernandez D, Chang YT, Gocke CD, McCray M, Varadhan R, Matsui WH, Jones RJ, Ghiaur G. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. The Journal of clinical investigation. 2016; 126:4460-4468.
15. Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, Gendron P, Romeo AA, Morris SJ, Cormack G, Jaquith JB, Cerchietti L, Cocolakis E, Amri A, Bergeron J, Leber B, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature. 2014; 511:90-93.
16. Basset-Seguin N, Sharpe HJ, de Sauvage FJ. Efficacy of Hedgehog pathway inhibitors in Basal cell carcinoma. Molecular cancer therapeutics. 2015; 14:633-641.
17. Nozawa YI, Lin C, Chuang PT. Hedgehog signaling from the primary cilium to the nucleus: an emerging picture of ciliary localization, trafficking and transduction. Current opinion in genetics & development. 2013; 23:429-437.
18. Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010; 29:469-481.
19. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, Gabrielson KL, Matsui W, Maitra A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer research. 2007; 67:2187-2196.
20. Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, Zhang K, Conner M, Landen CN. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clinical cancer research. 2012; 18:869-881.
21. Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM, Alvarez RD, Landen CN. Smoothened antagonists reverse taxane resistance in ovarian cancer. Molecular cancer therapeutics. 2012; 11:1587-1597.
22. Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. 2003; 22:7340-7358.
23. Kim DY, Kim JH, Lee SH, Kim TY, Heo DS, Bang YJ, Kim NK. Phase II study of oxaliplatin, 5-fluorouracil and leucovorin in previously platinum-treated patients with advanced gastric cancer. Annals of oncology. 2003; 14:383-387.
24. Allegrini G, Falcone A, Di Marsico R, Lencioni M, Masi G, Galli C, Cupini S, Pfanner E, Marcucci L, Conte PF. 5-fluorouracil administered as a 48-hour semiintermittent infusion in combination with leucovorin, cisplatin and epirubicin: phase II study in advanced gastric cancer patients. American journal of clinical oncology. 2004; 27:101-105.
25. Fujiwara Y, Takiguchi S, Nakajima K, Miyata H, Yamasaki M, Kurokawa Y, Okada K, Mori M, Doki Y. Neoadjuvant intraperitoneal and systemic chemotherapy for gastric cancer patients with peritoneal dissemination. Annals of surgical oncology. 2011; 18:3726-3731.
26. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature reviews Clinical oncology. 2015; 12:445-464.
27. Fox RG, Park FD, Koechlein CS, Kritzik M, Reya T. Musashi signaling in stem cells and cancer. Annu Rev Cell Dev Biol. 2015; 31:249-267.
28. Izrailit J, Reedijk M. Developmental pathways in breast cancer and breast tumor-initiating cells: therapeutic implications. Cancer Lett. 2012; 317:115-126.
29. Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001; 411:349-354.
30. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105-111.
31. Chen W, Zhang X, Chu C, Cheung WL, Ng L, Lam S, Chow A, Lau T, Chen M, Li Y, Nie Y, Wong BC, Pang R. Identification of CD44+ cancer stem cells in human gastric cancer. Hepato-gastroenterology. 2013; 60:949-954.
32. Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, Liu F, Liu J, Dai B, Chen X, Wang F, Zeng F, Xu H, et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell research. 2012; 22:248-258.
33. Fukamachi H, Shimada S, Ito K, Ito Y, Yuasa Y. CD133 is a marker of gland-forming cells in gastric tumors and Sox17 is involved in its regulation. Cancer science. 2011; 102:1313-1321.
34. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, Masuko T, Shimizu T, Ishikawa T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer cell. 2011; 19:387-400.
35. Wang T, Ong CW, Shi J, Srivastava S, Yan B, Cheng CL, Yong WP, Chan SL, Yeoh KG, Iacopetta B, Salto-Tellez M. Sequential expression of putative stem cell markers in gastric carcinogenesis. British journal of cancer. 2011; 105:658-665.
36. Jiang J, Zhang Y, Chuai S, Wang Z, Zheng D, Xu F, Zhang Y, Li C, Liang Y, Chen Z. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene. 2012; 31:671-682.
37. Rassouli FB, Matin MM, Saeinasab M. Cancer stem cells in human digestive tract malignancies. Tumour biology. 2016; 37:7-21.
38. Dong J, Li J, Liu SM, Feng XY, Chen S, Chen YB, Zhang XS. CD33(+)/p-STAT1(+) double-positive cell as a prognostic factor for stage IIIa gastric cancer. Medical oncology. 2013; 30:442.
39. Balic M, Schwarzenbacher D, Stanzer S, Heitzer E, Auer M, Geigl JB, Cote RJ, Datar RH, Dandachi N. Genetic and epigenetic analysis of putative breast cancer stem cell models. BMC cancer. 2013; 13:358.
40. Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002; 99:507-512.
41. Palapattu GS, Wu C, Silvers CR, Martin HB, Williams K, Salamone L, Bushnell T, Huang LS, Yang Q, Huang J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate. 2009; 69:787-798.
42. Buishand FO, Arkesteijn GJ, Feenstra LR, Oorsprong CW, Mestemaker M, Starke A, Speel EJ, Kirpensteijn J, Mol JA. Identification of CD90 as Putative Cancer Stem Cell Marker and Therapeutic Target in Insulinomas. Stem Cells Dev. 2016; 25:826-835.
43. Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clinical cancer research. 2006; 12:5615-5621.
44. Jiang Y, He Y, Li H, Li HN, Zhang L, Hu W, Sun YM, Chen FL, Jin XM. Expressions of putative cancer stem cell markers ABCB1, ABCG2, and CD133 are correlated with the degree of differentiation of gastric cancer. Gastric Cancer. 2012; 15:440-450.
45. Carina V, Zito G, Pizzolanti G, Richiusa P, Criscimanna A, Rodolico V, Tomasello L, Pitrone M, Arancio W, Giordano C. Multiple pluripotent stem cell markers in human anaplastic thyroid cancer: the putative upstream role of SOX2. Thyroid. 2013; 23:829-837.
46. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015; 17:1-15.
47. Mo W, Zhang JT. Human ABCG2: structure, function, and its role in multidrug resistance. International journal of biochemistry and molecular biology. 2012; 3:1-27.
48. Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chinese journal of cancer. 2012; 31:73-99.
49. Stacy AE, Jansson PJ, Richardson DR. Molecular pharmacology of ABCG2 and its role in chemoresistance. Molecular pharmacology. 2013; 84:655-669.
50. Yoon C, Park do J, Schmidt B, Thomas NJ, Lee HJ, Kim TS, Janjigian YY, Cohen DJ, Yoon SS. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clinical cancer research. 2014; 20:3974-3988.
51. He J, Sheng T, Stelter AA, Li C, Zhang X, Sinha M, Luxon BA, Xie J. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. The Journal of biological chemistry. 2006; 281:35598-35602.
52. Singh RR, Kunkalla K, Qu C, Schlette E, Neelapu SS, Samaniego F, Vega F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene. 2011; 30:4874-4886.
53. Xu J, Peng H, Chen Q, Liu Y, Dong Z, Zhang JT. Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer research. 2007; 67:4373-4381.
54. Gonzalez-Lobato L, Real R, Prieto JG, Alvarez AI, Merino G. Differential inhibition of murine Bcrp1/Abcg2 and human BCRP/ABCG2 by the mycotoxin fumitremorgin C. European journal of pharmacology. 2010; 644:41-48.
55. Yu B, Xie J. Identifying therapeutic targets in gastric cancer: the current status and future direction. Acta biochimica et biophysica Sinica. 2016; 48:90-96.
56. Liu Z, Xu J, He J, Zheng Y, Li H, Lu Y, Qian J, Lin P, Weber DM, Yang J, Yi Q. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood. 2014; 124:2061-2071.
57. Xu M, Gong A, Yang H, George SK, Jiao Z, Huang H, Jiang X, Zhang Y. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/ Musashi-1(+) gastric cancer stem cells. Cancer letters. 2015; 369:124-133.
58. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers. 2016; 8.
59. Sarkadi B, Ozvegy-Laczka C, Nemet K, Varadi A. ABCG2 --a transporter for all seasons. FEBS letters. 2004; 567:116-120.
60. Yoon C, Cho SJ, Aksoy BA, Park do J, Schultz N, Ryeom SW, Yoon SS. Chemotherapy Resistance in Diffuse-Type Gastric Adenocarcinoma Is Mediated by RhoA Activation in Cancer Stem-Like Cells. Clinical cancer research. 2016; 22:971-983.
61. Huang S, He J, Zhang X, Bian Y, Yang L, Xie G, Zhang K, Tang W, Stelter AA, Wang Q, Zhang H, Xie J. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis. 2006; 27:1334-1340.
62. Ma X, Chen K, Huang S, Zhang X, Adegboyega PA, Evers BM, Zhang H, Xie J. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis. 2005; 26:1698-1705.
63. Sheng T, Li C, Zhang X, Chi S, He N, Chen K, McCormick F, Gatalica Z, Xie J. Activation of the hedgehog pathway in advanced prostate cancer. Molecular cancer. 2004; 3:29.
64. Gu D, Fan Q, Zhang X, Xie J. A role for transcription factor STAT3 signaling in oncogene smoothened-driven carcinogenesis. The Journal of biological chemistry. 2012; 287:38356-38366.
65. Fan Q, He M, Sheng T, Zhang X, Sinha M, Luxon B, Zhao X, Xie J. Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. The Journal of biological chemistry. 2010; 285:36570-36576.
66. Fan Q, Gu D, Liu H, Yang L, Zhang X, Yoder MC, Kaplan MH, Xie J. Defective TGF-beta signaling in bone marrow-derived cells prevents hedgehog-induced skin tumors. Cancer research. 2014; 74:471-483.
67. Gu D, Liu H, Su GH, Zhang X, Chin-Sinex H, Hanenberg H, Mendonca MS, Shannon HE, Chiorean EG, Xie J. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Molecular cancer therapeutics. 2013; 12:1038-1048.
68. Yang L, He J, Huang S, Zhang X, Bian Y, He N, Zhang H, Xie J. Activation of hedgehog signaling is not a frequent event in ovarian cancers. Molecular cancer. 2009; 8:112.