
INTRODUCTION
Numerous approaches have been developed in attempts to selectively block tumor angiogenesis or induce collapse of tumor-associated blood vessels. While initial attempts such as development of endogenous inhibitors such as angiostatin and endostatin have failed, immunological means such as passive antibodies to VEGF (Avastin) have had success in terms of regulatory clearance and marketing approval. Drawbacks of Avastin include cardiotoxicity, development of resistance, has well as relatively poor survival advantage. Conceptually a more appealing method of inducing angiogenesis blockade would involve active immunization against several tumor endothelial associated antigens in the form of a polyvalent vaccine.
One major question that arises during attempts to induce active immunity to tumor associated endothelial is the “horror autotoxicus” potential of stimulating immunity towards non-malignant endothelium. We recently reviewed numerous works in which immunization to proliferating endothelial cells, whether syngeneic, allogeneic or xenogeneic results in selectivity of killing of tumors without damage to non-malignant tissues [1]. This is a fundamental point because numerous antigens found on tumor endothelial cells are also found on non-malignant cells, for example VEGFR is known to be associated with hematopoietic stem cell self-renewal. Despite this, as reviewed, immunization with VEGFR protein or plasmid does not result in ablation of hematopoietic stem cells, as would be expected. Accordingly multiple mechanisms must be at operation that discriminate tumor endothelial from cells expressing similar markers but are not under immunological attack and destruction as a result of the immunization. This is supported by clinical data in which immunization with HUVEC cells multiple times did not result in hematopoietic or other toxicities.
While numerous attempts have been made at immunizing tumor bearing mice to endothelial antigens, the mechanistic data behind development of immunity has not been elucidated. Wei et al, for example, demonstrated involvement of antibodies targeting alpha V beta III integrin in suppression of tumor angiogenesis following immunization of mice with xenogeneic HUVEC cells [2]. Other studies have implicated T cell responses [3–5]. Indeed, in some situations not only collaboration between T cells and B cells is required for successful antitumor immunity, but also epitope spreading is observed between initial immunity towards tumor endothelium, which is subsequently followed by immunity towards tumor antigens themselves. Thus there is a high degree of variability of biological mechanisms between different active immunotherapies which target the tumor vasculature.
ValloVax is a human placental endothelial cell derived product, which has demonstrated human safety in an initial pilot clinical trial [6], as well as being shown to effectively reduce tumor growth in lung cancer, melanoma, and breast cancer [7]. In contrast to other endothelium based vaccines, ValloVax has the unique properties of: a) Large donor supply. Since ValloVax is generated from placental endothelial cells, there exists a virtually unlimited supply of placentas, and additionally, each placenta is capable of generating a large number of doses; b) ValloVax is optimized for immunogenicity by pre-treatment with interferon gamma; and c) Placental endothelial is biologically naïve, thus allowing for a higher degree of plasticity. The enhanced plasticity allows for higher levels of surface marker manipulation subsequent to treatment with cytokines.
Initial experiments performed in this report sought to confirm whether interferon gamma treatment indeed increases immunogenicity of placental endothelial cells, which was assessed by quantification of Signal I molecules (HLA) and Signal II (CD40, CD80, and CD86). Subsequently function assessment was performed in terms of T cell allostimulatory activity as well as ability to elicit cytokines associated with tumor regression. The experiments subsequently assessed whether indeed selectivity of killing was occurring, specifically, whether in vitro generated tumor endothelium-like tissue was a target for antibodies generated in vivo from ValloVax immunized mice, and whether in vivo killing of endothelial cells was occurring. At a physiological level the assessment of tumor oxygenation was performed. From an immunological perspective, the dependence of ValloVax induced tumor regression on T cells or B cells was assessed through depletion and adoptive transfer studies. Furthermore, ability to synergize with clinically-used checkpoint inhibitors was performed. The current series of studies sought to establish a mechanistic basis for ValloVax efficacy, which will serve as the foundation for future clinical development. Based on previous immunological experiments describing mechanisms of allogeneic tumor vaccines, the following conceptual framework was used for designing the experiments (Figure 1)
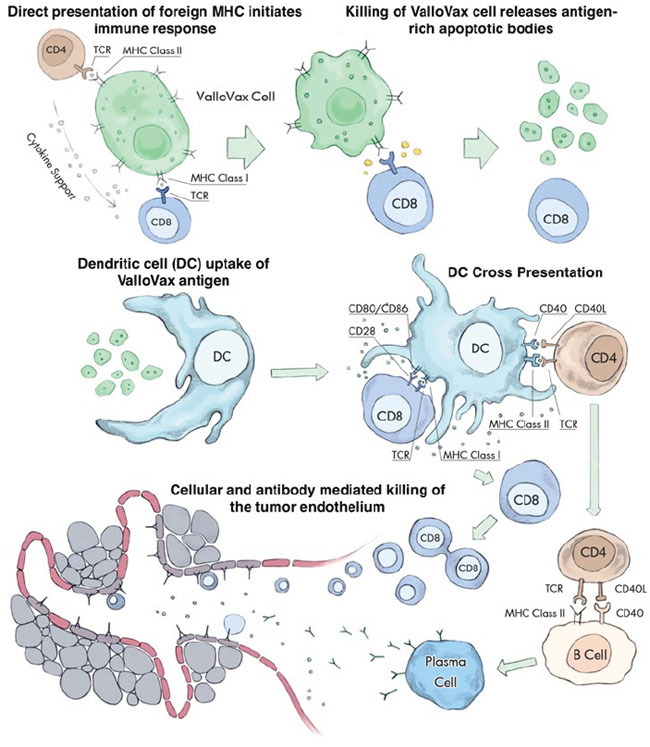
Figure 1: Multiple steps in the ValloVax-induced immune response. Initial recognition of MHC expressed on the ValloVax cell induces antigen presentation via indirect recognition. Engulfment of apoptotic bodies released from the ValloVax cells in the process of phagocytosis. Cross presentation of ValloVax antigens, inducing an antigen-specific cellular and humoral immune response. The homologies between angiogenesis targets expressed on the ValloVax cell and tumor endothelium result in a cross reaction where the immune response spreads to induce killing of the tumor vasculature.
RESULTS
Antitumor activity of ValloVax across histologically-distinct tumors
In line with the hypothesis that ValloVax induces immunity to tumor endothelium, studies were conducted to determine whether administration of the vaccine would induce antitumor responses across histological tumor types. Previously we demonstrated that ValloVax vaccination inhibits growth of lung cancer, melanoma, and breast cancer [7]. In this study we utilized the GL261 glioma model and demonstrated suppression of tumor growth (Figure 2A). Additionally, using the CT-26 colon cancer model we demonstrated regression of established tumor (Figure 2B). In this model, superior activity of ValloVax was observed as compared to inhibition of VEGFR2 inhibition, suggesting the possible potency of active immunization towards a plurality of tumor endothelium associated antigens as compared to passive antibody transfer against one tumor vasculature associated antigen. These data support the possibility that ValloVax acts either by immunizing against antigens shared by all tumors, or by targeting a process common to all tumors, such as tumor angiogenesis.
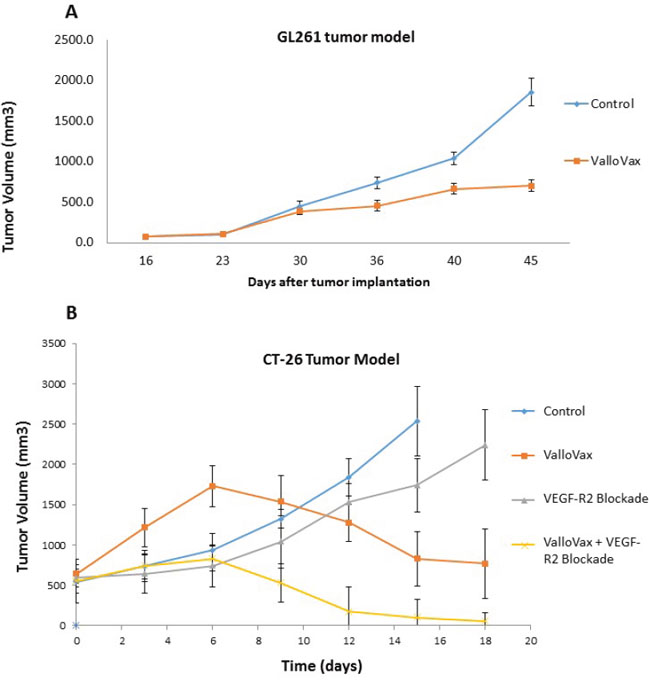
Figure 2: Efficacy of ValloVax across histologically distinct tumors. GL-261 A. or CT-26 B. tumor cells were inoculated at time of vaccination or 10 days prior to vaccination, respectively, at a concentration of 1.7 x 10(6) or 5 x 10(5) cells per mouse. For GL-261 experiments, ValloVax was administered weekly, whereas for CT-26 experiments, vaccination was administered at day 10 post tumor inoculation and day 17. Tumor growth was assessed at the indicated timepoints.
Interferon gamma pretreatment stimulates HLA and costimulatory molecule upregulation
Interferon gamma has previously demonstrated to induce upregulation of HLA I and HLA II on a variety of cell types [8–10]. In the creation of a cancer vaccine candidate the utility of allogeneic cells as immunogens has previously been reported in melanoma [11, 12], lung cancer [13, 14], renal cell cancer [15, 16], colon cancer [17, 18], and breast cancer [19–21]. It is known that correlations exist between HLA expression and immunogenicity of cancer cell vaccines [22]. Accordingly, while the rationale of pretreating cells with interferon is to upregulate immunogenicity, the actual determination of HLA upregulation, as well as effects on immunogenicity have not been reported. In a previous publication, antitumor efficacy of ValloVax treated with interferon gamma was demonstrated superior to untreated ValloVax [7], furthermore, increased expression of HLA I and II was demonstrated in endothelial cells pretreated with interferon gamma [23], but not in placental derived endothelial cells similar to ValloVax. As shown in Table 1, increased expression of HLA I and HLA II was observed in endothelial cells derived from all donor samples after treatment with interferon gamma for 48 hours. Interestingly upregulation of costimulatory molecules CD40, CD80, and CD86 was also observed.
Table 1: Upregulation of HLA and costimulatory molecules by IFN-gamma pretreatment
Flow Cytometric evaluation of markers upregulate by IFN-gamma treatment of EC |
||||||
|---|---|---|---|---|---|---|
Placenta Number |
Treatment |
HLA 1 |
HLA 2 |
CD40 |
CD80 |
CD86 |
1 |
Control |
ND |
+ |
+ |
+ |
ND |
1 |
50 U |
++ |
++ |
+++ |
++ |
+++ |
1 |
100 U |
+++ |
++ |
+++ |
++ |
+++ |
2 |
Control |
ND |
ND |
ND |
+ |
ND |
2 |
50 U |
++ |
++ |
+++ |
++ |
+ |
2 |
100 U |
+++ |
+++ |
+++ |
++ |
++ |
3 |
Control |
ND |
ND |
ND |
+ |
ND |
3 |
50 U |
++ |
++ |
+++ |
++ |
+ |
3 |
100 U |
+++ |
+++ |
+++ |
++ |
++ |
4 |
Control |
ND |
ND |
ND |
+ |
ND |
4 |
50 U |
+++ |
+++ |
+++ |
+++ |
+ |
4 |
100 U |
+++ |
+++ |
+++ |
+++ |
++ |
5 |
Control |
ND |
ND |
ND |
+ |
ND |
5 |
50 U |
++ |
+++ |
+++ |
++ |
+ |
5 |
100 U |
+++ |
+++ |
+++ |
++ |
++ |
Placental endothelial cells were isolated from 5 different donors and treated for 48 hours with dilution buffer (control) 50 or 100 IU interferon gamma. Flow cytometry was utilized to assess expression of HLA I, HLA II, as well as costimulatory molecules CD40, CD80, and CD86. Quantification was performed by mean fluorescent intensity. ND = Not Detectable compare to isotype control; + = >25% MFI compared to isotype control; ++ = > 50% MFI compared to isotype control; +++ = > 75% MFI compared to isotype control; Experiments were done in triplicate in 3 different flow cytometry runs.
Interferon gamma pretreatment of placental endothelial cells elicits hyperactive proliferative response from responding alloreactive T cells
It is known that HLA molecules present on stimulator cell are triggers of alloreactive responder T cell proliferation in mixed lymphocyte reaction [24]. Additionally, costimulatory molecule expression on stimulator cells of the mixed lymphocyte reaction also are known to enhance alloreactive T cell proliferation [25, 26]. As seen in Figure 3, significant increases in T cell proliferation were seen at all stimulator to responder ratios subsequent to 48 hour treatment of placental endothelial stimulator cells to responders.
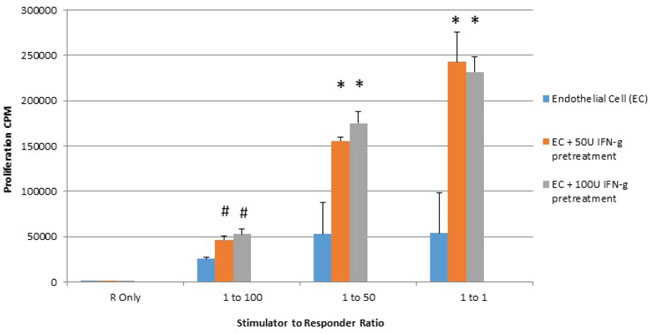
Figure 3: IFN-gamma pretreatment enhances proliferation of responding lymphocytes in mixed lymphocyte reaction (MLR). Placental endothelial cells were treated for 48 hours with dilution buffer (control) 50 or 100 IU interferon gamma and mitotically inactivated by irradiation. Cells were plated at the indicated stimulator:responder ratio in round bottom 96-well plates and cultured for 72 hours with tritiated thymidine added at the last 8 hours of culture. Proliferation was quantified by counts per minute. (*p < 0.01 as compared to control).
Interferon gamma pretreatment of placental endothelial cells elicits Th1 cytokines from responding alloreactive T cells
It is known that in mixed lymphocyte reaction, suppression of CD40, CD80, and CD86 is associated with reduction of T cell activation [27], stimulation of T regulatory cells [28], and upregulation of IL-10 [29]. Accordingly, given the upregulation of costimulatory molecules associated with interferon gamma pretreatment of placental endothelial cells, we sought to determine whether increased Th1 cytokines were produced when interferon gamma pretreated placental endothelial cells were utilized as stimulators. Interferon gamma pretreatment endowed placental endothelial cells ability to elicit upregulation of interferon gamma and IL-12 from responding T cells, as well as reduce production of IL-10. Interestingly, no modulation of IL-4 was observed (Figure 4).
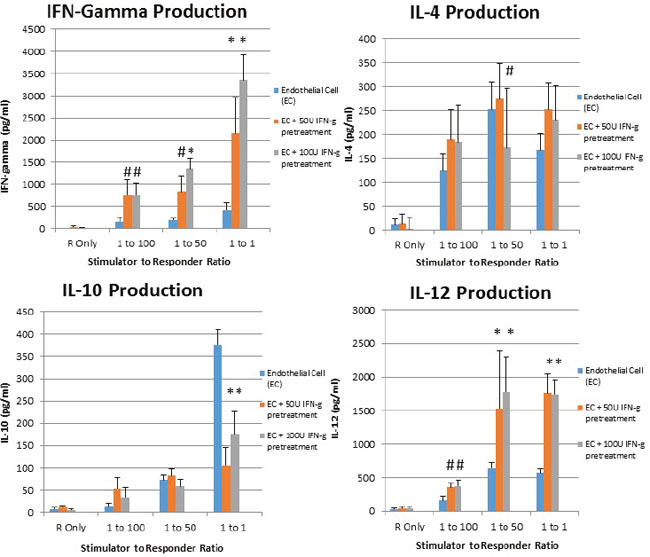
Figure 4: IFN-Gamma pretreated placental endothelial cells evoke Th1-Like response in MLR. Placental endothelial cells were treated for 48 hours with dilution buffer (control) 50 or 100 IU interferon gamma and mitotically inactivated by irradiation. Cells were plated at the indicated stimulator:responder ratio in round bottom 96-well plates and cultured for 48 hours, after which production of the indicated cytokines was assessed by ELISA (*p < 0.01 as compared to control; #p < 0.05 as compared to control)
Selectivity of immune response induced by ValloVax is specific to tumor endothelium in vitro
Several reports exist from in vitro and in vivo studies, including clinical studies that immunity can be selectively induced against tumor endothelium (reviewed in [1]). In order to assess whether immunization with ValloVax can induce immunity towards tumor endothelium, an in vitro model of tumor endothelium was created. Previous studies have reported that culture of murine endothelial cells with a cocktail of TGF-beta, IL-10 and PGE-2 was sufficient to endow TEM-1 and Fas ligand expression on non-malignant endothelium [30]. In order to replicate a tumor endothelial environment we utilized several cocktails of conditioned media from tumor cell lines (LLC (lung cancer), B16 (melanoma), RM-1 (prostate cancer), GL-261 (glioma), 4T1 (breast cancer)), as well as tumor associated cytokines (PGE-2, TGF-beta, and IL-10). Bone marrow endothelial cells cultured with these cytokines and conditioned media were assessed for similarity to tumor endothelium by quantification of expression of Fas ligand, TEM-1, and ability to induce apoptosis in PHA activated splenocytes. Interestingly, the only combination to elicit reproducible upregulation of characteristics of tumor endothelial cells was 4T1 conditioned media combined with TGF-beta. Specific criteria for identifying whether the non-malignant endothelial cells actually resembled tumor endothelium included: a) Enhanced FasL expression (MFI >2000); b) Upregulated TEM-1 expression (MFI >2000); and c) 200% increased apoptosis of PHA and antiCD3 anti CD28 activated splenocytes compared to non-treated.
To assess whether sera from immunized mice displayed preferential cytotoxicity towards in vitro generated tumor endothelium, mice were immunized 4 times with 500,000 cells of ValloVax, or control immunization with hen egg white lysozyme (HEL) at 10 ug/mouse. Immunization occurred on days 0, 7, 14, and 21, with mice being sacrificed at day 30. Sera was collected from control unimmunized mice, ValloVax immunized mice, and control HEL immunized mice. As seen in Figure 5A, no toxicity of sera was detected against healthy endothelial cells, whereas toxicity was observed against tumor-like endothelial cells in Figure 5B. Interestingly, a dose-dependent toxicity was only observed with sera from immunized but not either of the control mice.
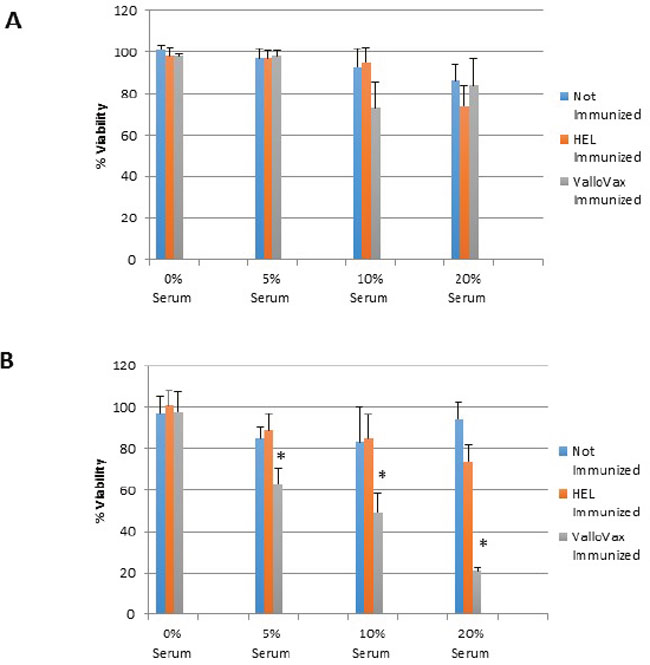
Figure 5: Selective killing of tumor endothelial cells by sera from ValloVax immunized mice. Sera was collected from C57/BL6 mice that were either: unimmunized; immunized with HEL; or immunized with ValloVax. Sera was incubated with either healthy bone marrow derived endothelium A. or bone marrow derived tumor-like endothelium B. Viability was represented as percentage of absorbance with untreated cells. Six mice per group were utilized and in vitro culture was representative of triplicate experiments performed at least 3 times (*p < 0.01 as compared to control).
Humoral responses were assessed using an in house generated ELISA for antibody responses to VEGFR1, TEM1, CD105, ROBO, FGFR2 and EGF-R. Although varying degrees of antibody responses were seen, all animals demonstrated a significant induction of antibody generation as seen in Figure 6. We confirmed expression of these antigens on placental endothelial cells prior to treatment with interferon gamma (Figure 7), and subsequent to treatment (Figure 8). In order to identify whether antibodies generated in immunized mice actually bound to ValloVax cells, sera from immunized mice was collected at various timepoints post immunization and incubated with ValloVax cells. Characterization of antibody isotype binding was performed using anti-IgG and anti-IgM secondary antibodies. As seen in Figure 9, significant binding of IgG was noted, and antibody generation was time-dependent.
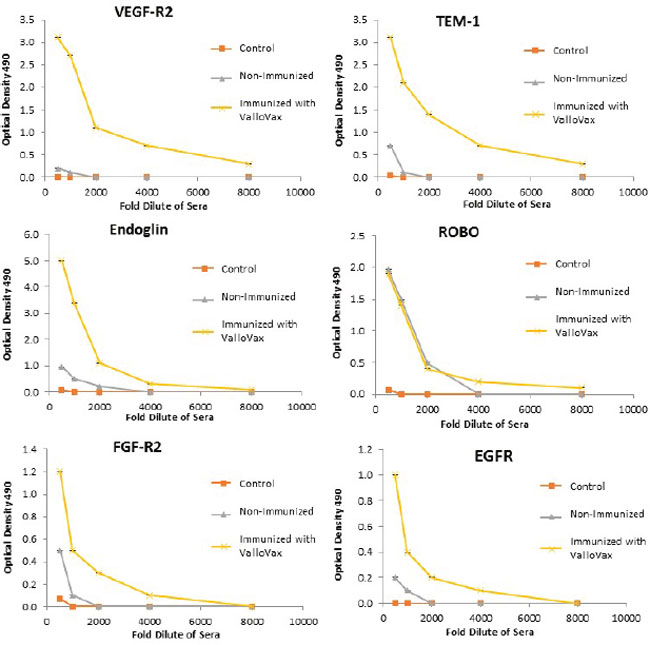
Figure 6: Antibody responses to tumor endothelial markers. Sera from ValloVax immunized mice was used as a source of antibodies for quantification using ELISA detecting binding to indicated protein. Results are from sera of 6 mice per group and performed in triplicate.
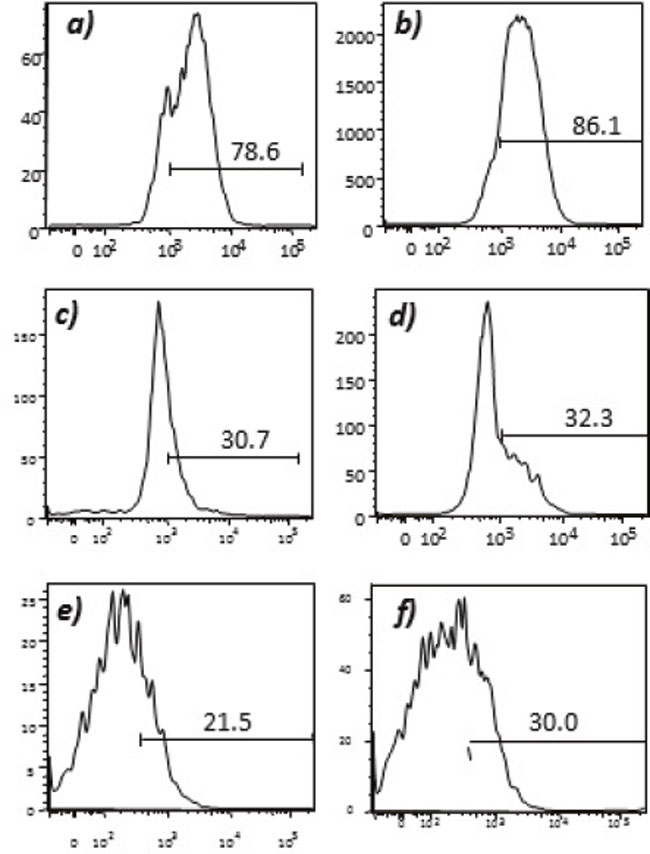
Figure 7: Expression of tumor endothelial markers on umbilical endothelial cells prior to interferon gamma treatment. Purified CD31 endothelial cells were stained with; A. anti-VEGFR2; B. TEM-1; C. Endoglin; D. ROBO; E. FGF-R2; and F. EGF-R. Cells were gated on FSC and SSC and analyzed for the respective markers.
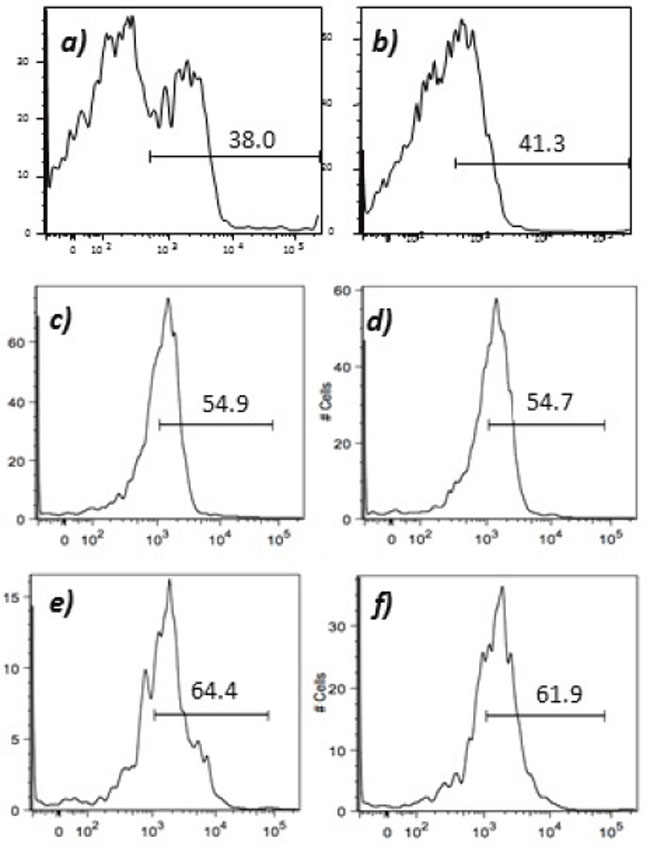
Figure 8: Expression of tumor endothelial markers on umbilical endothelial cells subsequent to interferon gamma treatment. Purified CD31 endothelial cells were cultured for 48 hours in the presence of 100 IU/ml of interferon gamma and subsequently stained with; A. anti-VEGFR2; B. TEM-1; C. Endoglin; D. ROBO; E. FGF-R2; and F. EGF-R. Cells were gated on FSC and SSC and analyzed for the respective markers.
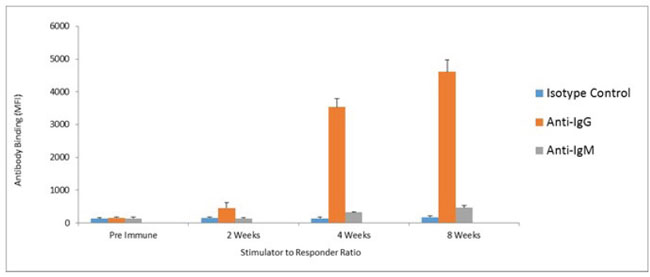
Figure 9: Assessment of class switching. ValloVax cells where fixed with paraformaldehyde 0.5% and bound to 96 well ELISA plates. Plates where incubated with sera from immunized mice, and goat anti-mouse anti-IgG and goat anti-mouse IgM where used as secondary antibodies.
In vivo killing of tumor endothelial cells
Although previous studies have demonstrated inhibition and regression of tumor growth, mechanisms of tumor regression were investigated. Animals implanted with LLC tumors were immunized and sections where made to assess apoptosis with TUNEL staining. As seen in Figure 10A, an increased number of apoptotic cells were seen within the tumor tissue. Morphologically, apoptotic cells appeared to line the area adjacent to the blood supply of the tumor. Furthermore, quantification of tumor endothelial cells by CD31 staining revealed a reduced number of endothelial cells in the tumors, which correlated with tumor growth in Figure 10B. In order to definitively elucidate whether inhibition of tumor growth was occurring as a result of reduced oxygen supply, oximetry studies were conducted. Figure 10C illustrates reduction in oxygen content of ValloVax treated tumor bearing mice as compared to control.
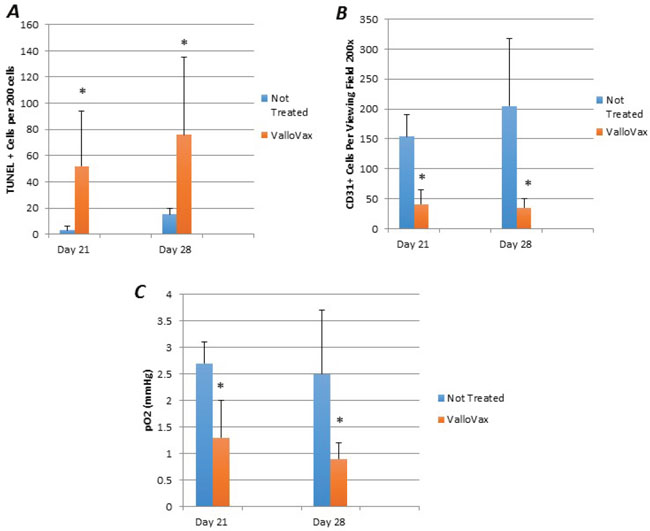
Figure 10: ValloVax induces reduction of tumor endothelial cells and reduces oxygenation. Mice bearing LLC tumors were immunized with ValloVax and apoptosis was assessed by TUNEL staining A., Endothelial cell content was assessed by CD31 staining B., and tumor oxygen content was assessed by oximetry C. Values represent 12 mice per group (*p < 0.01 as compared to control).
ValloVax mediated tumor regression requires B cells
Previous studies have demonstrated that antibodies may mediate anti-tumor endothelial responses mediated by immunization with endothelial cell antigens [31]. Accordingly, we assessed whether cytotoxicity to ValloVax cells in vitro was mediated by antibodies. To confirm whether effects were mediated by antibodies, T cells and B cells were depleted by antibody administration. As shown in Figure 11, tumor regression mediated by ValloVax was abrogated by B cell depletion but not T cell depletion, although a reduction of tumor inhibitory effects was observed in T cell depleted mice. These data suggest that there is some T cell contribution to the tumor inhibitory effect, which is in line with the classical immunological notion that B cells require T cell cytokine and costimulatory molecule support.
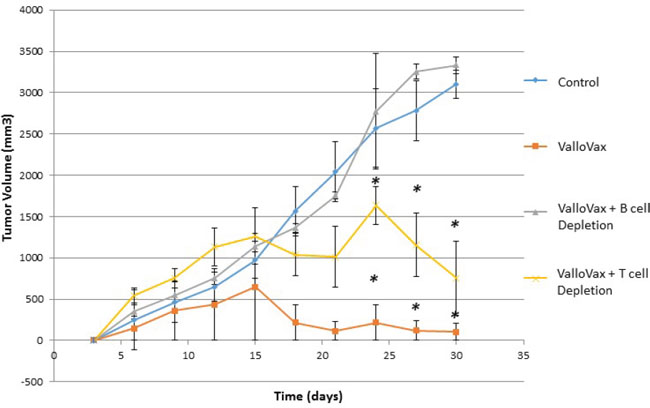
Figure 11: ValloVax mediated tumor regression is dependent on intact B cell compartment. Mice where depleted of T cells or B cells by anti-CD3, and anti-CD20 depletion prior to and subsequent to LLC tumor administration. Mice were immunized with ValloVax and tumor regression was quantified. Numbers represent 12 mice per group (*p < 0.01 as compared to control).
T cells are required for adoptive transfer of tumor endothelial immunity
Despite the need for antibodies in the induction of immune mediated tumor regression subsequent to ValloVax immunization, adoptive transfer experiments in 3 tumor models (B16, LLC, and 4T1) showed that CD3+ T cell transfer was capable of transferring immunity to all three tumors(Figure 12a–12c). This indicates that there may be a downstream activation of antigen-specific B cells subsequent to the adoptive transfer.
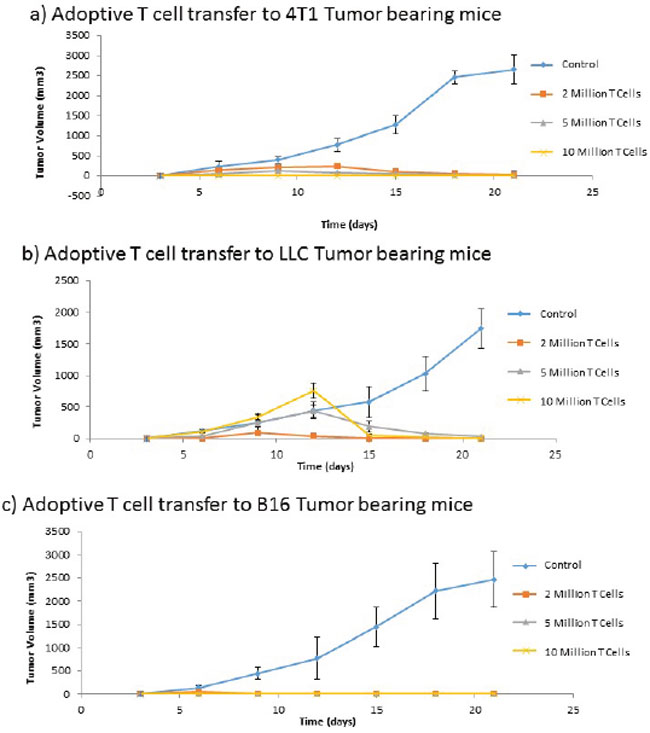
Figure 12: Adoptive transfer of immunity to ValloVax. Purified T cells were extracted from immunized non-tumor bearing mice and transferred to naive mice that were subsequently inoculated with the designated tumor (A. 4T1, B. LLC, C. B16). Tumor regression was quantified. Numbers represent 12 mice per group.
Memory T cell responses to selected tumor endothelial antigens
In order to assess whether memory T cells were generated in response to ValloVax, and whether the recall proliferative response resided in the memory or the naïve compartment, lymph node and spleen cells were harvested and separated into CD44high cells, which represent memory compartment and CD44low cells which represent naïve T cells. As seen in Figure 13, with exception of CD105 restimulation, all recall responses where higher in the memory T cell compartment for the antigens tested, which were, ValloVax, VEGFR1, VEGR2, FGFR, CD105, CD51, CD61, and TEM 1. To conclusively demonstrate induction of immunological memory subsequent to adoptive transfer, mice were rechallenged with tumor cells after initial clearance of tumors, in all cases tumors did not reestablish (Figure 14).
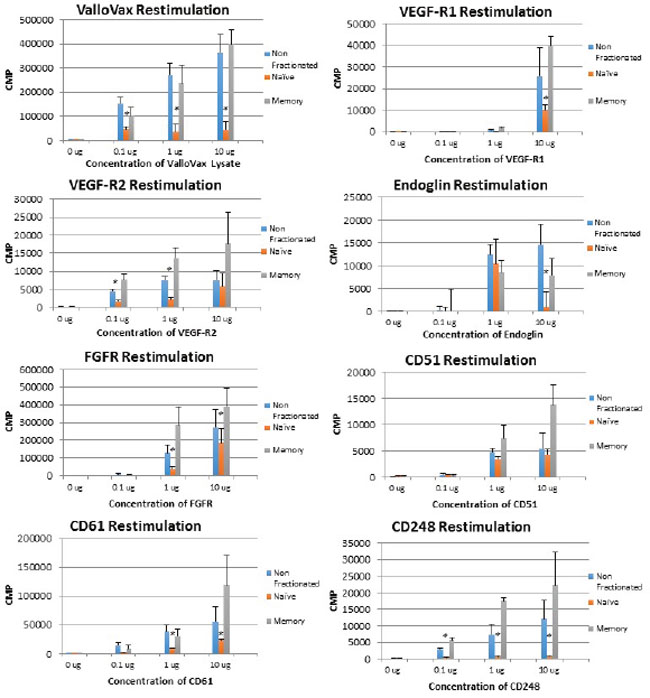
Figure 13: Memory T cell proliferation responses to tumor endothelial markers. T cells were extracted from ValloVax immunized mice and separated into memory and naïve phenotype. Proliferation in response to indicated antigen is provided in CPM. Results are from lymphatic tissue of 6 mice per group and performed in triplicate (*p < 0.01 as compared to control).
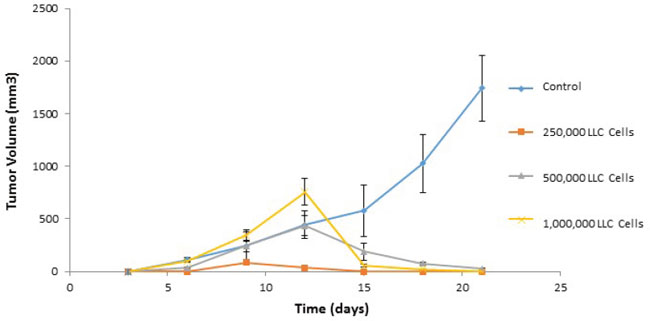
Figure 14: Adoptive transfer of immunity to naïve mice. Immunized cells were collected from ValloVax treated mice as described in Materials and Methods. Recipient mice received 2, 5, and 10 million cells intravenously at time of tumor inoculation. For tumor rechallenge experiments, immunized mice that were tumor free on day 45-50 where challenged with 250,000, 500,000 or 1,000,000 tumor cells. Tumor growth was quantified.
Stimulation of established tumor regression by combination of ValloVax with checkpoint inhibitors
Recent clinical trials have established the potency of checkpoint inhibitors against CTLA4 and PD1L in treatment of clinical tumors. Unfortunately checkpoint inhibitors possess the problem of lack of specificity to tumor tissue, thus in some cases causing autoimmunity. We sought to examine whether checkpoint inhibitors may potentiate ValloVax activity as assessed in an LLC model of established tumors. As seen in Figure 15, blockade of either CTLA4 or PD1 was not sufficient to induce regression of established tumors, whereas a synergy of effect was observed when combined with ValloVax.
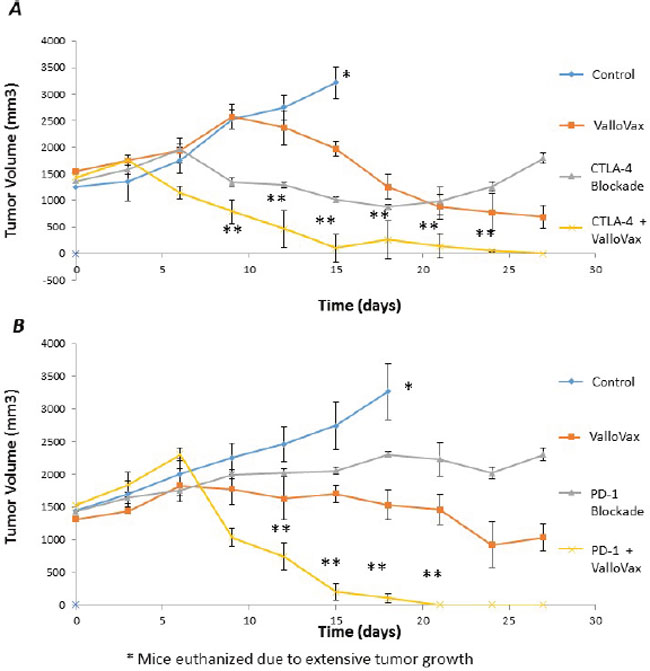
Figure 15: Enhancement of ValloVax activity by checkpoint inhibition. Mice were implanted with LLC tumors, which were allowed to establish prior to immunization with ValloVax together with anti-CTLA4 A. or Anti-PD1 B. Tumor regression was assessed and each number indicates 12 animals per group. (*Animals were euthanized because of excessive tumor growth. ** p < 0.01 as compared to control, # p < .05 as compared to control).
DISCUSSION
ValloVax is a GMP manufactured tumor endothelial-targeting immunotherapy candidate currently in clinical development by Batu Biologics for treatment of Non-Small Cell Lung Cancer (Investigational New Drug Application #16296). Previous studies have demonstrated antitumor efficacy of ValloVax in mouse models of breast and lung cancer, as well as melanoma. Confirmation of efficacy of ValloVax in tumors originating from histologically diverse tissues, particularly colon cancer and glioma, was performed in the current report. The reproducibility of this “universal vaccine” nature of ValloVax prompted us to evaluate mechanisms of ValloVax immunological activity, in part to identify cellular biomarkers that could be utilized in upcoming clinical trials, as well as to possibly explore means of augmentation of therapeutic effects. Although when compared to other tumor inhibitory immune vaccines [32–34], ValloVax appears more potent at reduction of tumor mass, translation of murine data to the clinical situation is usually characterized by loss of efficacy, and accordingly, we desired to possess possible means to augment efficacy during the clinical development of this candidate.
The concept behind generating ValloVax from placental endothelial cells was the involvement of these cells in not only creation of the vascular infrastructure supporting the maternal-fetal interphase, but also the unique immunological environment in the placenta, which resembles tumor vasculature from the perspective of both expressing FasL [35, 36], IL-10 [37, 38], TGF-beta [39], and other immune modulatory enzymes such as indolamine 2,3 deoxygenase [40]. Although other reports of endothelial cell vaccination have demonstrated immunity in humans without use of adjuvant, as quantified by antibodies to inoculation preparation [41], we sought to optimize immunogenicity of our endothelial vaccine by pretreatment with interferon gamma. In the current study found that placental endothelial cell treatment with interferon gamma increased immunogenicity of placental endothelial cells in vitro. Additionally, in vitro studies using artificially generated tumor endothelial lead to the finding of selective killing, which appeared to by IgG mediated, implying isotype and immunoglobulin class switching. The possibility of in vivo generation of antibodies suggests the feasibility of further epitope identification, as well as peptide vaccination.
We have identified several proteins that are found on ValloVax to which antibody responses are generated. These proteins comprised have previously been demonstrated to be present on tumor endothelium and have been used as targets of tumor endothelium vaccination: VEGFR2 [42–45], TEM-1 [46–48], Endoglin [49, 50], ROBO [51], FGF-R2 [5, 52], and EGF-R [53, 54]. The therapeutic induction of immune response toward the polyvalent mixture found in ValloVax suggests the possibility of synergy. Indeed, Miller et al noticed synergy between vaccination to EGF-R and VEGF-R2 at inhibiting tumor neovascularization [54]. Other have reported similar synergies in various animal models between different tumor endothelial specific antigens [55, 56]. In a previous publication we reported a pilot study of 3 solid tumor patients immunized with ValloVax. Antibody responses to VEGFR2, TEM-1, Endoglin, ROBO, FGF-R2 and EGF-R were observed. Most importantly, no elevations of toxicity associated enzymes where reported. This is of particular importance because of the theoretically possible concern of non-specific toxicity towards non-malignant endothelium or development of antigen-antibody complexes. Enzymes assessed included kidney enzymes, which were in normal ranges post immunization, which is reassuring, given that other immunotherapies which have off-target toxicity have been shown to elevate creatinine [57].
The concept of inducing immunity to a multi-valent or polyvalent immunization inoculum is superior from an immunological perspective to single antigen mediated vaccination from the perspective of tumor endothelial targeting given that recent evidence is suggesting not only tumor cells, but also tumor endothelium is capable of mutating. One study demonstrated that tumors with higher metastatic activity possess endothelial cells with higher mutating activity [58]. In some studies horizontal transfer of genetic material to endothelial cells has been demonstrated, thus causing variability of gene expression in the tumor vasculature [59].
In vivo studies demonstrated selective killing of tumor endothelial cells, through what appears to be apoptosis as demonstrated by TUNEL staining. Given that other forms of immunologically mediated cell death exist, such as autophagy [60, 61], the question is still open as to how such massive levels of tumor cell death are occurring although the TUNEL staining revealed relatively little death. One possibility is that the selective killing of endothelial cells results in regression of tumor through oxygen starvation, a possibility supported by our studies in which physiological levels of oxygen were severely reduced in tumor bearing mice.
We found that antibodies elicited to ValloVax cross reacted with major well known tumor endothelial cell antigens, thus demonstrating conclusively that potency and multivalent nature of the immune response generated. Supporting the concept of class switching during antibody development are the findings that memory T cells were obtained. Studies are underway to identify whether epitopes of the memory T cells are similar amino acid sequences to the B cell epitopes. The possibility remains that by linking T and B cell peptides together a synthetic vaccine can be generated that lacks the need for placental isolation and cell culture. One interesting observation was the transference of tumor immunity by the T cell compartment. This possibility indicates that tumor endothelial antigens are released at a basal rate from the tumor mass, which synergize with the adoptively transferred T cells. This suggests that processes already exist for immunological recognition of tumor endothelium, however T cell support is needed. Indeed other studies have shown the critical importance of T cells in supporting existing humoral tumor immune responses [62, 63]. Further support for immunological memory is the demonstration that mice which were rechallenged with tumor cells failed to develop tumors, even at concentrations of tumors as high as 10 million cells, which was observed in LLC, B16 and 4T1 systems.
Perhaps most striking were the tumor regression studies in which established tumors underwent considerable shrinkage subsequent to treatment with combination of checkpoint inhibitors and ValloVax. These data highly suggest the need to enter clinical trials with ValloVax alone, as well as in combination with classical checkpoint inhibitors. The possibility of enhancing specificity of checkpoint inhibitors while concurrently targeting the immune response to destroy tumor endothelium provides for enticing therapeutic possibilities. Another developmental possibility would be combination of ValloVax with CAR-T cell based approaches in order to augment efficacy in the treatment of solid tumors. The rationale would be that ValloVax induced anti-tumor endothelial responses would allow for enhanced intratumoral penetration of the CAR-T cells.
In conclusion, the current paper provides a mechanistic basis for utilization of ValloVax as a “second generation” Avastin in that an ongoing T cell response directed against proliferating tumor endothelium will lack the need for continued immunization, as well as possess ability to generate immunological memory and diversity, which should provide a significant therapeutic advantage to the current antiangiogenic treatments that are on the market now.
MATERIALS AND METHODS
Animals and cells
Female C57BL/6 and BALB/c mice aged 8–12 weeks were purchased from The Jackson Laboratory. Animals were housed under conventional conditions at the Animal Care Facility, University of Western Ontario, and were cared for in accordance with the guidelines established by the Canadian Council on Animal Care. A murine melanoma cell line established from a C57BL/6 mouse and designated B16F10 was obtained from the American Type Culture Collection (ATCC) and was maintained in RPMI 1640 medium (Sigma-Aldrich) with 10% FBS, l-glutamine, penicillin, and streptomycin at 37°C in 5% CO2. The murine mammary carcinoma 4 T1 cells (ATCC) were grown DMEM medium (Sigma-Aldrich) with 10% FBS, l-glutamine, penicillin, and streptomycin (complete DMEM) at 37°C in 5% CO2. Lewis Lung Carcinoma (LLC) is a murine lung carcinoma originating from C57/BL6 mice. The cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM glutamine (Gibco-BRL, Life Technologies, Inc.). The cell line was cultured at 37°C in a 5% incubator. GL261 cells murine glioblastoma cells (H-2b haplotype) were obtained from ATCC and cultured in complete DMEM media. Cells were administered into C57/BL6 mice heterotopically into the subcutaneous tissue of the mouse flank at a concentration of 1.7 x 10(6) per inoculation. CT-26 murine colorectal cancer cells (ATCC) where grown in DMEM media and administered similarly as GL261 cells, with exception that inoculation was performed in BALB/c mice.
Mixed lymphocyte reaction
Stimulator cells where placental endothelial cells that were treated for 48 hours with dilution buffer (control) 50 or 100 IU interferon gamma and mitotically inactivated by irradiation at 10 Gy. Cells were plated at the indicated stimulator:responder ratio, with 100,000 stimulators in round bottom 96-well plates and cultured for 72 hours with 1uC of tritiated thymidine added at the last 8 hours of culture. Proliferation was quantified by counts per minute.
For cytokine assessments, Quantikine ELISA kits (R & D Systems, Mannasses VA) were utilized. Conditioned media from MLR at 48 hours was used to assess cytokine production by responding cells.
GMP preparation of vaccine
ValloVax was produced under Good Manufacturing Practices (GMP) at iBiologics. Full term human placentas were collected from delivery room under informed consent. Fetal membranes were manually peeled back and the villous tissue is isolated from the placental structure. Villous tissue was subsequently washed with cold saline to remove blood and scissors used to mechanically digest the tissue. Lots of 25 grams of minced tissue were incubated with approximately 50 ml of HBSS with proprietary enzymatic digestion mixture. The supernatant collected from all three incubations was then pooled and is poured through approximately four layers of sterile gauze and through one layer of 70 micrometer polyester mesh. The suspension was subsequently mixed with 10 ml of 90% Percoll to give a final density of 1.027 g/ml and centrifuged at 550 g for 10 minutes with the centrifuge brake off. The pellet was then washed in HBSS and cells incubated for 48 hours in complete DMEM media. After 3–4 passages cells were incubating in media containing 100 IU of IFN-gamma per ml. Subsequent to incubation cells were either used: a) unmanipulated; b) used as a lysate, with 10 freeze thaw cycles in liquid nitrogen, subsequent to which lysate was filtered through a 0.2 micron filter; c) mitotically inactivated by irradiation at 10 Gy; or d) inactivated by fixation in 0.5% formalin and subsequently washed.
Immunization schedules and tumor assessment
For induction of tumor growth, 5 × 105 B16, LLC, or 4 T1 cells, American Type Culture Collection (Manassas, VA) cells were injected subcutaneously into the hind limb flank. Four weekly vaccinations of 5 × 105 test cells were administered subcutaneously on the contralateral side to which tumors were administered. Vaccination was performed on the day of tumor inoculation and on days 7, 14, and 21.
For GL261 studies, vaccination was performed on the day of tumor inoculation, followed by weekly administration. Vaccination dose was 1.7 x 10(6) cells per vaccination. For CT-26 studies, mice were immunized 10 days prior to tumor inoculation, as well as on days 10 and 17 subsequent to inoculation. Concentration of ValloVax cells was 5 x 10(5) cells per vaccination.
Tumor growth was assessed every 3 days by two measurements of perpendicular diameters by a caliper, and animals were sacrificed when tumors reached a size of 1 cm in any direction. Tumor volume was calculated by the following formula: (the shortest diameter2 × the longest diameter)/2.
Adoptive transfer and tumor rechallenge
Donor mice syngeneic to recipient mice were immunized twice with ValloVax at days 0 and 7 at a concentration of 5 x 10(5) cells per injection. Mice were euthanized at day 14 by carbon dioxide asphyxiation followed by cervical dislocation. Lymph nodes where dissected mechanically and dissociated over a .45 sterile mesh in a solution of PBS with DNase 0.05 ug/ml. Cells were centrifuged twice in 5 ml of PBS and subsequently counted for viability. Recipient mice received 2, 5, and 10 million cells intravenously at time of tumor inoculation. For tumor rechallenge experiments, immunized mice that were tumor free on day 45-50 where challenged with 250,000, 500,000 or 1,000,000 tumor cells. Tumor growth was quantified.
Memory cell assessment
C57/BL6 mice were immunized with Vallovax on days 1, 7, 14, and 21 and sacrificed on day 30. T cells were purified from single-cell suspensions of pooled lymphoid tissues (spleens and cervical, axillary, and inguinal lymph nodes). Naıve cells obtained by negative selection on CD4 T cell columns (R and D Systems), using the manufacturer’s directions modified by adding anti-CD44 antibody to the antibodies supplied with the kit. Anti-CD44 antibodies selectively bind to CD44 hi memory activated cells and allow them to be retained in the column and removed from the suspension. Memory cells were purified by adding 5 mg/ml of sodium azide-free anti-CD62L antibodies (PharMingen) to those supplied with the CD4 column kit. CD62L is expressed selectively on naıve cells, and this marker was used to remove the CD62L naïve cells from the suspension. BM derived DC where added at a concentration of 100,000 cells per well from B6 mouse in 96 well plate, antigen (ValloVax lysate or protein) was added at concentrations of 0.1, 1, 10 ug. Furthermore memory (CD44+) or naïve cells (CD44-) T cells were placed at 100,000 cells per well. The mixture was incubated for 48 hours and proliferation was assessed by tritiated thymidine (1 microcurie/well) incorporation for 6 hours. Quantification of thymidine uptake was performed by measurement of counts per minute using scintillation counting (1205 Betaplate Liquid Scintillation Counter, WALLAC Inc., Gaithersburg, MD).
Generation of in vitro tumor endothelium
BM CD31 selected cells were grown in Lonza EC media, and treated with TGF-beta (100 ng/ml) with 25% 4T1 supernatant. After culture for 48 hours, assessment of FasL and ability to induce apoptosis in PHA and anti-CD3/CD28 activated T cells was performed and compared to control BM CD31 selected cells, grown in Lonza EC media without TGF or 4T1 supernatant. These “tumor-like” EC where subsequently incubated for 48 hours with sera from immunized mice, 4 immunizations, sacrificed at day 30, used HEL immunization, same schedule as control. Assessed with ProMega Viability assay as percentage of control viability. Numbers are from 6 mice per group, in vitro done in triplicate x 3
Immunohistochemistry
4-μm paraffin sections were deparaffinized in xylol and rehydrated in graded alcohol series. Endogenous peroxidase was inhibited using 3% H2O2 in methanol. The sections were then washed in distilled water and heated in a microwave oven (in citrate buffer 10 mM, pH 6, for CD31, and EDTA 1 mM, pH 7.5, for factor VIII) 15 min for epitope retrieval. No pretreatment was needed for D2-40. Slides were incubated first with normal horse serum (1/30 avidin 10%) for CD31 and with normal goat serum (10% avidin) for factor VIII and Fli-1 for 5 min and then in biotin for 10 min. Endogenous biotin was inhibited with a Vector Blocking kit (Vector Laboratories; Burlingame, CA). The slides were then incubated at 20C for 40 min with monoclonal antibodies for CD31, the slides were incubated overnight at 8C. The slides were incubated with anti-mouse/rabbit biotinylated bridging antibodies (dilution 1/200) for 30 min. Sections were then washed and incubated with standard avidin-biotin complex (ABC; DakoCytomation, Glostrup, Denmark) for 30 min. Antibody binding was revealed using H2O2 as a substrate and diaminobenzidine as chromogen. Counterstaining was performed with hematoxylin.
Flow cytometry
Cells were suspended in phosphate buffered saline (PBS) and then incubated for 30 min at 4°C with the antibodies conjugated with FITC or PE against HLA I, HLA II, CD31, CD40, CD80, CD86, VEGFR-2, TEM-1, Endoglin, ROBO-4, FGFR-2, EGF-R and FasL. Flow cytometry analyses were performed using a FACSCalibur (BD Biosciences, San Jose, CA, USA). Acquired data were then analyzed by utilizing the Win-MDI software.
ELISA for mouse immunizations and isotype analysis
Blood was extracted from the mice (~100 μl) by puncturing the radial venous with a 26G needle (Terumo Medical Corporation, Tokyo, Japan), and using heparinized micro-hematocrit tubes (Drummond Scientific Company, Broomall, PA, USA). The blood was centrifuged at 3,200 × g for 15 min to obtain the serum, which was stored at −20°C until analysis. For ELISA high-binding Nunc™ 96-well polystyrene plates (Thermo Fisher Scientific, Inc., Waltham, MA, USA), were coated overnight with 10 μg/ml of protein (VEGFR-1, TEM-1, CD105, ROBO-4, FGF-R2, EGF-R) at 4°C in carbonate/bicarbonate buffer (0.1 M; pH 9.6; Merck, Kenilworth, NJ, USA). Titrations where performed in PBS (0.1 M; pH 7.4) containing 2% bovine serum albumin (BSA; Sigma-Aldrich, St Louis, MO, USA). The plates were incubated for 3 h at 37°C. Subsequently, polyclonal rabbit anti-mouse immunoglobulin (Ig)G antibody conjugated to peroxidase was diluted to 1:10,000 in PBS (0.1 M; pH 7.4) and 2% BSA, was added to the plates. The antigen-antibody reaction was detected by addition of o-phenylenediamine (EMD Millipore) and a H2O2 substrate (EMD Millipore), which was dissolved in disodium phosphate (0.02 M, pH 5.0; EMD Millipore). The colorimetric reaction was read at 490 nm using a plate reader (Multiskan™ GO Microplate Spectrophotometer; Thermo Fisher Scientific, Inc.).
For assessment of antibody binding an isotype class switching, ValloVax cells where fixed with paraformaldehyde 0.5% and bound to 96 well ELISA plates. Plates where incubated with sera from immunized mice, and goat anti-mouse anti-IgG and goat anti-mouse IgM where used as secondary antibodies.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
The studies were funded by Batu Biologics Inc.
REFERENCES
1. Wagner SC, Ichim TE, Ma H, Szymanski J, Perez JA, Lopez J, Bogin V, Patel AN, Marincola FM, Kesari S. Cancer anti-angiogenesis vaccines: is the tumor vasculature antigenically unique? J Transl Med. 2015; 13:340.
2. Wei YQ, Wang QR, Zhao X, Yang L, Tian L, Lu Y, Kang B, Lu CJ, Huang MJ, Lou YY, Xiao F, He QM, Shu JM, et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat Med. 2000; 6:1160–66.
3. Zhang W, Liu JN, Tan XY. Vaccination with xenogeneic tumor endothelial proteins isolated in situ inhibits tumor angiogenesis and spontaneous metastasis. Int J Cancer. 2009; 125:124–32.
4. Luo Y, Wen YJ, Ding ZY, Fu CH, Wu Y, Liu JY, Li Q, He QM, Zhao X, Jiang Y, Li J, Deng HX, Kang B, et al. Immunotherapy of tumors with protein vaccine based on chicken homologous Tie-2. Clin Cancer Res. 2006; 12:1813–19.
5. He QM, Wei YQ, Tian L, Zhao X, Su JM, Yang L, Lu Y, Kan B, Lou YY, Huang MJ, Xiao F, Liu JY, Hu B, et al. Inhibition of tumor growth with a vaccine based on xenogeneic homologous fibroblast growth factor receptor-1 in mice. J Biol Chem. 2003; 278:21831–36.
6. Wagner SC, Riordan NH, Ichim TE, Szymanski J, Ma H, Perez JA, Lopez J, Plata-Munoz JJ, Silva F, Patel AN, Kesari S. Safety of targeting tumor endothelial cell antigens. J Transl Med. 2016; 14:90.
7. Ichim TE, Li S, Ma H, Yurova YV, Szymanski JS, Patel AN, Kesari S, Min WP, Wagner SC. Induction of tumor inhibitory anti-angiogenic response through immunization with interferon Gamma primed placental endothelial cells: ValloVaxTM. J Transl Med. 2015; 13:90.
8. Brémond A, Meynet O, Mahiddine K, Coito S, Tichet M, Scotlandi K, Breittmayer JP, Gounon P, Gleeson PA, Bernard A, Bernard G. Regulation of HLA class I surface expression requires CD99 and p230/golgin-245 interaction. Blood. 2009; 113:347–57.
9. Maubach G, Lim MC, Kumar S, Zhuo L. Expression and upregulation of cathepsin S and other early molecules required for antigen presentation in activated hepatic stellate cells upon IFN-gamma treatment. Biochim Biophys Acta. 2007; 1773: 219-31.
10. Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, Turley H, Dobbs N, Gatter K, Talbot DC, Harris AL, Ganesan TS. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin Cancer Res. 2003; 9:84–92.
11. Lipson EJ, Sharfman WH, Chen S, McMiller TL, Pritchard TS, Salas JT, Sartorius-Mergenthaler S, Freed I, Ravi S, Wang H, Luber B, Sproul JD, Taube JM, et al. Safety and immunologic correlates of Melanoma GVAX, a GM-CSF secreting allogeneic melanoma cell vaccine administered in the adjuvant setting. J Transl Med. 2015; 13:214.
12. Sosman JA, Unger JM, Liu PY, Flaherty LE, Park MS, Kempf RA, Thompson JA, Terasaki PI, Sondak VK, and Southwest Oncology Group. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: impact of HLA class I antigen expression on outcome. J Clin Oncol. 2002; 20:2067–75.
13. Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, van den Heuvel MM, Lal R, Kloecker GH, Eaton KD, Chu Q, Dunlop DJ, Jain M, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015; 51:2321–29.
14. Raez LE, Cassileth PA, Schlesselman JJ, Sridhar K, Padmanabhan S, Fisher EZ, Baldie PA, Podack ER. Allogeneic vaccination with a B7.1 HLA-A gene-modified adenocarcinoma cell line in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2004; 22:2800–07.
15. Flörcken A, Grau M, Wolf A, Weilemann A, Kopp J, Dörken B, Blankenstein T, Pezzutto A, Lenz P, Lenz G, Westermann J. Gene expression profiling of peripheral blood mononuclear cells during treatment with a gene-modified allogeneic tumor cell vaccine in advanced renal cell cancer: tumor-induced immunosuppression and a possible role for NF-κB. Int J Cancer. 2015; 136:1814–26.
16. Westermann J, Flörcken A, Willimsky G, van Lessen A, Kopp J, Takvorian A, Jöhrens K, Lukowsky A, Schönemann C, Sawitzki B, Pohla H, Frank R, Dörken B, et al. Allogeneic gene-modified tumor cells (RCC-26/IL-7/CD80) as a vaccine in patients with metastatic renal cell cancer: a clinical phase-I study. Gene Ther. 2011; 18:354–63.
17. Zheng L, Edil BH, Soares KC, El-Shami K, Uram JN, Judkins C, Zhang Z, Onners B, Laheru D, Pardoll D, Jaffee EM, Schulick RD. A safety and feasibility study of an allogeneic colon cancer cell vaccine administered with a granulocyte-macrophage colony stimulating factor-producing bystander cell line in patients with metastatic colorectal cancer. Ann Surg Oncol. 2014; 21:3931–37.
18. Habal N, Gupta RK, Bilchik AJ, Yee R, Leopoldo Z, Ye W, Elashoff RM, Morton DL. CancerVax, an allogeneic tumor cell vaccine, induces specific humoral and cellular immune responses in advanced colon cancer. Ann Surg Oncol. 2001; 8:389–401.
19. Emens LA, Armstrong D, Biedrzycki B, Davidson N, Davis-Sproul J, Fetting J, Jaffee E, Onners B, Piantadosi S, Reilly RT, Stearns V, Tartakovsky I, Visvanathan K, et al. A phase I vaccine safety and chemotherapy dose-finding trial of an allogeneic GM-CSF-secreting breast cancer vaccine given in a specifically timed sequence with immunomodulatory doses of cyclophosphamide and doxorubicin. Hum Gene Ther. 2004; 15:313–37.
20. Dols A, Smith JW 2nd, Meijer SL, Fox BA, Hu HM, Walker E, Rosenheim S, Moudgil T, Doran T, Wood W, Seligman M, Alvord WG, Schoof D, et al. Vaccination of women with metastatic breast cancer, using a costimulatory gene (CD80)-modified, HLA-A2-matched, allogeneic, breast cancer cell line: clinical and immunological results. Hum Gene Ther. 2003; 14:1117–23.
21. Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, Levi J, Daphtary MM, Biedrzycki B, Wolff AC, Stearns V, Disis ML, Ye X, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol. 2009; 27:5911–18.
22. Srivatsan S, Patel JM, Bozeman EN, Imasuen IE, He S, Daniels D, Selvaraj P. Allogeneic tumor cell vaccines: the promise and limitations in clinical trials. Hum Vaccin Immunother. 2014; 10:52–63.
23. Holzinger C, Weissinger E, Zuckermann A, Imhof M, Kink F, Schöllhammer A, Kopp C, Wolner E. Effects of interleukin-1, -2, -4, -6, interferon-gamma and granulocyte/macrophage colony stimulating factor on human vascular endothelial cells. Immunol Lett. 1993; 35:109–17.
24. Bach FH, van Rood JJ. The major histocompatibility complex—genetics and biology. (First of three parts). N Engl J Med. 1976; 295:806–13.
25. Van Gool SW, Vermeiren J, Rafiq K, Lorr K, de Boer M, Ceuppens JL. Blocking CD40 - CD154 and CD80/CD86 - CD28 interactions during primary allogeneic stimulation results in T cell anergy and high IL-10 production. Eur J Immunol. 1999; 29:2367–75.
26. Vermeiren J, Ceuppens JL, Van Ghelue M, Witters P, Bullens D, Mages HW, Kroczek RA, Van Gool SW. Human T cell activation by costimulatory signal-deficient allogeneic cells induces inducible costimulator-expressing anergic T cells with regulatory cell activity. J Immunol. 2004; 172:5371–78.
27. Jiang XF, Cui ZM, Zhu L, Guo DW, Sun WY, Lin L, Wang XF, Tang YF, Liang J. CD40-CD40L costimulation blockade induced the tolerogenic dendritic cells in mouse cardiac transplant. Int Surg. 2010; 95:135–41.
28. Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med. 2001; 193:1311–18.
29. Boussiotis VA, Chen ZM, Zeller JC, Murphy WJ, Berezovskaya A, Narula S, Roncarolo MG, Blazar BR. Altered T-cell receptor + CD28-mediated signaling and blocked cell cycle progression in interleukin 10 and transforming growth factor-beta-treated alloreactive T cells that do not induce graft-versus-host disease. Blood. 2001; 97:565–71.
30. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014; 20:607–15.
31. Morera Y, Bequet-Romero M, Ayala M, Lamdán H, Agger EM, Andersen P, Gavilondo JV. Anti-tumoral effect of active immunotherapy in C57BL/6 mice using a recombinant human VEGF protein as antigen and three chemically unrelated adjuvants. Angiogenesis. 2008; 11:381–93.
32. Loukinov D, Ghochikyan A, Mkrtichyan M, Ichim TE, Lobanenkov VV, Cribbs DH, Agadjanyan MG. Antitumor efficacy of DNA vaccination to the epigenetically acting tumor promoting transcription factor BORIS and CD80 molecular adjuvant. J Cell Biochem. 2006; 98:1037–43.
33. Mkrtichyan M, Ghochikyan A, Loukinov D, Davtyan H, Ichim TE, Cribbs DH, Lobanenkov VV, Agadjanyan MG. DNA, but not protein vaccine based on mutated BORIS antigen significantly inhibits tumor growth and prolongs the survival of mice. Gene Ther. 2008; 15:61–64.
34. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. 2015; 33:55–63.
35. Pongcharoen S, Searle RF, Bulmer JN. Placental Fas and Fas ligand expression in normal early, term and molar pregnancy. Placenta. 2004; 25:321–30.
36. Ohshima K, Nakashima M, Sonoda K, Kikuchi M, Watanabe T. Expression of RCAS1 and FasL in human trophoblasts and uterine glands during pregnancy: the possible role in immune privilege. Clin Exp Immunol. 2001; 123:481–86.
37. Soeters PB, Grimble RF. The conditional role of inflammation in pregnancy and cancer. Clin Nutr. 2013; 32:460–65.
38. Bennett WA, Lagoo-Deenadayalan S, Whitworth NS, Brackin MN, Hale E, Cowan BD. Expression and production of interleukin-10 by human trophoblast: relationship to pregnancy immunotolerance. Early Pregnancy. 1997; 3:190–98.
39. Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, Alitalo K. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem. 1994; 269:6271–74.
40. Théate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, Hervé C, Gutierrez-Roelens I, Marbaix E, Sempoux C, Van den Eynde BJ. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015; 3:161–72.
41. Tanaka M, Tsuno NH, Fujii T, Todo T, Saito N, Takahashi K. Human umbilical vein endothelial cell vaccine therapy in patients with recurrent glioblastoma. Cancer Sci. 2013; 104:200–05.
42. Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, Hicklin DJ, Jaffee EM, Emens LA. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007; 13:3951–59.
43. Schmitz-Winnenthal FH, Hohmann N, Niethammer AG, Friedrich T, Lubenau H, Springer M, Breiner KM, Mikus G, Weitz J, Ulrich A, Buechler MW, Pianka F, Klaiber U, et al. Anti-angiogenic activity of VXM01, an oral T-cell vaccine against VEGF receptor 2, in patients with advanced pancreatic cancer: A randomized, placebo-controlled, phase 1 trial. OncoImmunology. 2015; 4:e1001217.
44. Hazama S, Nakamura Y, Takenouchi H, Suzuki N, Tsunedomi R, Inoue Y, Tokuhisa Y, Iizuka N, Yoshino S, Takeda K, Shinozaki H, Kamiya A, Furukawa H, et al. A phase I study of combination vaccine treatment of five therapeutic epitope-peptides for metastatic colorectal cancer; safety, immunological response, and clinical outcome. J Transl Med. 2014; 12:63.
45. Okuyama R, Aruga A, Hatori T, Takeda K, Yamamoto M. Immunological responses to a multi-peptide vaccine targeting cancer-testis antigens and VEGFRs in advanced pancreatic cancer patients. OncoImmunology. 2013; 2:e27010.
46. Diaz LA Jr, Coughlin CM, Weil SC, Fishel J, Gounder MM, Lawrence S, Azad N, O’Shannessy DJ, Grasso L, Wustner J, Ebel W, Carvajal RD. A first-in-human phase I study of MORAb-004, a monoclonal antibody to endosialin in patients with advanced solid tumors. Clin Cancer Res. 2015; 21:1281–88.
47. Ugel S, Facciponte JG, De Sanctis F, Facciabene A. Targeting tumor vasculature: expanding the potential of DNA cancer vaccines. Cancer Immunol Immunother. 2015; 64:1339–48.
48. Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G, Sehgal S, Raj A, Matthaiou E, Coukos G, Facciabene A. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J Clin Invest. 2014; 124:1497–511.
49. Duffy AG, Ulahannan SV, Cao L, Rahma OE, Makarova-Rusher OV, Kleiner DE, Fioravanti S, Walker M, Carey S, Yu Y, Venkatesan AM, Turkbey B, Choyke P, et al. A phase II study of TRC105 in patients with hepatocellular carcinoma who have progressed on sorafenib. United European Gastroenterol J. 2015; 3:453–61.
50. Ćavar S, Jelašić D, Seiwerth S, Milošević M, Hutinec Z, Mišić M. Endoglin (CD 105) as a potential prognostic factor in neuroblastoma. Pediatr Blood Cancer. 2015; 62:770–75.
51. Wang B, Xiao Y, Ding BB, Zhang N, Yuan X, Gui L, Qian KX, Duan S, Chen Z, Rao Y, Geng JG. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003; 4:19–29.
52. Plum SM, Vu HA, Mercer B, Fogler WE, Fortier AH. Generation of a specific immunological response to FGF-2 does not affect wound healing or reproduction. Immunopharmacol Immunotoxicol. 2004; 26:29–41.
53. Villaruz LC, Socinski MA. Personalized therapy for non-small cell lung cancer: which drug for which patient? Semin Thorac Cardiovasc Surg. 2011; 23:281–90.
54. Miller MJ, Foy KC, Overholser JP, Nahta R, Kaumaya PT. HER-3 peptide vaccines/mimics: combined therapy with IGF-1R, HER-2, and HER-1 peptides induces synergistic antitumor effects against breast and pancreatic cancer cells. OncoImmunology. 2014; 3:e956012.
55. Yin X, Wang W, Zhu X, Wang Y, Wu S, Wang Z, Wang L, Du Z, Gao J, Yu J. Synergistic antitumor efficacy of combined DNA vaccines targeting tumor cells and angiogenesis. Biochem Biophys Res Commun. 2015; 465:239–44.
56. Pan J, Jin P, Yan J, Kabelitz D. Anti-angiogenic active immunotherapy: a new approach to cancer treatment. Cancer Immunol Immunother. 2008; 57:1105–14.
57. Bondanza A, Zimmermann VS, Dell’Antonio G, Dal Cin E, Capobianco A, Sabbadini MG, Manfredi AA, Rovere-Querini P. Cutting edge: dissociation between autoimmune response and clinical disease after vaccination with dendritic cells. J Immunol. 2003; 170:24–27.
58. Ohga N, Ishikawa S, Maishi N, Akiyama K, Hida Y, Kawamoto T, Sadamoto Y, Osawa T, Yamamoto K, Kondoh M, Ohmura H, Shinohara N, Nonomura K, et al. Heterogeneity of tumor endothelial cells: comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am J Pathol. 2012; 180:1294–307.
59. Goldenberg DM. Horizontal transmission of malignancy by cell-cell fusion. Expert Opin Biol Ther. 2012; 12 Suppl 1:S133-9.
60. Buffen K, Oosting M, Quintin J, Ng A, Kleinnijenhuis J, Kumar V, van de Vosse E, Wijmenga C, van Crevel R, Oosterwijk E, Grotenhuis AJ, Vermeulen SH, Kiemeney LA, et al. Autophagy controls BCG-induced trained immunity and the response to intravesical BCG therapy for bladder cancer. PLoS Pathog. 2014; 10:e1004485.
61. Yan Y, Liu N, Lu L, Zang CM, Shao B, Li Y, Wen Y, Wei Y, Cheng P. Autophagy enhances antitumor immune responses induced by irradiated hepatocellular carcinoma cells engineered to express hepatitis B virus X protein. Oncol Rep. 2013; 30:993–99.
62. Noguchi M, Uemura H, Naito S, Akaza H, Yamada A, Itoh K. A phase I study of personalized peptide vaccination using 14 kinds of vaccine in combination with low-dose estramustine in HLA-A24-positive patients with castration-resistant prostate cancer. Prostate. 2011; 71:470–79.
63. Wu J, Guo Z. Improving the antigenicity of sTn antigen by modification of its sialic acid residue for development of glycoconjugate cancer vaccines. Bioconjug Chem. 2006; 17:1537–44.