
INTRODUCTION
Calcium-activated chloride channels (CaCCs) are widely expressed in various tissues and involved in important physiological processes such as epithelial secretion, smooth muscle contraction and sensory transduction [1, 2]. ANO1, also known as TMEM16A, was identified as a bona fide CaCC that mediates endogenous calcium-activated chloride current [3–5]. ANO1 is a voltage-sensitive calcium activated chloride channel that is expressed in several epithelial and nonepithelial tissues, playing important physiological roles similar to native CaCCs [6–8]. Besides the physiological significance, ANO1 is also implicated in tumorigenesis since its discovery. ANO1 gene is located within the 11q13 amplicon, one of the most frequently amplified chromosomal regions in human cancers that is associated with a poor prognosis [9, 10]. Amplification or overexpression of ANO1 has been found in several cancers, including gastrointestinal stromal tumor (GIST), head and neck squamous cell carcinoma (HNSCC), prostate cancer, breast cancer and pancreatic cancer [11–17]. The upregulation of ANO1 has also recently been reported in colon cancer and lung adenocarcinoma [18, 19], and is correlated with poor prognosis of HNSCC and breast cancer [15, 20].
Although ANO1 is considered as a potential tumor biomarker, reports on its roles in tumor progression are inconsistent. It has been shown that ANO1 promotes cell proliferation and tumor growth in HNSCC and breast cancer by activating MAPK signaling pathway and activating EGFR and CAMK signaling respectively [15, 21]. Pro-survival effects have also been shown in some cell lines such as colon cancer cell line SW620 and lung cancer cell line GLC82 [18, 19]. In HNSCC cell lines BHY, HEp-2, SCC-25 and some pancreatic cancer cell lines, ANO1 overexpression or knockdown affects cell migration rather than proliferation [14, 17, 20]. In addition, some studies have also shown that ANO1 has no effect on either cell proliferation or migration [22, 23]. These findings imply that ANO1 effect might be mediated by either same or distinct signaling pathways or cell type-dependent mechanism. Then, the questions arise as to whether different expression levels of ANO1 in different epithelial cells of the same origin differentially affect the cell proliferation and viability, and whether suppressing ANO1 expression and function can have any impact on different epithelium-originated tumor cells.
In the present study, we selected several cell lines with high level of ANO1 expression, and investigated the effect of ANO1 on these cell lines by means of lentiviral knockdown and pharmacological inhibition. We found that silencing ANO1 inhibited cell proliferation and induced apoptosis in all tested cell lines. Treatment with ANO1 inhibitor CaCCinh-A01 reduced cell viability whereas inhibitor T16Ainh-A01 had a little effect on cell viability. Both inhibitors showed inhibitory effect on cell migration. Our findings demonstrate that upregulation of ANO1 promotes cell proliferation and migration; and the pro-survival properties of ANO1 are characterized by different types of epithelial cells, suggesting that effect of ANO1 on epithelial cancer cells is likely mediated by similar signaling pathways.
RESULTS
High expression of ANO1 in prostate and colon cancer cell lines
To investigate the biological function of ANO1, we started detecting the expression levels of ANO1 in several normal and cancer cell lines. The mRNA expression of ANO1 was very low in normal breast epithelial cells MCF 10A and normal bronchial epithelial cells BEAS-2B as examined by real-time PCR. Much higher ANO1 expression was found in human keratinocyte cell line HaCaT, prostate cancer cell line PC-3, and the three colon cancer cell lines SW480, HCT116 and HT-29. ANO1 expression in these cell lines increased more than 28-fold, as compared with MCF 10A cells (Figure 1A). The protein expression of ANO1 was also detected by Western blot (Figure 1B), and quantitative analysis showed about 6-fold elevation in HaCaT and four cancer cell lines, as compared with MCF 10A and BEAS-2B cells (Figure 1C). This result is consistent with the real-time PCR analysis, further confirming the relative high expression of ANO1 in HaCaT and prostate and colon cancer cell lines.
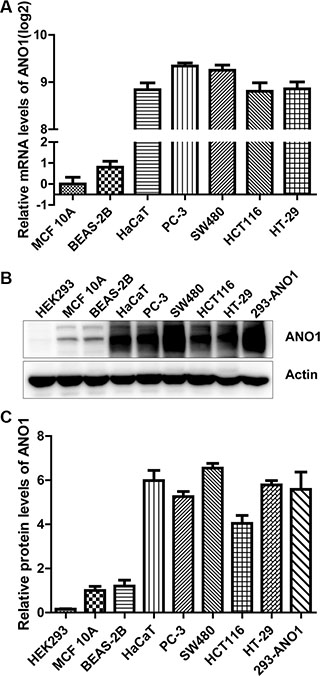
Figure 1: Comparison of ANO1 expression levels in multiple epithelial cell lines. (A) ANO1 mRNA expression in seven cell lines was determined by quantitative real-time PCR. The mRNA expression of ANO1 was much higher in human keratinocyte HaCaT, prostate cancer cell line PC-3, colon cancer cell lines SW480, HCT116 and HT-29 than that in normal breast epithelium MCF 10A and bronchial epithelial cell line BEAS-2B. (B) ANO1 protein expression in the cell lines was detected by Western blot. Higher expression of ANO1 was confirmed in cell lines of HaCaT, PC-3, SW480, HCT116 and HT-29 than MCF 10A and BEAS-2B. HEK293 cells transfected with ANO1 plasmid were used as positive control. (C) Quantitative analysis of ANO1 protein expression in cell lines from B.
Knockdown of ANO1 inhibits cell proliferation
Having screened the expression of ANO1 in the selected cell lines, we intended to evaluate its biological roles by gene silencing. Since lentivirus can transduce a wide range of cell types with high efficiency and integrate into the host genome, resulting in long-term expression of the transgene, we constructed lentiviral vectors expressing ANO1 shRNAs or control shRNA according to the sequences previously used in our lab. Lentiviral particles were used to silence the expression of ANO1 in these cell lines. Three days after infection, cells were re-plated into 96-well plates and cell proliferation was measured by CCK8 every day for six days. As shown in Figure 2A–2E, knockdown of ANO1 resulted in significant inhibition of cell proliferation in a time-dependent manner compared with control shRNA. Similar results were obtained in all cell lines tested, whether tumor cell lines or normal HaCaT cells.
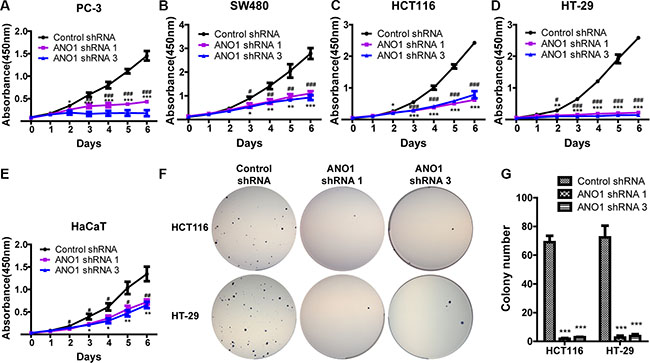
Figure 2: Silencing ANO1 inhibits cell proliferation. (A–E) Cell proliferation of the indicated cell lines was assessed by CCK8 assay. Three days after lentiviral infection of ANO1 shRNAs, cells were re-plated into 96-well plates and cell proliferation was measured every day for six days. Silencing ANO1 resulted in significant inhibition of cell viability in a time-dependent manner. Data are expressed as mean ± SEM; n = 4; #P < 0.05, ##P < 0.01 and ###P < 0.001 are for statistical comparisons of ANO1 shRNA 1 versus control shRNA; *P < 0.05, ** P < 0.01 and *** P < 0.001 are for statistical comparisons of ANO1 shRNA 3 versus control shRNA. (F) Representative images of colony formation in soft agar assay. Knockdown of ANO1 by ANO1 shRNA1 or shRNA3 decreased the colony-formation capacity of HCT116 and HT-29 as compared with control shRNA. (G) Quantitative analysis of colony numbers from F. Data are expressed as mean ± SEM; n = 3; ***P < 0.001.
To further confirm the effect of ANO1 on proliferation, we selected two colon cancer cell lines, HCT116 and HT-29, and observed the colony formation in soft agar after ANO1 silencing. Fewer colonies were found in groups expressing ANO1 shRNAs (Figure 2F). Quantitative analysis of colony number revealed that knockdown of ANO1 significantly reduced the colony formation ability of both HCT116 and HT-29 cells (Figure 2G).
Silencing ANO1 causes retardation of the cell cycle procession
The decrease of cell proliferation can result from either slow proliferation rate or increased cell death. To confirm which mechanism underlying the decrease of cell proliferation, we measured the cell cycle distribution using flow cytometry analysis. In colon cancer HT-29 cells, silencing ANO1 resulted in significant increase of the cell number at G1 phase and a decrease of the cell number at S phase. The percentage of cells in G1 phase increased from 68.08% to 84.07% and 76.02% by ANO1 shRNA 1 and shRNA 3, respectively, while the percentage of cells in S phase decreased from 24.6% to 11.72% and 12.42%, respectively (Figure 3A–3D). Similar results were obtained in PC-3 cells treated with ANO1 shRNAs (Figure 3E). These results indicated that knocking-down ANO1 arrested the cells at G1 phase of the cell cycle, thus caused the inhibitory effects on cell growth.
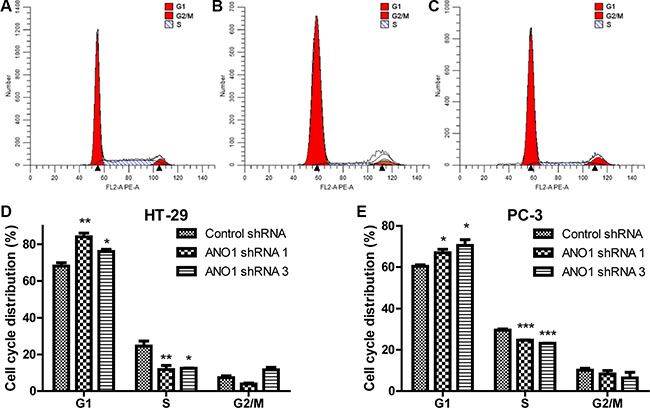
Figure 3: Flow cytometric analysis of the cell cycle distribution in colon cancer HT-29 and prostate cancer PC-3 cells. Flow cytometric analysis of the cell cycle distribution in colon cancer HT-29 cells infected with control shRNA virus (A), ANO1 shRNA 1 virus (B) and ANO1 shRNA 3 virus (C) respectively. Bar graphs showing an increase of G1 phase and a decrease of S phase in cell cycle for the percentage of indicated cells after ANO1 silencing in HT-29 (D) and PC-3 (E) cell lines. Data are expressed as mean ± SEM; n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
Cyclins and CDKs are key regulators of the cell cycle, we therefore detected the changes of these molecules at mRNA level by real-time PCR. In prostate cancer PC-3 cells, silencing ANO1 reduced the expression of cyclin D1 (CCND1), cyclin A2 (CCNA2), CDK1 and CDK2 (Figure 4A). As shown in Figure 4B, the expression of cyclin A2, CDK1 and CDK2 in colon cancer HT-29 cells was decreased upon the knockdown of ANO1 (Figure 4B). Similar reduction of cyclins and CDKs was found in colon cancer HCT116 cells (Figure 4C). We also detected the change of these molecules at protein level, and found that silencing ANO1 decreased protein expression of CDK2 and cyclin D1 in PC-3, HT-29 and HCT116 cells (Figure 4D).
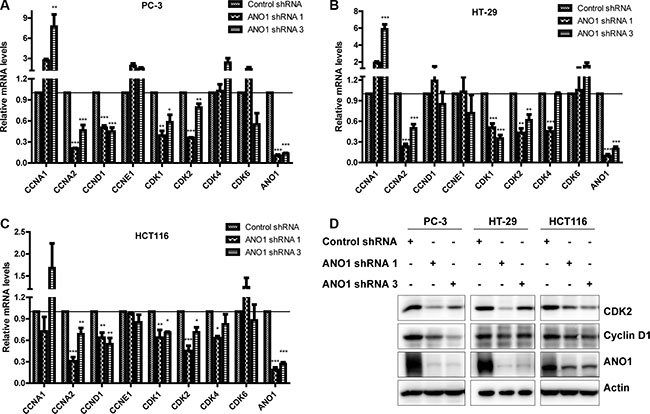
Figure 4: Analysis of mRNA and protein levels of cyclins and CDKs in PC-3, HT-29 and HCT116 cells after ANO1 knockdown. Four days after lentiviral infection of ANO1 shRNAs, the mRNA levels of several cyclins and CDKs were examined by real-time PCR. Bar graphs showing reduced expressions of cyclin D1 (CCND1), cyclin A2 (CCNA2), CDK1 and CDK2 in prostate cancer PC-3 cells (A), decreased expressions of cyclin A2, CDK1 and CDK2 in colon cancer HT-29 cells (B), and reduced expressions of cyclin D1, cyclin A2, CDK1 and CDK2 in colon cancer HCT116 cells (C). Data are expressed as mean ± SEM; n = 3–4; *P < 0.05; **P < 0.01; ***P < 0.001. (D) Western blot shows a decreased expression of CDK2 and cyclin D1 in the indicated cells after knockdown of ANO1.
Silencing ANO1 induces cell apoptosis in prostate cancer and colon cancer cells
To investigate the effects of ANO1 on cell apoptosis of cancer cell lines, we carried out an ELISA assay to determine the amount of nucleosomes in the cytoplasmic fraction of cell lysates on the fourth day after lentiviral infection of ANO1 shRNAs. As shown in Figure 5A–5D, knockdown of ANO1 induced apoptosis in prostate cancer PC-3 cells and colon cancer cell lines SW480, HCT116 and HT-29, as compared with cells expressing control shRNA. In Figure 5E, silencing ANO1 in skin HaCaT cells also resulted in an increase of apoptosis, suggesting the critical role of ANO1 in skin physiology. Caspases, a family of cysteine acid proteases, are central regulators of apoptosis. We detected the changes of some effector caspases by Western blot. We found an increased expression of full-length and cleaved caspase-7 upon knockdown of ANO1 in PC-3, HCT116 and HT-29 cells (Figure 5F). These results show that ANO1 functions as a protein that promotes cell survival.
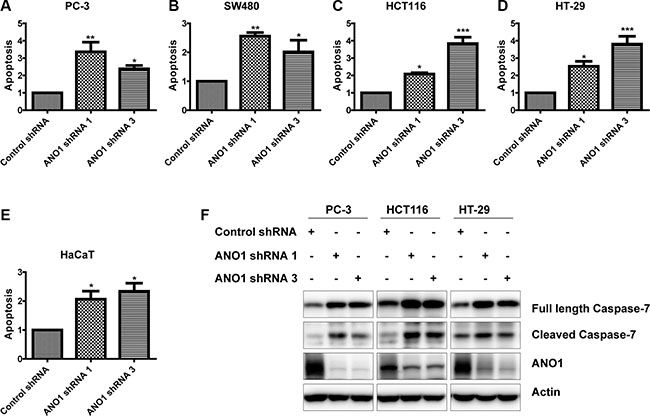
Figure 5: Knockdown of ANO1 induces cell apoptosis in prostate cancer and colon cancer cells. Four days post-infection, cell apoptosis was determined by ELISA in prostate cancer PC-3 cells (A) and colon cancer cells SW480 (B), HTC116 (C) and HT-29 (D) as well as skin HaCaT cells (E). Data are expressed as mean ± SEM; n = 3–4; *P < 0.05; **P < 0.01; ***P < 0.001. (F) Western blot showed increased Caspase-7 expression in the indicated cells after knockdown of ANO1.
ANO1 inhibitors decrease cell viability and suppress cell migration
Having shown that genetic ablation of ANO1 inhibited cell proliferation and induced cell apoptosis, we wondered whether ANO1 inhibitors could show any similar effects. Two ANO1 inhibitors, T16Ainh-A01 and CaCCinh-A01 were used to test whether the chloride channel activity of ANO1 is required for cell proliferation. Cells were treated with various concentrations of inhibitors for 72 h before cell viability was measured by CCK8 assay. The results demonstrated that T16Ainh-A01 had a little inhibitory effect on cell viability at a high concentration of 30 μM, while CaCCinh-A01 could reduce the viability of PC-3, HCT116 and HT-29 cells in a dose-dependent manner (Figure 6A–6C). The effects of CaCCinh-A01 and T16Ainh-A01 on ANO1 protein levels were examined by Western blot after treating different concentrations of ANO1 inhibitors for 72 h. CaCCinh-A01 markedly decreased ANO1 protein levels in concentration-dependent manner, whereas inhibitor T16Ainh-A01 had a weak effect (Figure 6D–6F).
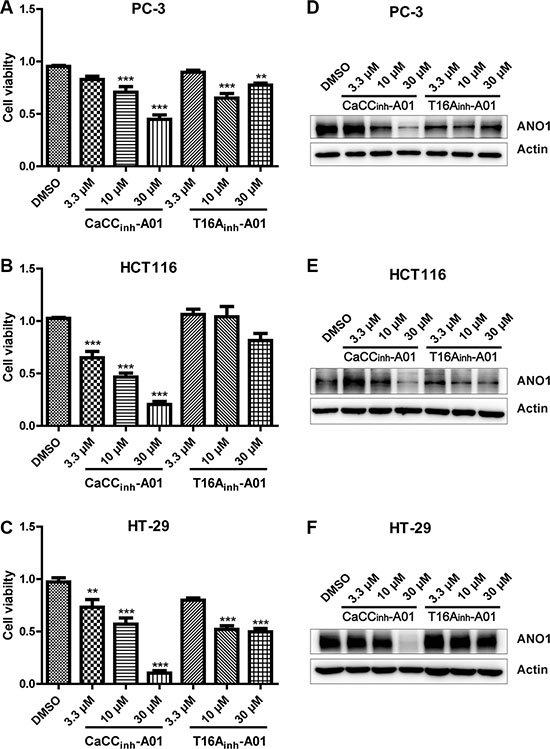
Figure 6: Effects of pharmacological inhibition of ANO1 on cell viability of PC-3, HCT116 and HT-29. Cells were treated with different concentrations of ANO1 blockers T16Ainh-A01 or CaCCinh-A01 for 72 h. Cell viability was measured by CCK8 assay. Inhibition of ANO1 with CaCCinh-A01 decreased the viability of PC-3 (A), HCT116 (B) and HT-29 (C) cells in a dose-dependent manner, whereas T16Ainh-A01 showed a weak inhibitory effect on cell viability compared with CaCCinh-A01. Data were normalized to the DMSO-treated samples. Data are expressed as mean ± SEM; n = 4; **P < 0.01; ***P < 0.001. Effects of CaCCinh-A01 and T16Ainh-A01 on ANO1 protein levels were examined by Western blot after treatment of different concentrations of inhibitors or DMSO for 72 h, and representative Western blots are shown (D–F).
In addition, we performed wound-healing assay to investigate the effects of ANO1 inhibitors on cell migration. Treatment of PC-3 cells with CaCCinh-A01 or T16Ainh-A01 inhibited the wound closure as compared to DMSO group (Figure 7A and 7B). Similar results were obtained in BEAS-2B cells (Figure 7C and 7D). Quantitative analysis of the relative migration distance showed that in PC-3 cells, 30 μM CaCCinh-A01 and 30 μM T16Ainh-A01 reduced cell migration by 80.5% and 68.4% at 36 h, while in BEAS-2B cells, the inhibitory rate was 55.6% and 50.3% respectively (Figure 7E–7H).
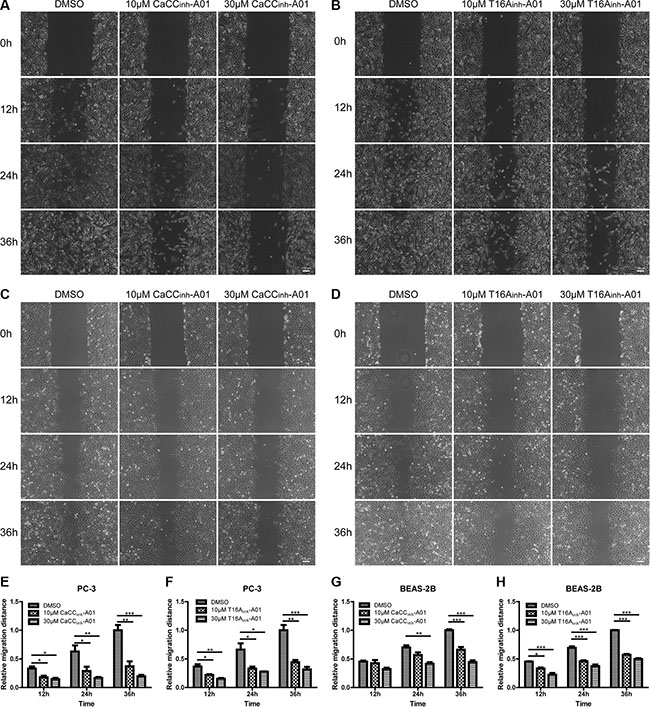
Figure 7: Suppression of cell migration of PC-3 and BEAS-2B cells by ANO1 inhibition in wound-healing assay. Cell migration was assessed by wound-healing assay in the presence of ANO1 inhibitor or DMSO control. Images of wound closure in prostate cancer PC-3 cells treated with different concentrations of CaCCinh-A01 (A) or T16Ainh-A01 (B) were taken at 0 h, 12 h, 24 h and 36 h. Representative images of bronchial epithelial BEAS-2B cells treated with CaCCinh-A01 (C) or T16Ainh-A01 (D) were presented. Scale bar: 100 μm. (E–H) Bar graphs of panel A–D showing treatment of PC-3 and BEAS-2B cells with CaCCinh-A01 or T16Ainh-A01 suppressed cell migration. Data are expressed as mean ± SEM; n = 3; * P < 0.05; ** P < 0.01;*** P < 0.001.
DISCUSSION
The purpose of this study was to investigate the role of calcium-activated chloride channel ANO1 in epithelium-originated cancer cells that highly express the channel. Previous investigations from others and ours have shown that ANO1 is amplified or overexpressed in various cancers including head and neck squamous cell carcinoma (HNSCC), prostate cancer, breast cancer, colon cancer and lung adenocarcinoma, making it a promising tumor marker [18, 19, 23]. However, there are also reports that ANO1 affects neither cell proliferation nor migration, and even inhibits cell migration [23, 24]. The inconsistent results suggest that ANO1 may exert biological effects that are cell type-dependent. In this study, we find that knockdown of ANO1 inhibits cell proliferation and induces cell apoptosis of four different epithelial tumor cell lines that highly express ANO1 channels. Consistent with our findings, ANO1 has been reported to be a regulator of proliferation in interstitial cells of Cajal (ICC), pacemaker cells of the gut. ANO1 is expressed abundantly and specifically in ICC and plays fundamental role in the generation of slow waves in gastrointestinal smooth muscles [7]. Mice lacking ANO1 had significantly fewer proliferating ICC in the small intestine, suggesting that ANO1 regulates proliferation of ICC [25]. Recently, Cha et al. found that ANO1 is essential for testosterone-induced prostate hyperplasia, and selective knockdown of ANO1 inhibits DHT-induced cell proliferation in vitro and reduces prostate enlargement in vivo [26]. This finding is consistent with our observations that ANO1 promotes proliferation of prostate cancer cells.
Although we have not extensively investigated the underling mechanism of ANO1 in proliferation, it is likely that some common signaling pathways are shared partially in these epithelium-originated cells in which ANO1 is highly expressed and promotes proliferation. Evidence from breast cancer shows that ANO1 is involved in oncogenic signaling by activating EGFR and CAMK pathways to promote cancer progression [15]. In the HNSCC cell line UM-SCC1 and colon cancer SW620 cells, ANO1 promotes cell proliferation by activating MAPK signaling for progression of cell cycle [18, 21]. Since cyclins and CDKs are key regulators of the cell cycle [27], we have also detected the changes of these molecules and the cell cycle distribution. Our results show decreased expression of some cyclins and CDKs and the cell cycle arrest at G1 phase after ANO1 knockdown, indicating that cell cycle arrest may contribute partially to the decrease of proliferation. Our observation also shows that ANO1 silencing induces apparent apoptosis, which is likely another reason responsible for decreased proliferation.
There is evidence that the chloride channel activity of ANO1 is required for its effects on cell viability. Treatment with channel inhibitors or mutations in critical residues for anion permeability abrogate cell proliferation in many cell lines including breast cancer and HNSCC [23]. However, contrary reports also show that inhibition of ANO1 function alone is not sufficient to diminish proliferation of ANO1-dependent cancer cells. The ANO1 inhibitor CaCCinh-A01 decreases cancer cell proliferation by facilitating degradation of ANO1 protein, while T16Ainh-A01, another small molecule inhibitor of ANO1, cannot inhibit the proliferation of these cell lines, although both compounds have been reported to inhibit ANO1 activity [28–31]. Structure-activity analysis reveals that both the carboxylic acid and hydrophobic bulk present in CaCCinh-A01 are necessary for its effect on cell proliferation [30]. Consistent with the latter view, we found that treatment with CaCCinh-A01 reduces cell viability in a dose-dependent manner whereas the inhibitor T16Ainh-A01 shows a little inhibitory effect on cell proliferation. Furthermore, a decrease in ANO1 protein level was also observed. It is reasonable to assume that in these cell lines CaCCinh-A01 also reduces ANO1 protein levels by facilitating endoplasmic reticulum-associated, proteasomal degradation of ANO1, thus inhibiting cell proliferation.
The different effects of the two ANO1 inhibitors on cell proliferation and migration suggest that distinct mechanism may be involved in the two processes. As discussed above, inhibition of the channel activity is not sufficient to affect cell proliferation, and ANO1 may exert its pro-survival properties via interaction with other proteins. Indeed ANO1 has been shown to associate with EGFR to facilitate the EGFR-signaling and regulate HNSCC cell proliferation [32]. The protein degradation of ANO1 induced by CaCCinh-A01 may impair this interaction, thus diminishing its pro-survival effects. The process of cell migration comprises cell swelling at the leading edge and subsequent cell shrinkage at the rear part of the cell. The increase in intracellular calcium will activate calcium-activated chloride channels and calcium-activated potassium channels at cell rear edge to release intracellular anion ions and potassium ions, resulting in shrinkage of cell volume [33, 34]. Because ANO1 is activated by cell swelling and involved in regulatory volume decrease (RVD) [35, 36], we propose that ANO1 may affect migration by regulating cell shape or volume, and inhibiting the channel activity impairs migration ability.
In summary, we have shown in this study that knockdown of ANO1 inhibits cell proliferation and induces cell apoptosis in different epithelium originated cancer cells that highly express ANO1 channels. Pharmacological inhibition of ANO1 with inhibitor CaCCinh-A01 reduces cell viability and suppresses cell migration. Our findings indicate that ANO1 promotes cell proliferation and migration, and the pro-survival properties of ANO1 are shared by different types of epithelial cancers, suggesting the critical role of calcium-activated chloride channel ANO1 in the pathophysiology of epithelial cells.
MATERIALS AND METHODS
Cell culture
Human prostate cancer cell line PC-3, human bronchial epithelial cell line BEAS-2B and colon cancer cell lines SW480, HCT116 and HT-29 were cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% FBS (Gibco). Human keratinocyte HaCaT cells and human embryonic kidney 293LTV cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS. Normal breast epithelium MCF 10A cells were maintained in DMEM/F-12 medium (Invitrogen) supplemented with 5% horse serum (Gibco), 20 ng/ml human recombinant EGF (Peprotech), 0.5 μg/ml hydrocortisone (M&C Gene Technology), 100 ng/ml cholera toxin (M&C Gene Technology), 10 μg/ml insulin (Sigma). The cells were cultured at 37°C with 5% CO2 in a humidified incubator. PC-3, 293LTV, MCF 10A cells, SW480, HCT116 and HT-29 cells were gifts from Dr. Hongquan Zhang at the Department of Anatomy, Histology and Embryology, Peking University Health Science Center. BEAS-2B cells were gifts from Dr. Bo Zhang at the Department of Pathology, Peking University Health Science Center. The HaCaT cell line was originally obtained from the American Type Culture Collection.
Constructions of lentiviral plasmids and preparations of lentivirus
The lentiviral vector pLKO.1 was used to construct ANO1 shRNAs and control shRNA plasmids. The shRNA oligos were designed according to the target sequences previously used in our lab [16]. The oligo sequences of ANO1 shRNAs and control shRNA were as follows: ANO1 shRNA 1 forward, CCGGGACGT GTACAAAGGCCAAGTACTCGAGTACTTGGCCTTTG TACACGTCTTTTTG; ANO1 shRNA 1 reverse, AATTC AAAAAGACTGTACAAAGGCCAAGTACTCGAGTAC TTGGCCTTTGTACACGTC; ANO1 shRNA 3 forward, CCGGGACGAAGAAGATGTACCACATCTCGAGATG TGGTACATCTTCTTCGTCTTTTTG; ANO1 shRNA 3 reverse, AATTCAAAAAGACGAAGAAGATGTACCA CATCTCGAGATGTGGTACATCTTCTTCGTC; control shRNA forward, CCGGGACGAGTGGTCTAGTTG AGAACTCGAGTTCTCAACTAGACCACTCGTCTTTT TG; control shRNA reverse, AATTCAAAAAGACGAGT GGTCTAGTTGAGAACTCGAGTTCTCAACTAGACC ACTCGTC. The oligos were annealed and cloned into the AgeI and EcoRI sites of pLKO.1 vector. For lentiviral production, 293LTV cells were cotransfected using Lipofectamine 2000 (Invitrogen) with pLKO.1 shRNA and packaging plasmids. The culture medium was replaced after 16 h, and supernatants were collected 48 h and 72 h after transfection. Target cells were infected with lentivirus in the presence of 8 μg/ml Polybrene (M&C Gene Technology).
RNA extraction and real-time PCR
Total RNA was isolated from cells by Trizol reagent (Invitrogen). Two micrograms of RNA was reverse transcribed to cDNA using the GoScript™ Reverse Transcription System (Promega). Real-time PCR was set up using SYBR Green mix (Promega) with the following PCR conditions: 95°C for 2 minutes; 95°C for 15 seconds, and 60°C for 1 minute, for 40 cycles. The primer sequences were as follows: ANO1 forward, GAGCCAAAGACATCGGAATCTG; ANO1 reverse, TGA AGGAGATCACGAAGGCAT; CCNA1 forward, ACATG GATGAACTAGAGCAGGG; CCNA1 reverse, GAGTGT GCCGGTGTCTACTT; CCNA2 forward, CGCTGGCG GTACTGAAGTC; CCNA2 reverse, GAGGAACGGTG ACATGCTCAT; CCND1 forward, AGAGGCGGAGGA GAACAAAC; CCND1 reverse, GGCACAGAGGGCAAC GAAG; CCNE1 forward, TCGGCCTTGTATCATTTC TCGTC; CCNE1 reverse, GCTCCCCGTCTCCCTTATAA CC; CDK1 forward, GGATGTGCTTATGCAGGATTCC; CDK1 reverse, CATGTACTGACCAGGAGGGATAG; CDK2 forward, GTGGGCCCGGCAAGATTTTAG; CDK2 reverse, GCCGAAATCCGCTTGTTAGGG; CDK4 forward, CTGGTGTTTGAGCATGTAGACC; CDK4 reverse, GATCCTTGATCGTTTCGGCTG; CDK6 forward, CCAGATGGCTCTAACCTCAGT; CDK6 reverse, AACTTCCACGAAAAAGAGGCTT; Actin forward, AGAAGGATTCCTATGTGGGCG; Actin reverse, GGATAGCACAGCCTGGATAGCA. Expression of various genes was determined by the comparative Ct method (2–ΔΔCt). All genes were normalized to Actin levels.
Western blot analysis
Cell lysates were prepared using Triton X-100 lysis buffer (150 mM NaCl, 20 mM Tris, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 10 mM EDTA) containing cocktail protease inhibitor (Roche Applied Science). Protein samples were denatured at 95°C for 5 minutes, separated by SDS-PAGE and then transferred to PVDF membranes (Millipore). After blocking with 5% milk for 1 hour, the membranes were incubated with primary antibodies against ANO1 (1:1000; ab53212, Abcam), Caspase-7 (1:1000; 12742, Cell Signaling Technology), CDK2 (1:500; sc-6248, Santa Cruz), Cyclin D1 (1:1000; 2978, Cell Signaling Technology) or actin (1:2000; TA-09, Zhongshan golden bridge) overnight at 4°C. After extensive washing in TBS buffer, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. The signal was detected using an ECL Western blotting detection system (Millipore).
Cell proliferation assay
Cells were re-plated into 96-well plates at a density of 2000 cells per well 3 days after ANO1 shRNA infection, and cell proliferation was measured after cell adherence and on the following six days by Cell Counting Kit-8 (Dojindo Laboratories, Japan). Briefly, 10 μl of the Cell Counting Kit solution was added into each well and the cells were further incubated for 2 hours at 37°C. Then the absorbance was measured at 450 nm with a microplate reader (Thermo Scientific). For ANO1 inhibitors, cell viability was determined 72 hours after treatment with different concentrations of T16Ainh–A01 (Sigma) or CaCCinh–A01 (Sigma).
Soft agar colony formation assay
Colony formation assay in soft agar was carried out in 6-well plates. The base layer was made by mixing 1.2% LMP agarose (Invitrogen) and equivalent volume of 2× medium supplemented with 20% FBS. Then cells expressing ANO1 shRNA or control shRNA were harvested and suspended in medium containing 0.4% agarose and plated over the base layer in triplicate at a density of 3000 cells per well. Three weeks later, cells were stained with NBT (Sigma) overnight in the incubator when colonies were visible. Images were taken after staining and the number of blue colonies was counted.
Cell-cycle analysis
Cells were detached using trypsin-EDTA, washed with PBS and fixed overnight in 70% ethanol at –20°C. The fixed cells were washed twice with PBS, treated with 10 μg/ml RNase A for 30 minutes at 37°C before stained with 50 μg/ml PI. The stained cells were analyzed by flow cytometry (Beckman).
Apoptosis detection by ELISA
Three days after lentiviral infection, cells were re-plated into 12-well plates at the density of 7 × 104 per well, and further incubated for one day. Then cell apoptosis was detected by Cell Death Detection ELISA kit (Roche) according to the manufacturer’s instructions. Cells were lysed with 0.5 ml of lysis buffer at room temperature for 30 minutes. Then cell lysates were centrifuged at 200 × g for 10 minutes at 4°C. Transfer 20 μl of the supernatant into the streptavidin coated MP, add 80 μl of the immunoreagent containing Anti-DNA-POD and Anti-histone-biotin to each well and incubate at room temperature for 2 hours with shaking. Wash the wells for three times, then add 100 μl ABTS and incubate for 10–20 minutes before the absorbance was measured at 405 nm.
Wound-healing assay
Cell migration was measured by in vitro wound-healing assay. Cells were seeded in 6-well plates until confluent and starved for 12 hours in serum free medium. Cells were scraped with a sterile 200 μl tip and washed twice with PBS. The cells were photographed at 0 h, 12 h, 24 h and 36 h under an inverted microscope (Olympus). The relative migration distance was determined by a ratio of average migration distance in cells treated with ANO1 inhibitors versus that in DMSO group.
Statistical analysis
All experiments were repeated for at least three times, and data were presented as mean ± SEM. Statistical analyses were performed in GraphPad Prism 5 using one-way ANOVA. P value of less than 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by research grants to K.W.W. from the Ministry of Science and Technology of China (2013CB531302 and 2014ZX09507003-006). K.W.W. wishes to thank J. M. Wang for her consistent support during this research.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005; 67:719–758.
2. Huang F, Wong X, Jan LY. International Union of Basic and Clinical Pharmacology. LXXXV: calcium-activated chloride channels. Pharmacol Rev. 2012; 64:1–15.
3. Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008; 322:590–594.
4. Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008; 134:1019–1029.
5. Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008; 455:1210–1215.
6. Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci USA. 2009; 106:21413–21418.
7. Hwang SJ, Blair PJ, Britton FC, O’Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM, Ward SM. Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol. 2009; 587:4887–4904.
8. Huang F, Zhang H, Wu M, Yang H, Kudo M, Peters CJ, Woodruff PG, Solberg OD, Donne ML, Huang X, Sheppard D, Fahy JV, Wolters PJ, et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci USA. 2012; 109:16354–16359.
9. Ormandy CJ, Musgrove EA, Hui R, Daly RJ, Sutherland RL. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat. 2003; 78:323–335.
10. Schuuring E. The involvement of the chromosome 11q13 region in human malignancies: cyclin D1 and EMS1 are two new candidate oncogenes—a review. Gene. 1995; 159:83–96.
11. Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1, that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci U S A. 2002; 99:11369–11374.
12. West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, Zhu S, Ball CA, Nielsen TO, Patel R, Goldblum JR, Brown PO, Heinrich MC, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004; 165:107–113.
13. Huang X, Godfrey TE, Gooding WE, McCarty KS, Jr. Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006; 45:1058–1069.
14. Ayoub C, Wasylyk C, Li Y, Thomas E, Marisa L, Robe A, Roux M, Abecassis J, de Reynies A, Wasylyk B. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br J Cancer. 2010; 103:715–726.
15. Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa MO, Lilley S, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci USA. 2013; 110:E1026–1034.
16. Liu W, Lu M, Liu B, Huang Y, Wang K. Inhibition of Ca(2+)-activated Cl(–) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012; 326:41–51.
17. Sauter DR, Novak I, Pedersen SF, Larsen EH, Hoffmann EK. ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflugers Arch. 2015; 467:1495–1508.
18. Sui Y, Sun M, Wu F, Yang L, Di W, Zhang G, Zhong L, Ma Z, Zheng J, Fang X, Ma T. Inhibition of TMEM16A Expression Suppresses Growth and Invasion in Human Colorectal Cancer Cells. PLoS One. 2014; 9:e115443.
19. Jia L, Liu W, Guan L, Lu M, Wang K. Inhibition of Calcium-Activated Chloride Channel ANO1/TMEM16A Suppresses Tumor Growth and Invasion in Human Lung Cancer. PLoS One. 2015; 10:e0136584.
20. Ruiz C, Martins JR, Rudin F, Schneider S, Dietsche T, Fischer CA, Tornillo L, Terracciano LM, Schreiber R, Bubendorf L, Kunzelmann K. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One. 2012; 7:e43265.
21. Duvvuri U, Shiwarski DJ, Xiao D, Bertrand C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K, Schreiber R, Seethala RS, Egloff AM, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012; 72:3270–3281.
22. Ubby I, Bussani E, Colonna A, Stacul G, Locatelli M, Scudieri P, Galietta L, Pagani F. TMEM16A alternative splicing coordination in breast cancer. Mol Cancer. 2013; 12:75.
23. Qu Z, Yao W, Yao R, Liu X, Yu K, Hartzell C. The Ca(2+) -activated Cl(−) channel, ANO1 (TMEM16A), is a double-edged sword in cell proliferation and tumorigenesis. Cancer Med. 2014; 3:453–461.
24. Shiwarski DJ, Shao C, Bill A, Kim J, Xiao D, Bertrand CA, Seethala RS, Sano D, Myers JN, Ha P, Grandis J, Gaither LA, Puthenveedu MA, et al. To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin Cancer Res. 2014; 20:4673–4688.
25. Stanich JE, Gibbons SJ, Eisenman ST, Bardsley MR, Rock JR, Harfe BD, Ordog T, Farrugia G. Ano1 as a regulator of proliferation. Am J Physiol Gastrointest Liver Physiol. 2011; 301:G1044–1051.
26. Cha JY, Wee J, Jung J, Jang Y, Lee B, Hong GS, Chang BC, Choi YL, Shin YK, Min HY, Lee HY, Na TY, Lee MO, et al. Anoctamin 1 (TMEM16A) is essential for testosterone-induced prostate hyperplasia. Proc Natl Acad Sci USA. 2015.
27. Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003; 36:131–149.
28. De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008; 73:758–768.
29. Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011; 286:2365–2374.
30. Bill A, Hall ML, Borawski J, Hodgson C, Jenkins J, Piechon P, Popa O, Rothwell C, Tranter P, Tria S, Wagner T, Whitehead L, Gaither LA. Small Molecule-facilitated Degradation of ANO1 Protein: A new targeting approach for anticancer therapeutics. J Biol Chem. 2014; 289:11029–11041.
31. Bradley E, Fedigan S, Webb T, Hollywood MA, Thornbury KD, McHale NG, Sergeant GP. Pharmacological characterization of TMEM16A currents. Channels (Austin). 2014; 8:308–320.
32. Bill A, Gutierrez A, Kulkarni S, Kemp C, Bonenfant D, Voshol H, Duvvuri U, Gaither LA. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget. 2015; 6:9173–9188. doi: 10.18632/oncotarget.3277.
33. Schwab A. Ion channels and transporters on the move. News Physiol Sci. 2001; 16:29–33.
34. Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Arch. 2007; 453:421–432.
35. Almaca J, Tian Y, Aldehni F, Ousingsawat J, Kongsuphol P, Rock JR, Harfe BD, Schreiber R, Kunzelmann K. TMEM16 proteins produce volume-regulated chloride currents that are reduced in mice lacking TMEM16A. J Biol Chem. 2009; 284:28571–28578.
36. Benedetto R, Sirianant L, Pankonien I, Wanitchakool P, Ousingsawat J, Cabrita I, Schreiber R, Amaral M, Kunzelmann K. Relationship between TMEM16A/anoctamin 1 and LRRC8A. Pflugers Arch. 2016.